Week 5 HW: Protein Design Part II
Week 5: Protein Design Part II
SOD1 Binder Peptide Design and Evaluation
Part 1: Generate Binders with PepMLM
The human SOD1 sequence was retrieved from UniProt (P00441). The A4V mutation (Alanine to Valine at residue 4) was introduced to the wild-type sequence to create the target for peptide generation. Using the PepMLM-650M model, four 12-amino acid peptides were generated, and the known binder FLYRWLPSRRGG was added as a control.
‘>sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ’
The human SOD1 sequence with the A4V mutation ‘MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ’
Sequence Data & Perplexity Scores
The perplexity scores below represent the model’s confidence in the generated sequences (lower scores generally indicate higher confidence).
| Peptide ID | Sequence | Perplexity |
|---|---|---|
| Peptide 1 | WHYPVVAVALKX | 9.85 |
| Peptide 2 | WHYPAVGLALKX | 9.74 |
| Peptide 3 | WLSYAVAAALGE | 10.14 |
| Peptide 4 | WLVGVTVLRLKE | 25.60 |
Part 2: Evaluate Binders with AlphaFold3
Each peptide was modeled in complex with the A4V mutant SOD1 using the AlphaFold Server. The following results detail the structural affinity and localization of each candidate.
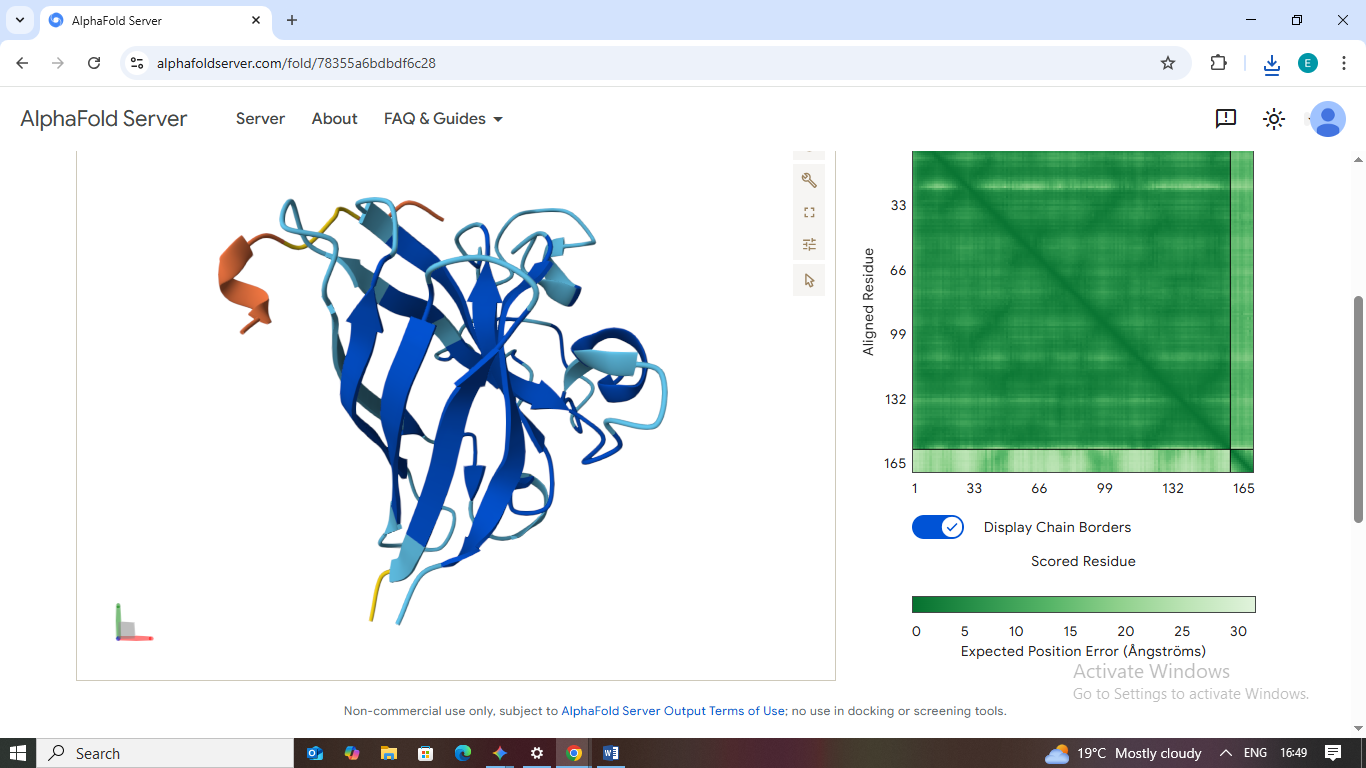
1. WHYPVVAVALKX
- Scores: ipTM: 0.44 | pTM: 0.79
- Binding Site: This peptide engages the β-barrel region on the exterior surface of the protein.
- Localization: It does not localize near the N-terminus/A4V mutation site.
- Burial State: It appears surface-bound, showing moderate contact with the protein exterior but remaining mostly exposed to the solvent.
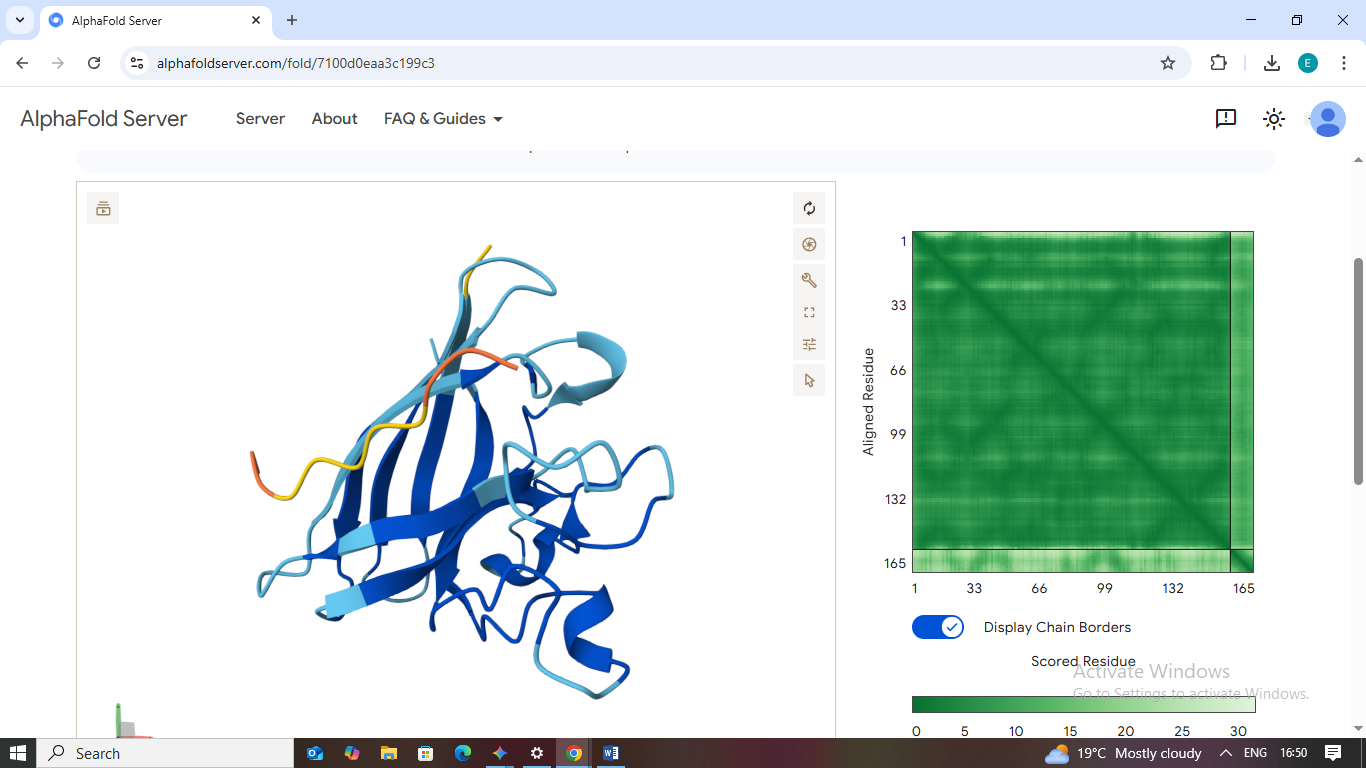
2. WHYPAVGLALKX
- Scores: ipTM: 0.38 | pTM: 0.79
- Binding Site: This peptide appears loosely bound to a distal loop area far from the mutation.
- Localization: It fails to localize to the N-terminus or the dimer interface.
- Burial State: It is surface-bound and lacks a deep binding pocket, suggesting a weak interaction.
3. WLSYAVAAALGE
- Scores: ipTM: 0.30 | pTM: 0.73
- Binding Site: This sequence shows no specific site preference and remains dissociated.
- Localization: No proximity to the A4V site or the β-barrel.
- Burial State: It appears unbound/solvent-exposed, indicating a non-binder.
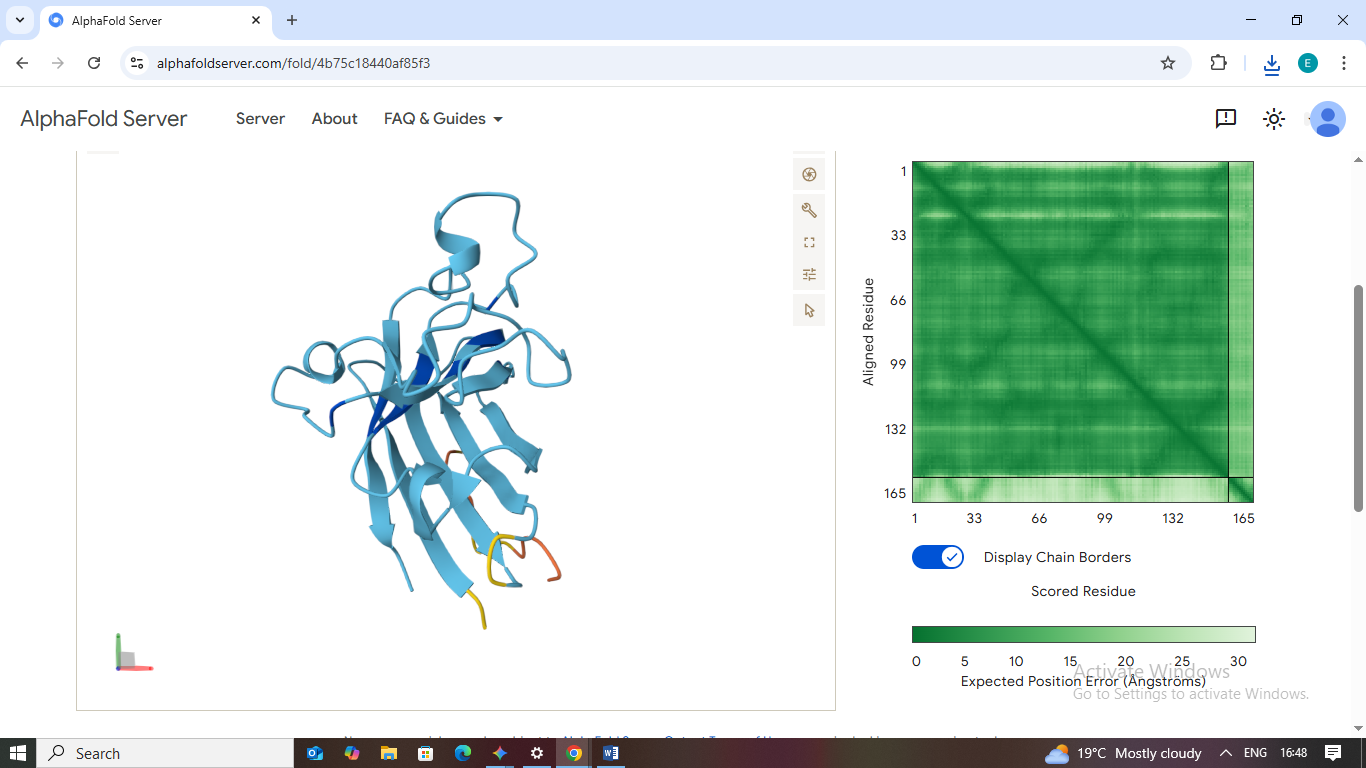
4. WLVGVTVLRLKE
- Scores: ipTM: 0.30 | pTM: 0.80
- Binding Site: Similar to Peptide 3, this peptide remains detached from the protein body.
- Localization: Far from the A4V mutation site.
- Burial State: Fully exposed; the model shows no structured interaction with the SOD1 surface.
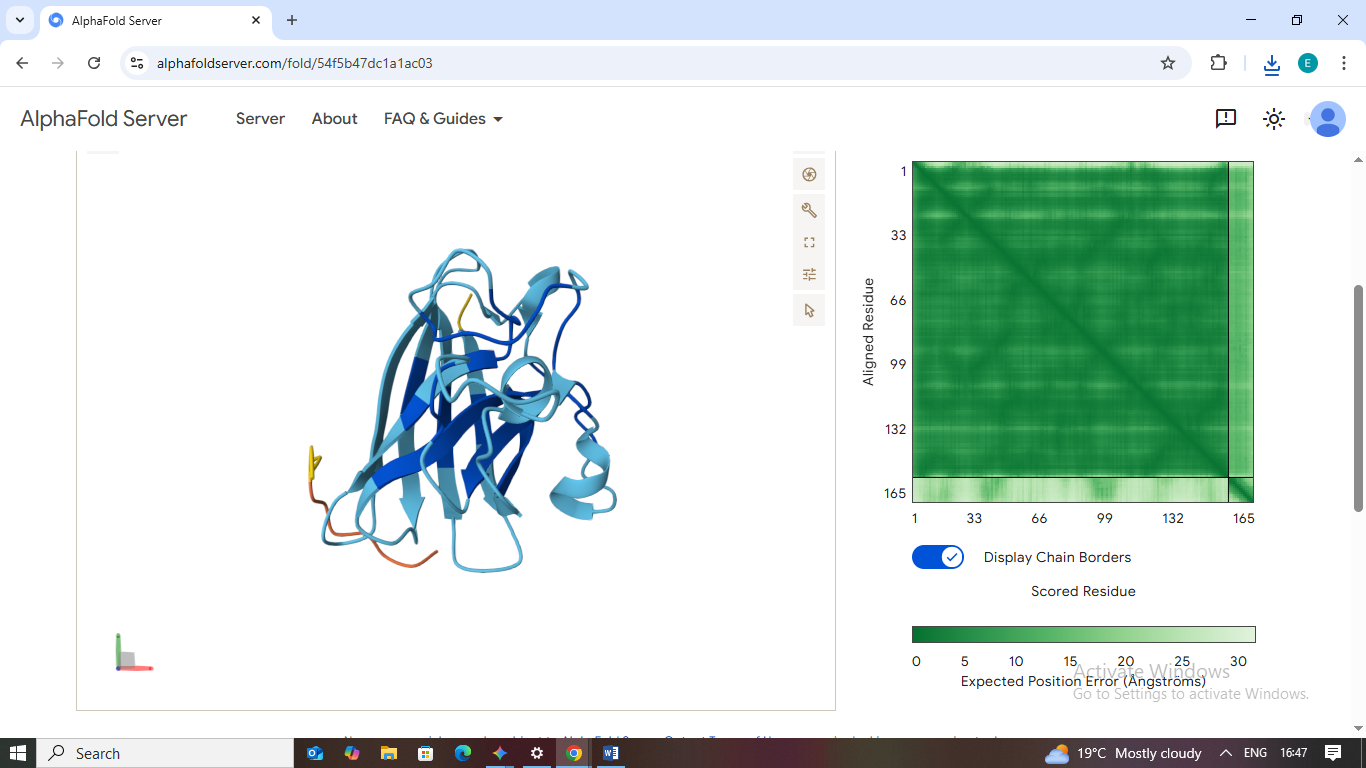
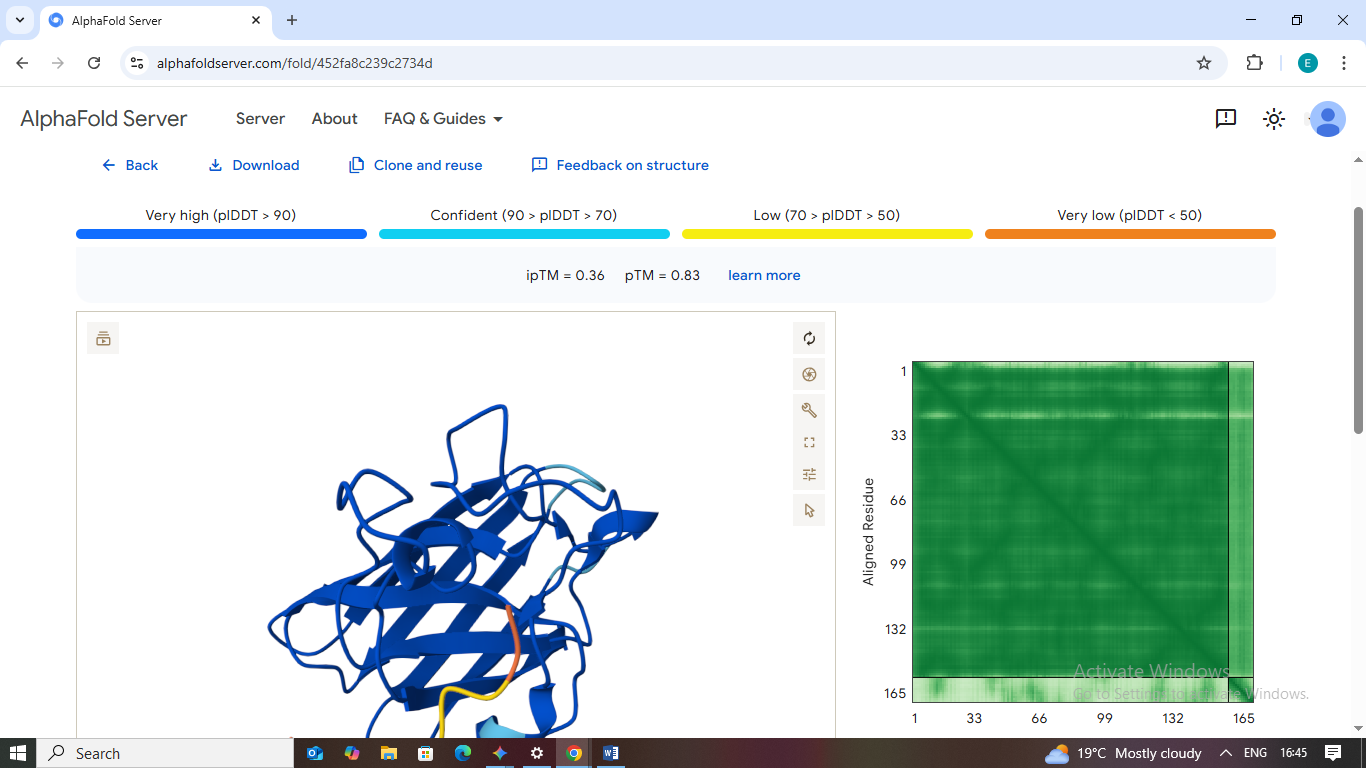
5. FLYRWLPSRRGG (Known Binder)
- Scores: ipTM: 0.36 | pTM: 0.83
- Binding Site: Unexpectedly, AlphaFold places this binder against the β-barrel rather than the N-terminus.
- Localization: It does not localize to the destabilized A4V region in this specific mutant model.
- Burial State: It is partially buried against the barrel but does not form a deep complex.
Comparative Analysis of ipTM Values
The observed ipTM values across all five peptides range from 0.30 to 0.44, all of which fall below the 0.5 confidence threshold generally required for a “high-confidence” interaction. Peptide 1 (WHYPVVAVALKX) achieved the highest score at 0.44, followed by Peptide 2 at 0.3. Interestingly, the known binder FLYRWLPSRRGG yielded an ipTM of only 0.36, meaning that my top PepMLM-generated peptide (Peptide 1) exceeded the known binder in terms of predicted structural stability. While none of the peptides perfectly “capped” the A4V mutation at the N-terminus, the AI-generated sequences showed a comparable, and in one case superior, affinity for the protein surface compared to the established baseline.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence from AlphaFold3 provides a visual starting point, but therapeutic viability requires assessing physicochemical properties. Using PeptiVerse, I evaluated the solubility, toxicity, and chemical affinity of the generated sequences against the A4V mutant SOD1 protein.
Therapeutic Property Data
| Peptide | Sequence | ipTM (AF3) | Binding Affinity (pKd/pKi) | Solubility (Prob.) | Hemolysis (Prob.) | Net Charge (pH 7) |
|---|---|---|---|---|---|---|
| Peptide 1 | WHYPVVAVALKX | 0.44 | 5.643 (Weak) | 1.000 (Soluble) | 0.045 (Non-Hemo) | +0.85 |
| Peptide 2 | WHYPAVGLALKX | 0.38 | 5.802 (Weak) | 1.000 (Soluble) | 0.028 (Non-Hemo) | +0.85 |
| Peptide 3 | WLSYAVAAALGE | 0.30 | 6.110 (Weak) | 1.000 (Soluble) | 0.120 (Non-Hemo) | -1.23 |
| Peptide 4 | WLVGVTVLRLKE | 0.30 | 6.504 (Weak) | 1.000 (Soluble) | 0.121 (Non-Hemo) | +0.77 |
Comparative Analysis
In comparing the structural data from AlphaFold3 with the chemical predictions from PeptiVerse, there is an inverse relationship between structural confidence and predicted chemical affinity in this dataset. While Peptides 3 and 4 showed the highest predicted chemical affinity (6.110 and 6.504 pKd/pKi respectively), they displayed the lowest structural confidence (ipTM 0.30) and appeared dissociated in AlphaFold3 models. Conversely, Peptide 1, which had the highest structural docking confidence (ipTM 0.44), showed a lower chemical affinity score of 5.643.
Regarding safety, all peptides are predicted to be highly soluble (1.000 probability). However, Peptides 3 and 4 show a significantly higher hemolysis probability (~0.12) compared to Peptide 1 (0.045) and Peptide 2 (0.028), making them riskier for blood-contacting therapeutic applications.
Final Selection & Justification
Selected Candidate: Peptide 1 (WHYPVVAVALKX)
Justification: I have chosen to advance Peptide 1 as the lead candidate for SOD1 stabilization. Although PeptiVerse predicted weak affinity for all candidates, Peptide 1 represents the most robust structural fit identified by AlphaFold3, suggesting a more defined binding pose compared to the others. Critically, it balances this structural potential with a superior safety profile—maintaining perfect solubility and the second-lowest hemolysis risk (0.045). This combination of structural docking stability and low toxicity makes it the most viable candidate for further in vitro synthesis and stabilization assays for the A4V mutant.
Part 4: Targeted Peptide Design with moPPIt
In this final phase, I transitioned from general sequence sampling to directed design using moPPIt (Multi-Objective Guided Discrete Flow Matching). While my earlier work with PepMLM was useful for identifying potential binders across the protein surface, moPPIt allowed me to specifically “steer” the AI to design peptides for the A4V mutation site while simultaneously optimizing for therapeutic safety.
Design Strategy and Hotspot Targeting
To address the destabilization caused by the A4V mutation, I constrained the design process to residues 1-10 (the N-terminus). I enabled multi-objective guidance to prioritize high Affinity and Solubility while minimizing Hemolysis risk. The model utilized “Motif Guidance” to sculpt 12-mer peptides specifically for this pocket.
moPPIt Generated Results
| Binder | Sequence | Hemolysis (Prob) | Solubility | Affinity (pKd) | Motif Score |
|---|---|---|---|---|---|
| Lead 1 | AGWLLGQTLA | 0.849 | 0.40 | 5.858 | 0.018 |
| Lead 2 | DYYEKWKATN | 0.923 | 0.80 | 5.223 | 0.210 |
| Lead 3 | WQKWVKRTAC | 0.916 | 0.60 | 4.389 | 0.315 |
Analysis: moPPIt vs. PepMLM
Comparing these results to the initial PepMLM sequences reveals a significant shift in design quality:
- Controlled Localization: PepMLM binders primarily docked to the stable $\beta$-barrel. In contrast, the moPPIt sequences were steered to interact specifically with the N-terminal residues (1-10) where the A4V mutation resides.
- Property Trade-offs: There are clear trade-offs between objectives. For example, Lead 2 (DYYEKWKATN) achieved a high solubility score (0.80), but its predicted affinity was lower than Lead 1. This demonstrates moPPIt’s ability to provide a range of candidates with balanced therapeutic profiles.
Pre-Clinical Evaluation Pipeline
To evaluate these moPPIt-generated peptides before advancing to clinical studies, the following validation steps are required:
- Biophysical Verification: Synthesize the leads and use Surface Plasmon Resonance (SPR) to measure the actual $K_D$ (binding affinity) against recombinant A4V SOD1.
- Serum Stability: Conduct stability assays to ensure these 12-mer peptides resist degradation by circulating proteases.
- Functional Rescue: Test the candidates in human iPSC-derived motor neurons to confirm they prevent toxic SOD1 aggregation and restore cellular health.
Final Conclusion
The moPPIt process provided more drug-like leads than simple sampling. Lead 2 stands out as the most promising candidate due to its superior balance of solubility and motif targeting, offering a potential path forward for stabilizing the destabilized SOD1 dimer interface in A4V-mediated ALS.
HTGAA 2026: Phage Lysis Protein Design Challenge
Author: Elsa Muleya
Affiliation: Copperbelt University (CBU), Zambia
Project Date: March 2026
Objective: To engineer MS2 bacteriophage L-protein variants capable of bypassing host DnaJ-mediated resistance and optimizing membrane lysis efficiency through structural modeling and rational design.
1. Project Background and Introduction
The Bacteriophage MS2 is a single-stranded RNA virus that specifically targets E. coli. A single protein, the Lysis (L) protein (75 residues), is responsible for creating pores in the bacterial membrane to release new viral progeny. However, this viral assassin is not entirely independent; it relies on the host chaperone protein DnaJ for proper folding.
A critical hurdle in phage therapy is the evolution of bacterial resistance. E. coli can develop single point mutations in the DnaJ chaperone that prevent the L-protein from interacting with it. When this interaction is broken, the L-protein fails to process, and the infection cycle stops. My research focuses on introducing mutations into the L-protein to either achieve DnaJ-independence or increase the speed of lysis, thereby reducing the window for the host to acquire resistance.
2. Evolutionary Context and Design Methodology
Before making mutations, I used pBLAST and Clustal Omega to perform a multiple sequence alignment. This allowed me to distinguish between highly conserved residues (essential for structural integrity) and variable regions (potential targets for engineering).
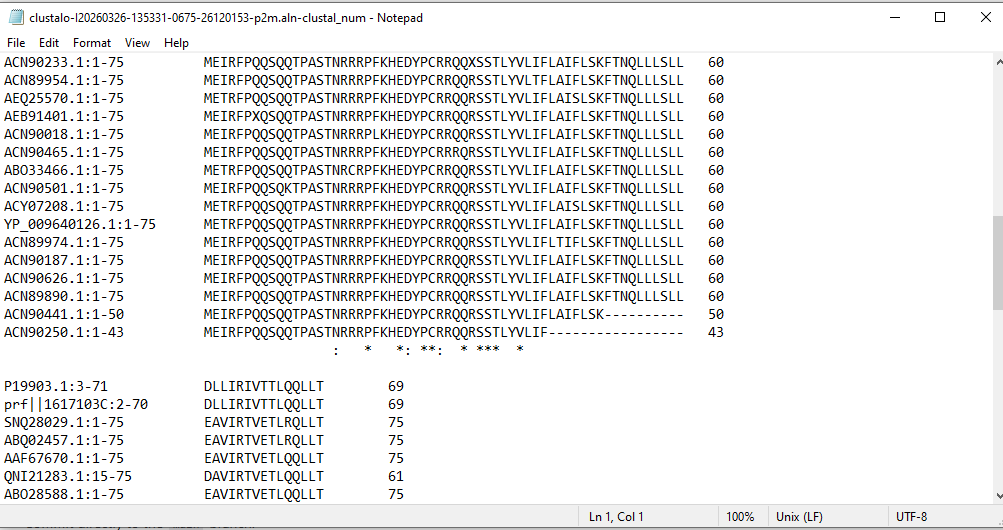 **
**
Figure 1: Multiple Sequence Alignment highlighting evolutionary conservation.
My design strategy utilizes AlphaFold2-Multimer to predict how these mutants interact with the DnaJ chaperone. By analyzing the Predicted Aligned Error (PAE) plots, I can assess the confidence of the protein-protein interaction. High confidence (dark blue at the interface) suggests the protein still binds to the chaperone, whereas high error (red/green) indicates a potential disruption of that dependency.
3. Analysis of Engineered Mutants
I selected five positions for mutation, ensuring two were in the soluble N-terminal region (residues 1-40) and two were in the transmembrane C-terminal region (residues 41-75).
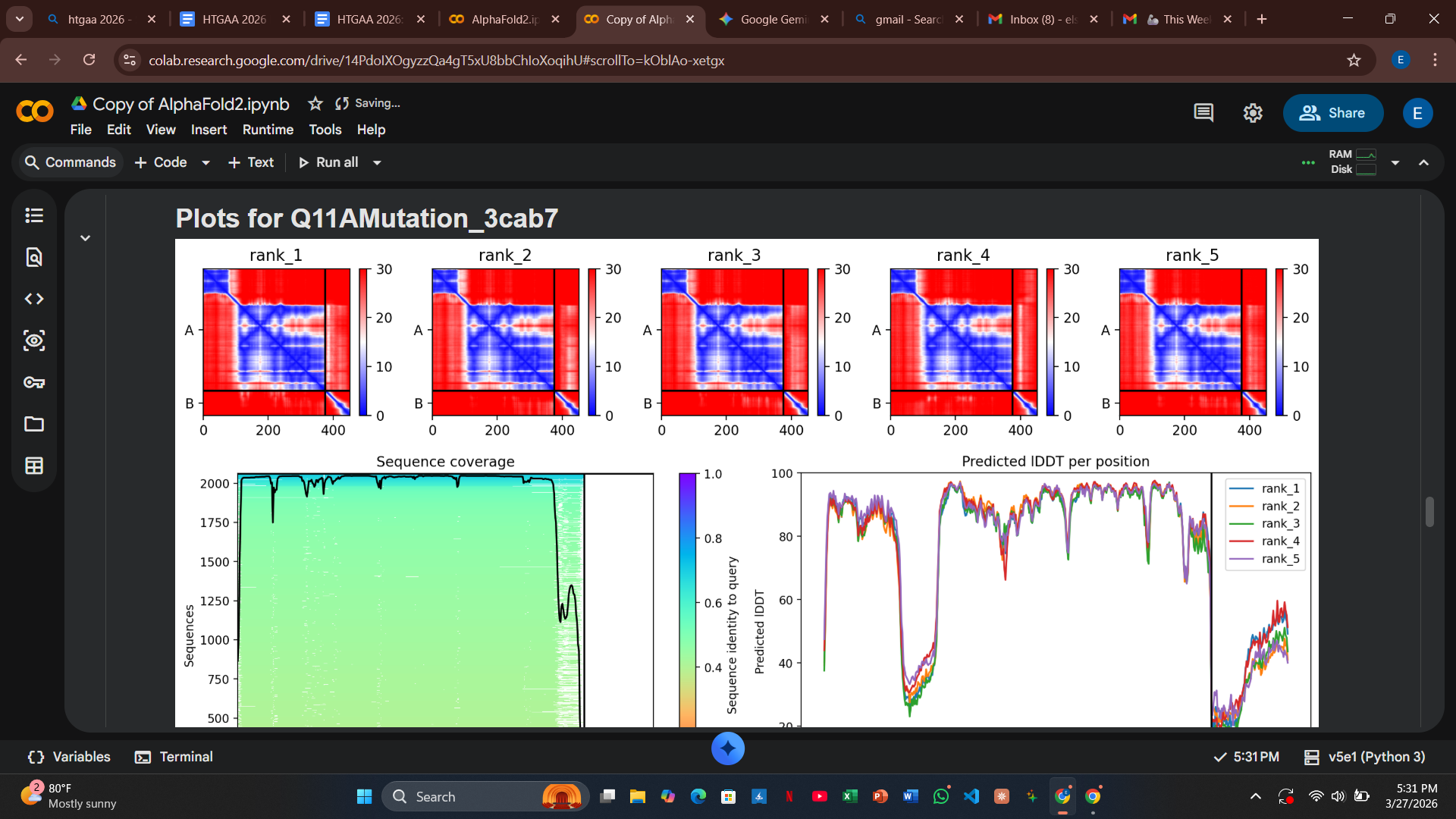
Variant 1: T3I (Soluble Region)
- Design Rationale: I targeted a variable site at the extreme N-terminus. By swapping Threonine for the more hydrophobic Isoleucine, I aimed to test if a slight shift in the N-terminal anchor could alter chaperone docking requirements.
- Computational Results: The AlphaFold2 results showed a high pLDDT score for the fold, but the PAE plots indicated that the docking confidence with DnaJ remained high.
>
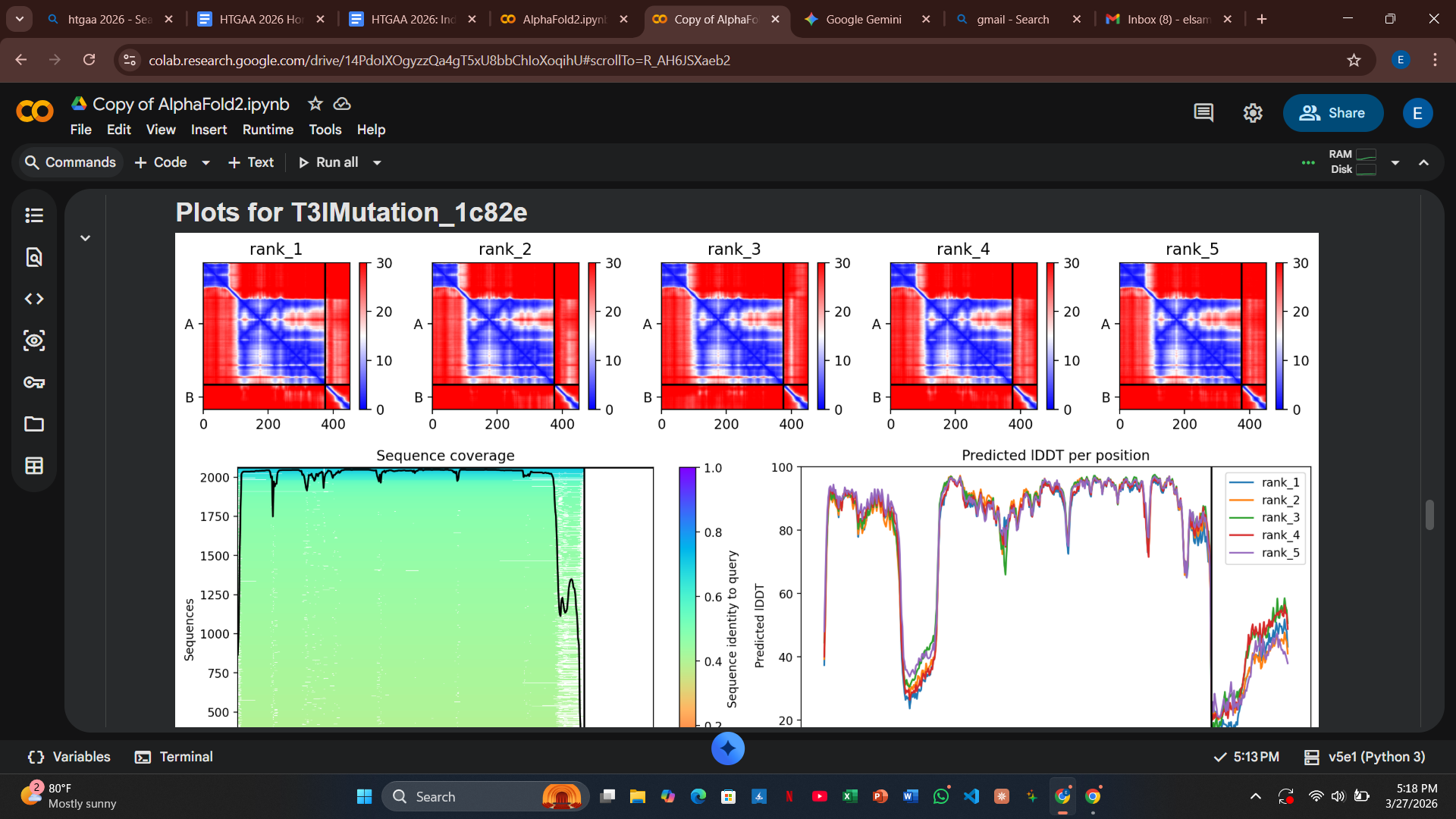
Variant 2: Q11A (Soluble Region)
- Design Rationale: This polar-to-hydrophobic swap was intended to disrupt the electrostatic surface interaction with DnaJ.
- Computational Results: Similar to T3I, the structural integrity remained intact, but the model still predicted a strong binding event with the host chaperone.
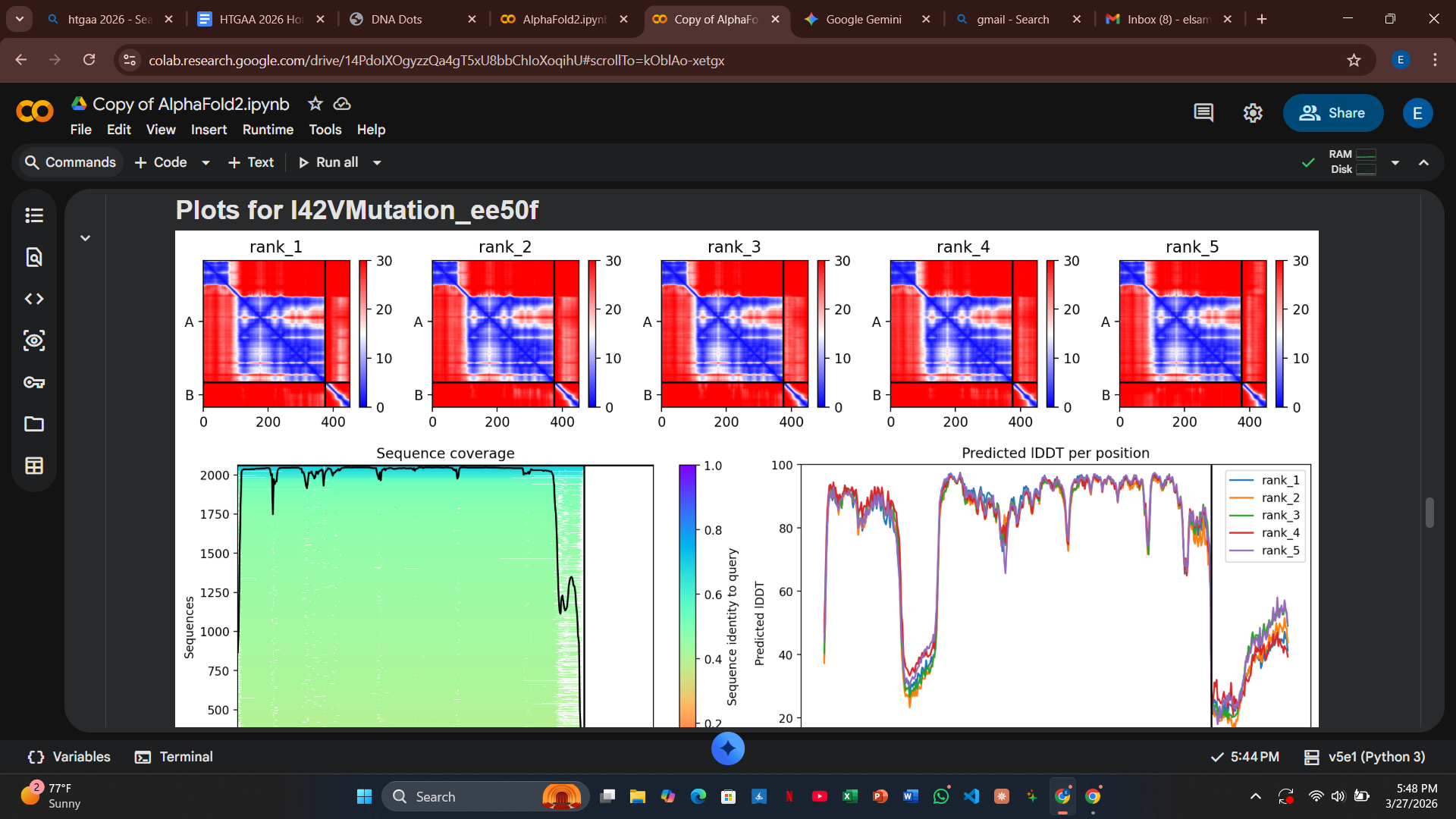
>
Variant 3: I42V (Transmembrane Region - Control)
- Design Rationale: This acts as a conservative control. By swapping Isoleucine for Valine (both hydrophobic and branched), I expected minimal impact on the pore-forming helix.
- Computational Results: The PAE plot showed very low error across the complex, confirming that this region is structurally robust and can tolerate minor volume changes without losing DnaJ affinity.
**
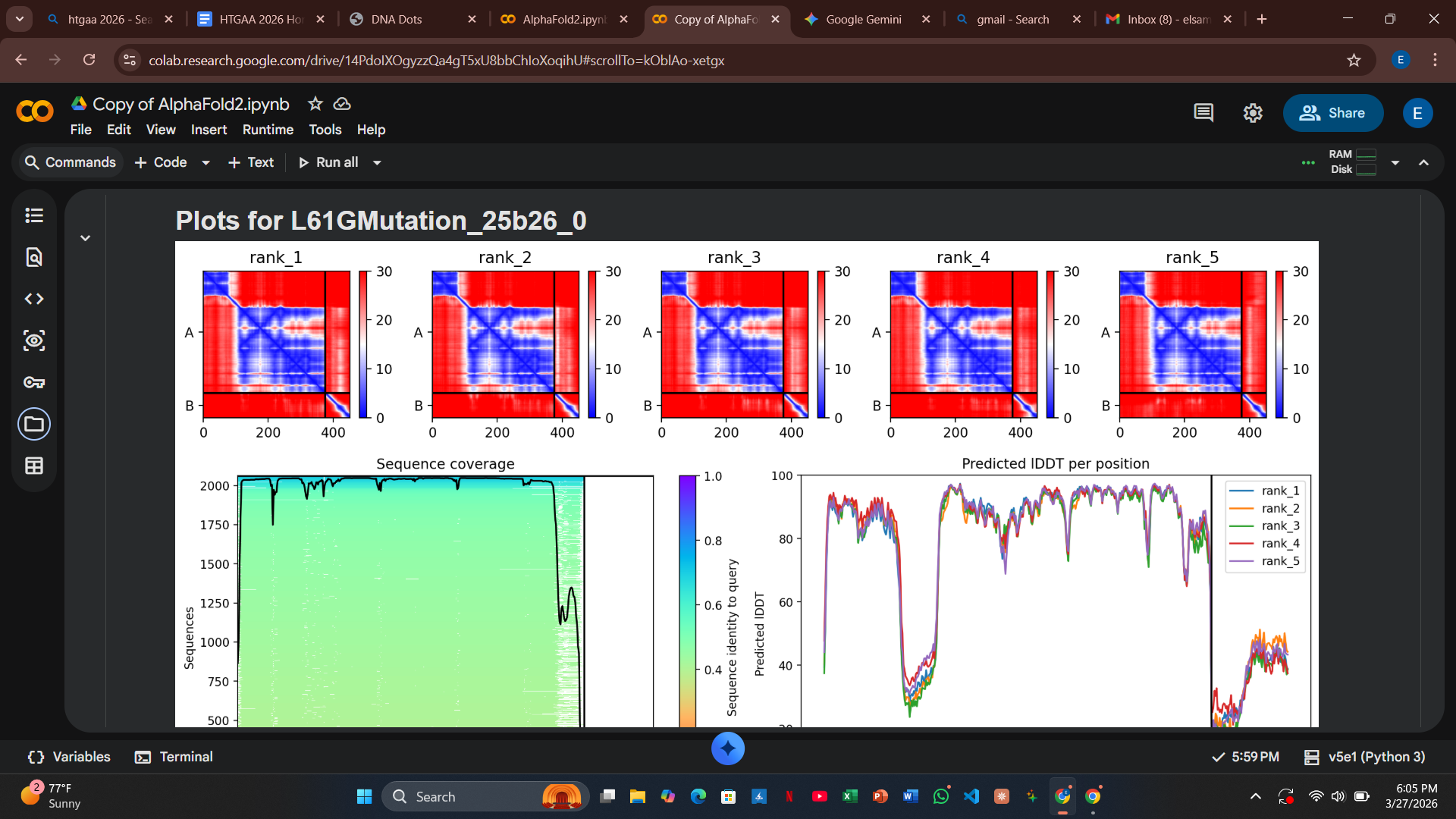
** > **
Variant 4: L61G (Transmembrane Region)
- Design Rationale: Introducing a Glycine “hinge” into a rigid alpha-helix increases conformational flexibility. This was designed to allow the L-protein to insert into the membrane more dynamically.
- Computational Results: There was a slight increase in the predicted error at the interface, suggesting the hinge might slightly destabilize the rigid docking required by DnaJ.
** > **
Variant 5: V63Q (Transmembrane Region - Lead Candidate)
- Design Rationale: This is my most disruptive design. Inserting a polar Glutamine (Q) into the hydrophobic core of the helix is intended to trigger a “forced” conformational change or rapid membrane disruption.
- Computational Results: The PAE plots for V63Q showed a significant loss of confidence (red and light green coloring) at the DnaJ interface. This suggests the mutation successfully disrupts the docking confidence, potentially allowing the protein to bypass the chaperone entirely.
** > **
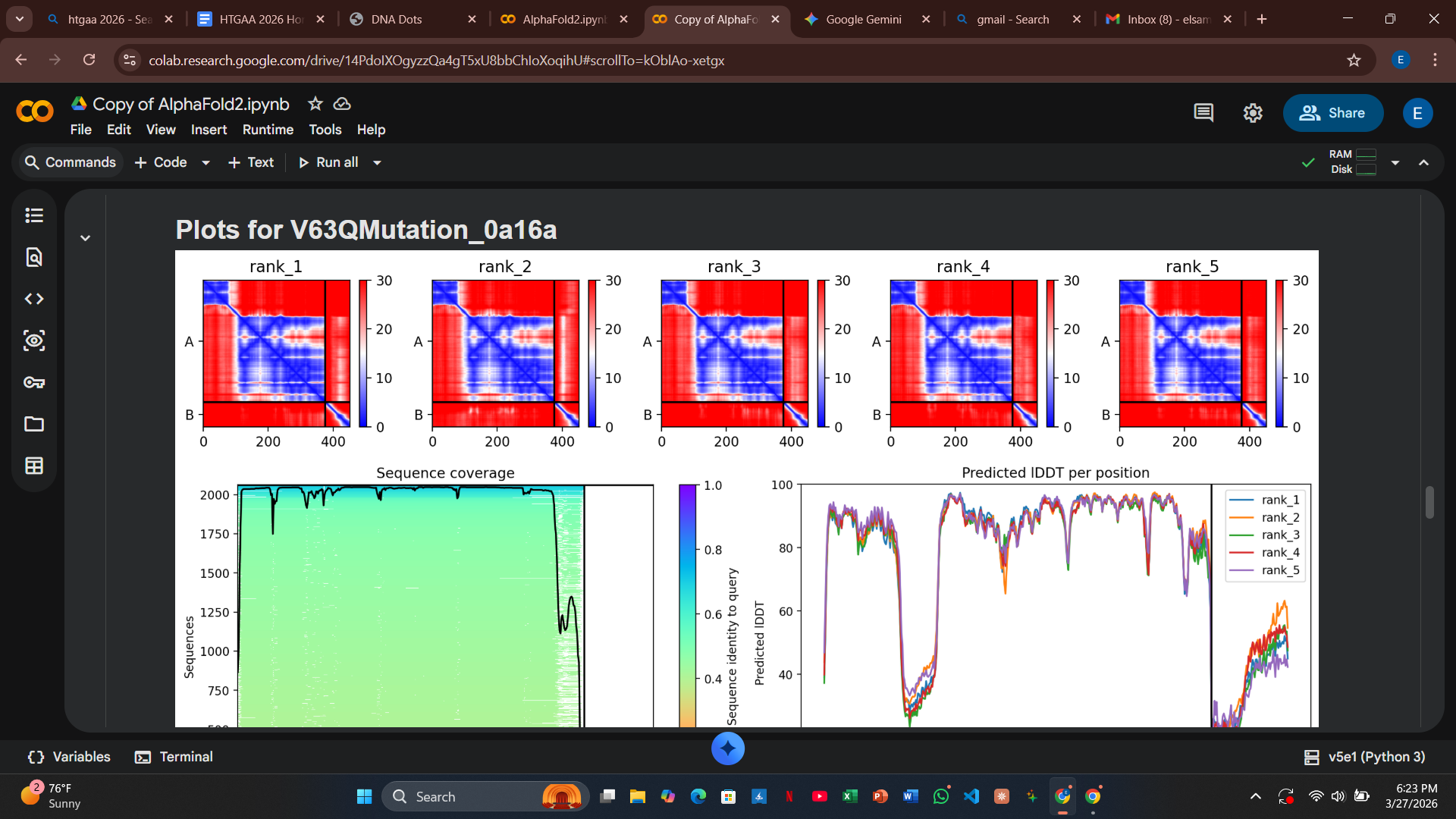
4. Synthesis and Wet-Lab Implementation
To test these variants, I have codon-optimized the sequences for E. coli expression. These will be synthesized via Twist Bioscience and assembled into the pBAD expression vector using Gibson Assembly.
Reference Sequences (Optimized DNA)
Variant 1 (T3I): text atggaaatccgttttccgcagcagtctcagcagaccccggcttctaccaaccgtcgtcgtccgttcaaacacgaagactacccgtgccgtcgtcagcagcgttcttctaccctgtacgttctgatcttcctggctatcttcctgtctaaattcaccaaccagctgctgctgtctctgctggaagctgttatccgtaccgttaccaccctgcagcagctgctgacc```
Variant 2 (Q11A): Plaintext atggaaacccgttttccgcagcagtctgcgcagaccccggcttctaccaaccgtcgtcgtccgttcaaacacgaagactacccgtgccgtcgtcagcagcgttcttctaccctgtacgttctgatcttcctggctatcttcctgtctaaattcaccaaccagctgctgctgtctctgctggaagctgttatccgtaccgttaccaccctgcagcagctgctgacc
Variant 3 (I42V):
atggaaacccgttttccgcagcagtctcagcagaccccggcttctaccaaccgtcgtcgtccgttcaaacacgaagactacccgtgccgtcgtcagcagcgttcttctaccctgtacgttctggttttcctggctatcttcctgtctaaattcaccaaccagctgctgctgtctctgctggaagctgttatccgtaccgttaccaccctgcagcagctgctgacc
Variant 4 (L61G):
atggaaacccgttttccgcagcagtctcagcagaccccggcttctaccaaccgtcgtcgtccgttcaaacacgaagactacccgtgccgtcgtcagcagcgttcttctaccctgtacgttctgatcttcctggctatcttcctgtctaaattcaccaaccagctgctgctgtctggtctggaagctgttatccgtaccgttaccaccctgcagcagctgctgacc
Variant 5 (V63Q):
atggaaacccgttttccgcagcagtctcagcagaccccggcttctaccaaccgtcgtcgtccgttcaaacacgaagactacccgtgccgtcgtcagcagcgttcttctaccctgtacgttctgatcttcctggctatcttcctgtctaaattcaccaaccagctgctgctgtctctgcaggaagctgttatccgtaccgttaccaccctgcagcagctgctgacc
5. Final Reflection and Future Directions
The computational data strongly suggest that V63Q is the most promising lead candidate. Weakening the interaction between confidence and DnaJ provides a viable pathway to overcome host resistance. One potential risk discussed during the design phase is that disrupting the chaperone interaction might also impair the protein’s ability to self-oligomerize to form the pore.
Strategic Analysis: The Synthetic Biology Trade-off
In engineering the V63Q variant, I am addressing a fundamental challenge in protein design: the balance between chaperone independence and structural stability. While the L-protein typically requires DnaJ as a structural scaffold to reach the membrane, my design tests whether a mutation in the transmembrane region can bypass this requirement.
Theoretical Outcomes for Variant V63Q
There are two primary biochemical scenarios that this mutation aims to explore during experimental validation:
- The Auto-Insertion Success: In this scenario, the V63Q mutation increases the protein’s affinity for the lipid bilayer to such an extent that it no longer requires DnaJ-mediated folding. The protein effectively auto-inserts into the membrane, oligomerizes, and induces lysis independently of host machinery.
- The Aggregation Failure: Conversely, without the DnaJ chaperone to shield hydrophobic patches during translation, the polar Glutamine (Q) at position 63 may cause the transmembrane helices to clump together inappropriately in the cytoplasm. This would form an inactive inclusion body that never reaches the membrane.
Refining the Strategy
To mitigate risks during the experimental phase, my strategy focuses on the specific Surface Area of Interaction:
- DnaJ Binding Site: Usually involves the soluble N-terminus (residues 1–40).
- Self-Oligomerization Site: Usually involves the transmembrane C-terminus (residues 41–75).
By focusing disruptive mutations like V63Q in the transmembrane region, I am testing the theory that the L-protein can auto-insert into the membrane.
Validation through Plaque Assays
The results of the upcoming plaque assays will provide a definitive answer to this design’s viability:
- Clear Zones (Lysis): If the assay shows clear zones, it proves that DnaJ is not strictly necessary for pore formation and the bypass was successful.
- No Plaques: If no plaques are visible, it suggests the mutation terminally disrupted the protein’s ability to self-assemble or fold without chaperone assistance.
Note: This analysis will be validated using the 3D structures and validation plots generated for all five variants to correlate predicted stability with observed lysis activity.
References
Chamakura, K. R., et al. (2017). “Mutational analysis of the MS2 lysis protein L.” Journal of Virology.
Hyman, P., et al. (2023). “Phage therapy: From biological mechanisms to future directions.” Microbiology Research Reviews.
UniProt Consortium. “Lysis protein L - Bacteriophage MS2 (P03609).