Week 2 HW: DNA Read, Write, & Edit
1-Benchling-in-silico-gel-art
Using Benchling.com, Lambda DNA, Paul Vanouse’s Latent Figure Protocol artworks, and Ronan’s website as references, and incorporating creative design principles, simulations of restriction enzyme digests of the Lambda genome were performed using EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI:

Figure 1. Virtual restriction digest of Lambda DNA.
Once the banding patterns were characterized, images inspired by the previously mentioned works were created:
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Part 3: DNA Design Challenge
3.1. Choosing a protein:
Protein Chosen: INS Homo sapiens (Insulin): This is a known protein, short sequence, and without stop codons: AAP35454.1 insulin [Homo sapiens] MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQVGQVELGGGPGAGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN
Obtaining the DNA sequence in tblastn:
In tblastn with the Human INS sequence (AAP35454.1), the search was on for the DNA sequence with the highest identity.
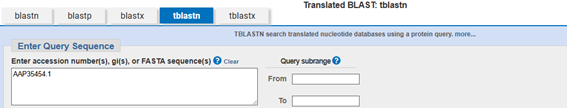

Figure 2. tblast of the selected sequence.
Once the result is obtained, by entering the sequence ID (AB587580.1) the gene sequence is obtained:

Figure 3. DNA selected sequence.
BT006808.1 Homo sapiens insulin mRNA, complete cds ATGGCCCTGTGGATGCGCCTCCTGCCCCTGCTGGCGCTGCTGGCCCTCTGGGGACCTGACCCAGCCGCAGCCTTTGTGAACCAACACCTGTGCGGCTCACACCTGGTGGAAGCTCTCTACCTAGTGTGCGGGGAACGAGGCTTCTTCTACACACCCAAGACCCGCCGGGAGGCAGAGGACCTGCAGGTGGGGCAGGTGGAGCTGGGCGGGGGCCCTGGTGCAGGCAGCCTGCAGCCCTTGGCCCTGGAGGGGTCCCTGCAGAAGCGTGGCATTGTGGAACAATGCTGTACCAGCATCTGCTCCCTCTACCAGCTGGAGAACTACTGCAACTAG
3.2. Reverse translation: Protein sequence (amino acids) to DNA sequence (nucleotides)
The central dogma, discussed and recited in class, describes the process by which the DNA sequence is transcribed and translated into protein. The central dogma provides the framework for working in reverse from a given protein sequence and inferring the DNA sequence from which it is derived. Using one of the tools discussed in class, the NCBI (tblastn) or online tools (search “reverse translation tools” on Google), determine the nucleotide sequence that corresponds to the protein sequence you chose earlier.
3.3. Codon optimization
Codon optimization: on the web site NovoProLabs modifying the nucleotide sequence of a gene to maximize protein production in a specific organism by adapting codon usage to the host’s codon bias. Gene expression in a different organism may require adjusting codon usage to match the host’s translational preferences. Restriction enzymes EcoRI and XhoI are not involved in the codon optimization itself, but are used to facilitate cloning of the optimized gene into an expression vector. During sequence design, the optimization tool adds the specific recognition sites of these enzymes to the ends of the gene, so that, by digesting both the insert and the plasmid with the same enzymes, compatible cohesive ends are generated that allow the gene to be inserted in the correct orientation within the vector. Instead, the changes observed in Relative Adaptiveness and GC content come exclusively from the codon optimization process, which modifies the nucleotide sequence without altering the amino acid sequence to improve translation efficiency in the host organism.

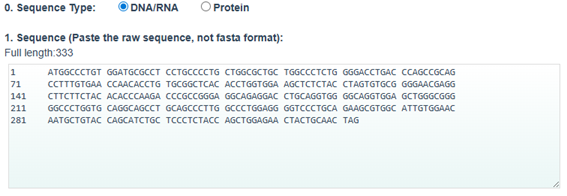
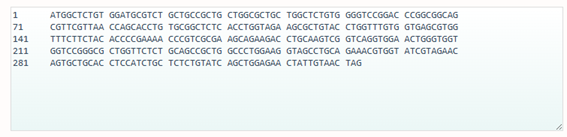

Codon optimization of the insulin coding sequence for E. coli expression increased the Codon Adaptation Index (CAI) from 0.48 to 0.90, indicating improved compatibility with the host’s codon usage preferences and a higher expected translation efficiency. At the same time, GC content was adjusted from 64.56% to 60.00%, bringing it closer to a balanced range that can improve mRNA stability and transcriptional performance. Overall, the optimization modifies the nucleotide sequence without changing the amino acid sequence, with the goal of enhancing recombinant protein expression in the bacterial system.
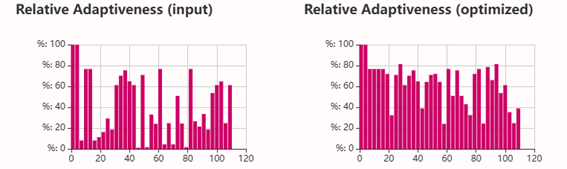
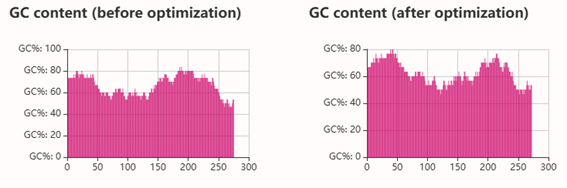
3.4 Optimized protein transcription and translation
To produce this protein from DNA, the optimized gene can be inserted into a recombinant expression vector and introduced into a host such as *E. coli *using recombinant DNA technology and heterologous expression systems. Inside the cell, the DNA sequence is first transcribed into mRNA by RNA polymerase, and the mRNA is then translated by ribosomes into the protein according to the genetic code. Alternatively, the same DNA template can be used in cell-free expression systems (in vitro transcription - translation, IVTT), which contain purified enzymes, ribosomes, tRNAs, and energy sources that allow transcription and translation to occur outside living cells. Both approaches rely on the central dogma of molecular biology- DNA => RNA => protein - but differ in whether protein production occurs in living cells or in a controlled biochemical system.
3.5 Alternative splicing
A single gene can produce multiple proteins at the transcriptional level mainly through alternative splicing (alternative splicing). During pre-mRNA processing, different exons can be included or excluded in different combinations, generating multiple mature mRNAs from the same gene. Each of these mRNAs can be translated into different protein isoforms, with variations in their structure and function. Furthermore, mechanisms such as alternative promoters or alternative polyadenylation sites can also produce different transcripts from the same gene locus.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account, and Benchling account
4.2. Build Your DNA Insert Sequence
In Benchling, select New DNA/RNA sequence. Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
4.5. Vector Selection
The sequence of plasmid PP533546.1 was downloaded from GenBank and used as cloning backbone. Then, a synthetic INS coding sequence from Homo sapiens was designed in Benchling. After preparing both sequences, virtual DNA assembly was performed to insert the INS construct into the plasmid backbone, generating a recombinant circular plasmid containing the expression cassette (promoter, RBS, INS CDS, His tag, and terminator). This in-silico cloning step allowed visualization and verification of the final construct.
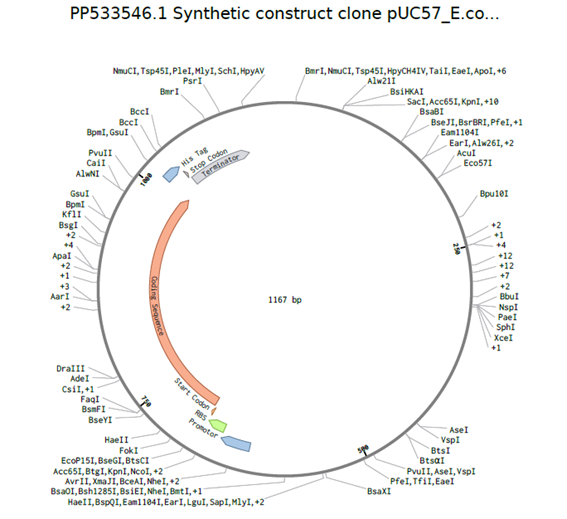
pUC57- E. coli -ISN Hs plasmid
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
Sanger sequencing is a first-generation DNA sequencing method based on chain termination. During DNA synthesis, modified nucleotides (dideoxynucleotides, ddNTPs) are incorporated randomly, stopping elongation. The resulting fragments of different lengths are separated by capillary electrophoresis, and the sequence is read from fluorescent labels. It is highly accurate and ideal for validating plasmids or specific genes, but it is low-throughput and typically limited to ~700 - 1000 bp per read. Nanopore sequencing is a third-generation method that sequences DNA in real time by passing single DNA molecules through a biological nanopore. As nucleotides move through the pore, they disrupt an ionic current in characteristic ways, allowing base identification. It can generate very long reads (even entire plasmids or genomes in one fragment) and works without PCR amplification, but raw accuracy can be slightly lower than Sanger, though it has improved significantly. An example of next-generation sequencing (NGS) is Illumina sequencing. It uses massively parallel sequencing by synthesis, where millions of DNA fragments are immobilized on a flow cell and sequenced simultaneously through cyclic incorporation of fluorescently labeled nucleotides. This approach provides extremely high throughput and is commonly used for whole-genome sequencing, transcriptomics (RNA-seq), and large-scale variant analysis.
(ii) What technology or technologies would you use to perform your DNA sequencing and why?
Genes of the gut microbiota. To sequence gut microbiota genes, the most widely used and appropriate technology would be second-generation sequencing (NGS), especially platforms like Illumina. Third-generation sequencing, such as Oxford Nanopore Technologies, could also be considered, depending on the objective (taxonomic resolution, or complete assembly). Responda también las siguientes preguntas:
Is your method first, second, or third generation, or something else? What does that mean?
Recommended primary technology: Illumina (NGS). What generation is it? It’s second generation. It’s characterized by performing millions of reads in parallel (massively parallel sequencing) with short fragments and high accuracy. For gut microbiota studies (e.g., 16S rRNA gene sequencing or metagenomics), Illumina is ideal due to its high accuracy, low cost per sample, high sequencing depth, and excellent bioinformatics support. Nanopore would be useful if you are looking for long reads or whole genome assembly.
What is your opinion? How do you prepare your information (e.g., fragmentation, adapter ligation, PCR)? List the essential steps.
Sample Preparation (e.g., 16S rRNA or metagenomics). Essential Steps:
- DNA extraction from the fecal sample.
- Fragmentation (if metagenomics; not always necessary for 16S rRNA).
- PCR amplification
- For 16S rRNA: amplification of variable regions (V3–V4).
- Ligation of adapters and indices (barcodes).
- Purification and quantification.
- Loading into the flow cell of the sequencer.
What are the essential steps of the sequencing technology you have chosen and how does it decode the bases of your DNA sample (base calling)?
How does Illumina technology work?
- DNA with adapters is attached to a flow cell.
- A cluster is generated by bridge amplification.
- Reversibly fluorescent terminator nucleotides are incorporated.
- Each cycle adds one base.
- A camera detects the fluorescence.
- The terminator is removed, and the cycle repeats. How are the bases decoded? Each base (A, T, C, G) emits a different fluorescence. The system records the optical signal and converts it into a digital sequence (base calling).
What is the outcome of the chosen sequencing technology?
The end result is millions of short reads (FASTQ files). Each read includes base sequence and quality scores (Phred score). Subsequently, bioinformatics analysis, taxonomic identification, bacterial abundance profiling, and alpha and beta diversity are performed.
5.2 DNA writing
What DNA would you like to synthesize (e.g., write) and why?
Genes that are expressed in inflammatory settings. Genes that are artificially expressed so that in IBD contexts, they are expressed in cell cultures, spheroids, or organoids. It should serve as a reliable indicator, expressed through the presence of butyrate, SCFAs, or various bacterial metabolites, or oxidative stress scenarios. It should be activated by the presence of a common marker metabolite. A promoter sensitive to an inflammatory or metabolic signal controls the expression of a reporter gene (e.g., GFP, luciferase, mCherry, etc.). When the stimulus appears (butyrate, ROS, NF-κB, etc.), the promoter is activated and the reporter gene is expressed.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
To design and validate a promoter sensitive to inflammatory or metabolic signals, it is first necessary to identify endogenous regulatory regions that respond to the stimulus of interest using NGS technologies such as RNA-seq to detect stimulus-induced genes, and ATAC-seq or ChIP-seq to map open chromatin regions or binding sites of specific transcription factors (e.g., NF-κB). Complementarily, techniques such as STARR-seq allow for the functional evaluation of promoter activity across thousands of sequences simultaneously, identifying those that actually activate transcription under the stimulus. By combining these data, synthetic promoters can be designed to drive the specific and quantifiable expression of a reporter gene (GFP, luciferase, mCherry) in response to the desired signal.
Please also answer the following questions:
What are the essential steps of the sequencing methods you have chosen?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, and scalability?
The essential steps of RNA-seq are: (1) isolation of total RNA or messenger RNA (mRNA) from the samples of interest; (2) fragmentation and library preparation, which includes conversion to cDNA, ligation of adapters, and sometimes size selection; (3) sequencing of the libraries on an NGS platform to obtain short reads; (4) data processing, which involves quality control, filtering, alignment of the reads to the reference genome or transcriptome, and quantification of gene expression; and (5) differential analysis to identify genes whose expression level changes between conditions, followed by functional annotation and pathway analysis. The main limitations of RNA-seq in terms of speed, accuracy, and scalability are: Speed: Library preparation and sequencing can take anywhere from several hours to several days, especially if biological replicates and multiple conditions are required; bioinformatics analyses can also be slow if the datasets are large. Accuracy: Quantification can be affected by amplification bias, reverse transcription efficiency, fragmentation differences, or ambiguous mapping of reads to homologous genes or isoforms; genes with low expression are difficult to detect reliably. Scalability: Processing many samples simultaneously increases costs and complexity, and storing and analyzing large volumes of data requires robust computational infrastructure; furthermore, high-resolution methods such as single-cell RNA-seq exponentially increase the amount of data and the complexity of the analysis.
5.3 DNA Editing
(i) Which DNA would you like to edit and why?
CRISPR editing. I believe it’s a technique with a very promising future.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your preferred technology edit DNA? What are the essential steps?
CRISPR edits DNA using a Cas enzyme guided by a guide RNA that recognizes a specific sequence in the genome. Cas cuts the DNA at that point, and then the cell repairs the break, which can result in insertions, deletions, or allow the incorporation of a new sequence if a repair template is provided.
What preparation is needed (e.g., design steps) and what information (e.g., DNA template, enzymes, plasmids, primers, guides, cells) is required for editing?
A guide RNA that recognizes the target sequence needs to be designed, and, if insertion is desired, a DNA template with the sequence to be incorporated. Information and materials include: Cas9 enzyme (or similar), plasmids or delivery vectors, guide RNA, verification primers, the repair template if applicable, and the cells to be edited.
What are the limitations of your editing methods (if any) in terms of efficiency or accuracy?
CRISPR has limitations in efficiency, since not all cells correctly receive the editing machinery or repair DNA in the desired way, and in precision, because off-target effects or unforeseen insertions/deletions can occur at the cutting site. Furthermore, efficiency and precision depend on cell type, guide RNA design, and genomic context.
title: ‘Week 3 HW: Lab Automation’ weight: 30
Assignment: Python Script for Opentrons Artwork
Based on the Lissajous function, the figure to be created on the agar will be the following:

Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
Paper: Automation of biochemical assays using an open-sourced, inexpensive robotic liquid handler
Moukarzel et al. 2024
https://doi.org/10.1016/j.slast.2024.100205
The study aimed to evaluate the feasibility of using an open-sourced, low-cost robotic liquid handler—specifically the Opentrons OT-2—for automating biochemical assays that are traditionally run on expensive industrial liquid handling platforms. High-throughput screening is a core process in pharmaceutical development, but the cost and training requirements of conventional robotic systems can be prohibitive. The authors set out to determine whether a lightweight, Python-programmable robot could perform common assay workflows with sufficient precision and reliability to be useful in early-stage assay development and method transfer.
To test this, the team programmed the OT-2 to perform two standard biochemical assays—PicoGreen for DNA quantification and Bradford for protein concentration—using custom Python protocols that controlled pipetting and reagent transfers across microplates. These automated workflows were run repeatedly and compared to runs on a more expensive Tecan EVO liquid handler to benchmark performance. The study measured pipetting accuracy, variability, and overall assay consistency to assess how well the OT-2 handled the tasks relative to the industrial system.
The results showed that the OT-2 delivered accurate pipetting with low covariance across replicates, demonstrating performance close to that of the Tecan EVO despite its substantially lower cost and simpler hardware. Although limitations such as the absence of a crash detection system and a relatively small deck space were noted, the robot’s affordability and flexibility were highlighted as significant advantages. The authors concluded that the OT-2 represents a cost-effective, medium-throughput automation solution well suited for early-stage assay development and method transfer without requiring large capital investments.