Week 4 HW: protein desing part 1
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
If we assume that meat contains approximately 20% protein, then 500 g of meat provides around 100 g of protein. Since the average molecular weight of an amino acid is approximately 100 Daltons (approx 100 g/mol), that 100 g corresponds to approximately 1 mole of amino acids. One mole contains approx 6.02 × 10²³ molecules, so you would ingest approximately 6 × 10²³ molecules of amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because when we eat meat, the animal’s proteins (from cows, fish, etc.) are not incorporated intact into our bodies. Instead, they are digested in the gastrointestinal tract into their basic components: amino acids and small peptides. These are absorbed, and then our own cells reuse them to synthesize human proteins according to the information encoded in our DNA. In other words, we don’t incorporate the “biological identity” of the animal we eat, but rather molecular raw material that our genome reorganizes according to the instructions specific to the human species.
3. Why are there only 20 natural amino acids?
There are only 20 standard “natural” amino acids because the universal genetic code that has evolved encodes precisely these 20 building blocks for protein synthesis (with rare exceptions such as selenocysteine and pyrrolysine). This selection is not chemical but evolutionary: among many possible molecules, these 20 offered an optimal balance between structural diversity (charges, sizes, polarity, hydrophobicity), chemical stability, and biosynthetic efficiency. With this set, an enormous variety of protein structures and functions can be generated, so evolution did not need to significantly expand the basic alphabet to support biological complexity.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible to create non-natural amino acids both chemically and by expanding the genetic code in biological systems. From a design perspective, it is sufficient to maintain the α-amino acid backbone (amino group, carboxyl group, and chiral α-carbon) and modify the side chain to introduce new physicochemical properties. For example, one could design (1) an amino acid with a bulky fluorinated side group to increase hydrophobic stability and resistance to degradation, (2) one with a photoreactive side chain (such as an azide or diazirine group) to allow light-induced cross-linking, (3) an amino acid with a chelating metal group to create artificial catalytic sites, or (4) one with a redox-active side chain capable of participating in electron transfer. In fact, synthetic biology has already incorporated hundreds of non-natural amino acids into proteins through reassigned codons or modified tRNA/synthetase systems, functionally expanding the protein “alphabet” beyond the standard 20.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes and cellular life existed, amino acids could have formed through abiotic prebiotic chemistry. Classic experiments like the Miller-Urey experiment demonstrated that, under conditions simulating the early atmosphere (simple gases such as methane, ammonia, water vapor, and electrical discharges), several amino acids can be spontaneously synthesized. Furthermore, amino acids have been found in meteorites such as the Murchison meteorite, indicating that they can also form in space through interstellar chemistry and then reach Earth via impacts. Other possible environments include oceanic hydrothermal vents and mineral surfaces that catalyze organic reactions. Taken together, the evidence suggests that amino acids arose through natural physicochemical processes before the emergence of enzymes and were part of the molecular inventory that preceded the origin of life.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If you build an α-helix exclusively from D-amino acids, you would expect it to adopt a left-handed helix. In natural proteins made from L-amino acids, the stable α-helix is typically right-handed due to stereochemical constraints of the α-carbon and the allowed φ and ψ angles in conformational space. By inverting the chirality (using D instead of L), the geometric preference for folding is also reversed, producing the mirror image: a stable α-helix but with the opposite orientation.
7. Can you discover additional helices in proteins?
Yes, in principle, additional helices beyond the classical conformations can be discovered or engineered. Helical conformation depends on the allowed φ/ψ angles, hydrogen bonding patterns, and the chemistry of the peptide backbone. Modifying these variables, for example, by using non-natural amino acids, changing the backbone length, or applying specific steric constraints, can lead to the emergence of new, stable helical geometries. In fact, helical architectures not commonly found in natural proteins have been observed in synthetic peptides and foldamers. However, within proteins composed of the 20 standard amino acids and the natural peptide backbone, the repertoire of helices is strongly restricted by the stereochemistry and physics of the peptide bond, so additional variants tend to be rare or less stable.
8. Why are most molecular helices right-handed?
Most molecular helices are right-handed because they are built from chiral building blocks with a predominant stereochemical configuration. In terrestrial biology, almost all amino acids are L-shaped, and this chirality imposes specific geometric constraints on the angles of the peptide backbone, favoring a right-handed α-helix as the most energetically and sterically stable conformation. In other words, the molecular asymmetry of the monomers is amplified at the macroscopic level in the secondary structure. If life had predominantly adopted D-amino acids, the predominant helices would most likely have been left-handed.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their geometry exposes repeating patterns of amide and carbonyl groups in the backbone that can form extensive networks of intermolecular hydrogen bonds. When several polypeptide chains adopt extended conformations, they can readily align and stabilize each other through these bonds, forming larger sheets and, in extreme cases, amyloid-like fibrils. The primary driving force for aggregation is the minimization of the system’s free energy: the cooperative formation of hydrogen bonds, along with hydrophobic interactions between side chains and the burial of nonpolar surfaces, offsets the entropic cost of ordering the chains. Taken together, geometric complementarity and intermolecular stabilization make β-sheets particularly prone to aggregation when they are partially unfolded or misfolded.
Part B: Protein Analysis and Visualization
Protein selected: PyMOL>fetch 6BHU
Cryo-EM structure of ATP-bound, outward-facing bovine multidrug resistance protein 1 (MRP1)
Longitud:
PyMOL>get_extent
cmd.extent: min: [ 73.772, 111.074, 115.447]
cmd.extent: max: [ 206.780, 198.350, 201.157]
The most frequent amino acid:
PyMOL>stored.residues=[]
PyMOL>iterate name CA, stored.residues.append(resn)
Iterate: iterated over 1465 atoms.
PyMOL>count_atoms 6BHU and name CA
count_atoms: 1465 atoms
PyMOL>print(max(set(stored.residues), key=stored.residues.count))
LEU
Con Colab:
The length of the protein is: 1345 aminoacids.
The most common amino acid is: L, which appears 158 times.
• How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs
PyMOL>print(cmd.get_fastastr(“6BHU”))
Does your protein belong to any protein family? ABC Family

Figure 1. Chosen protein sequence.

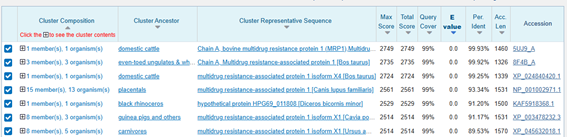
Figure 2 and 3. BLASTp alignment results.
3. Identify the structure page of your protein in RCSB
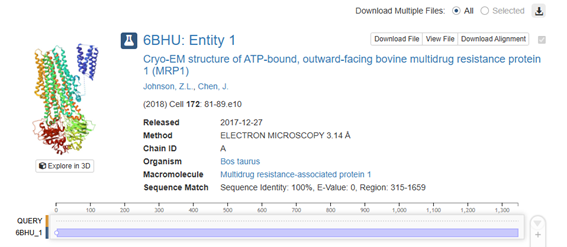
Figure 4. Protein structure selected in RCSB.
• When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Quality: In terms of traditional crystallography, 3.50 Å is considered low-to-medium resolution. However, for a membrane protein as large as this one (MRP1) resolved by cryo-electron microscopy (cryo-EM), it is a structure of acceptable quality.
• Are there any other molecules in the solved structure apart from protein?
Yes, there are several. In addition to the protein chain, the resolved structure contains small molecules (ligands) that are crucial for its function and for the process of obtaining the structure:
•ATP (Adenosine-5’-triphosphate): Appears as “ATP”. There are two molecules, one at each nucleotide-binding site (NBD). It is the fuel the protein uses for pumping.
Magnesium ions (Mg2+): Appear as “MG”. They are necessary for ATP to bind correctly.
• Cholesterol (CLR): Lipid molecules that remain attached to the protein during purification.
• LMT (Dodecyl-beta-D-maltoside): A detergent used to keep the protein stable outside the cell membrane.
• Does your protein belong to any structure classification family?
Yes, it is perfectly classified in international hierarchies (CATH and SCOP):
• Superfamily: ABC transporters (ATP-Binding Cassette). It is one of the largest and oldest families of membrane proteins.
• Specific family: MRP (Multidrug Resistance-associated Proteins).
Functional classification: It is an exporter.
• Structural architecture (CATH): It is classified as an Alpha-Beta type structure, since it combines transmembrane helices (alpha) with cytoplasmic domains that have beta sheets where ATP binds.
4. Open the structure of your protein in any 3D molecule visualization software:
• PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
• Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
• Color the protein by secondary structure. Does it have more helices or sheets?
PyMOL>as cartoon
PyMOL>util.cbss(“6BHU”, “red”, “yellow”, “green”)
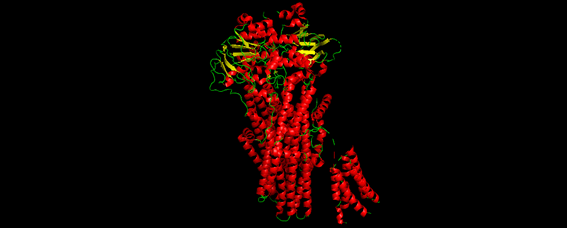
Figure 5. Chosen protein structure.
• Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
PyMOL># Hidrofóbicos (En naranja/rojo: los que están dentro de la membrana)
PyMOL>color orange, resn ala+val+leu+ile+met+phe+trp+pro+gly
Executive: Colored 4865 atoms.
PyMOL># Hidrofílicos polares (En azul: los que tocan el agua o el citoplasma)
PyMOL>color marine, resn ser+thr+asn+gln+tyr+cys
Executive: Colored 2342 atoms.
PyMOL># Hidrofílicos cargados (En azul oscuro: muy polares)
PyMOL>color slate, resn arg+his+lys+asp+glu
Executive: Colored 2455 atoms.
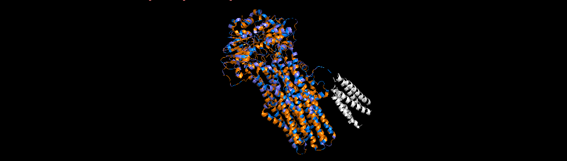
Figure 6. Chosen protein structure.
• Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Hole visualize:
PyMOL># 1. Mostrar superficie sólida: PyMOL>show surface, 6BHU
PyMOL># 2. Hacerla un poco transparente para ver el interior: PyMOL>set transparency, 0.4
Setting: transparency set to 0.40000.
PyMOL># 3. Resaltar los ligandos (ATP) para ver dónde están los huecos principales: PyMOL>show spheres, organic
PyMOL>color brightorange, organic
Executive: Colored 146 atoms.
• Does it have binding pockets?
Yes, and they are essential to its function. When observing the surface of 6BHU, you will notice two main types of cavities:
The Large Central Vestibule: Because 6BHU is a transporter in an outward-facing conformation, you will see a large hole or “funnel” at the top (the side facing the cell exterior). This is the pathway through which substrates exit.
Nucleotide Binding Sites (NBDs): On the cytoplasmic (lower) side, there are two deep pockets where ATP molecules are held. If you used the transparency command, you will see the ATP “stuck” in these pockets.
Internal Substrate Cavity: In the center of the membrane protein, there is a highly flexible pocket that, in this structure, is designed to release the cargo.
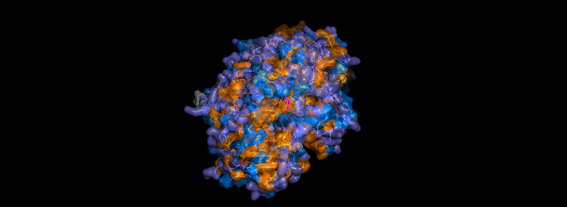
Figure 7. ABC protein pockets.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
The heat map displays the Model Scores, which in this case are the Log Likelihood Ratios (LLRs) of a specific mutation at a given position, compared to the wild-type (WT) amino acid. The ‘Viridis’ color scale used in the visualization assigns:
Darker colors (blues/purples): Lower, more negative LLR values. This indicates that the mutation to that amino acid at that position is unfavorable compared to the WT.
Lighter colors (greens/yellows): Higher, more positive LLR values. This indicates that the mutation to that amino acid at that position is favorable compared to the WT, or at least more likely.
A conserved site is a position in the protein where the wild-type amino acid is crucial for function or structure, and any mutation to a different amino acid would be detrimental or very unlikely. In terms of LLRs, this would mean that all possible mutations at that position (i.e., the entire column for that position, excluding the wild-type amino acid) would have very low and negative LLR values (dark colors on the heatmap).
Therefore, the yellow dots (positive or high LLRs) on the heatmap indicate positions where certain mutations are favorable or better accepted by the model, not where the site is conserved. Conserved sites are represented by entire columns of the heatmap that are predominantly dark in color.
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
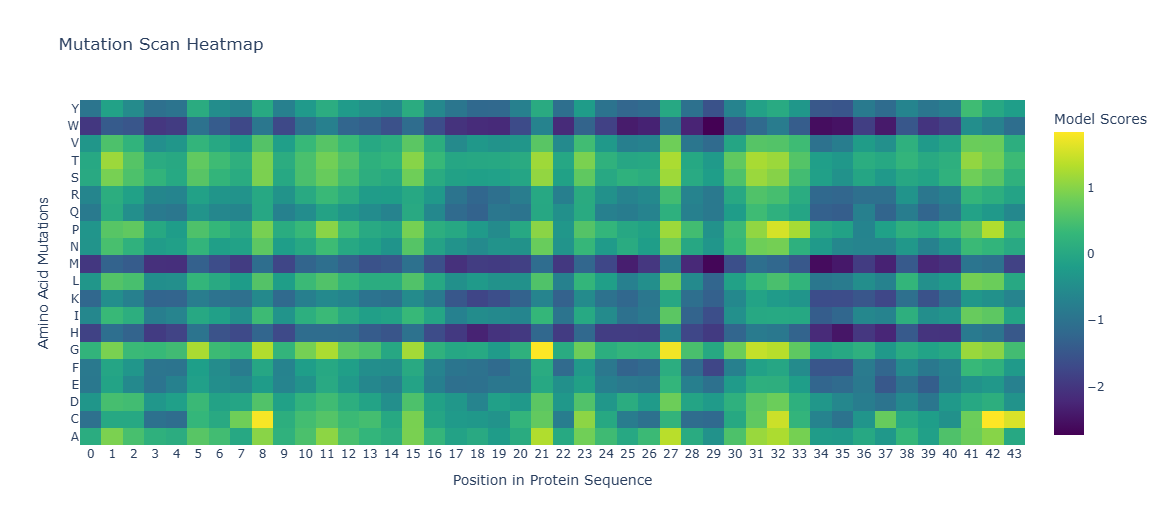
Figure 8. Amino Acids Mutation Position in Protein Sequence.
Latent Space Analysis
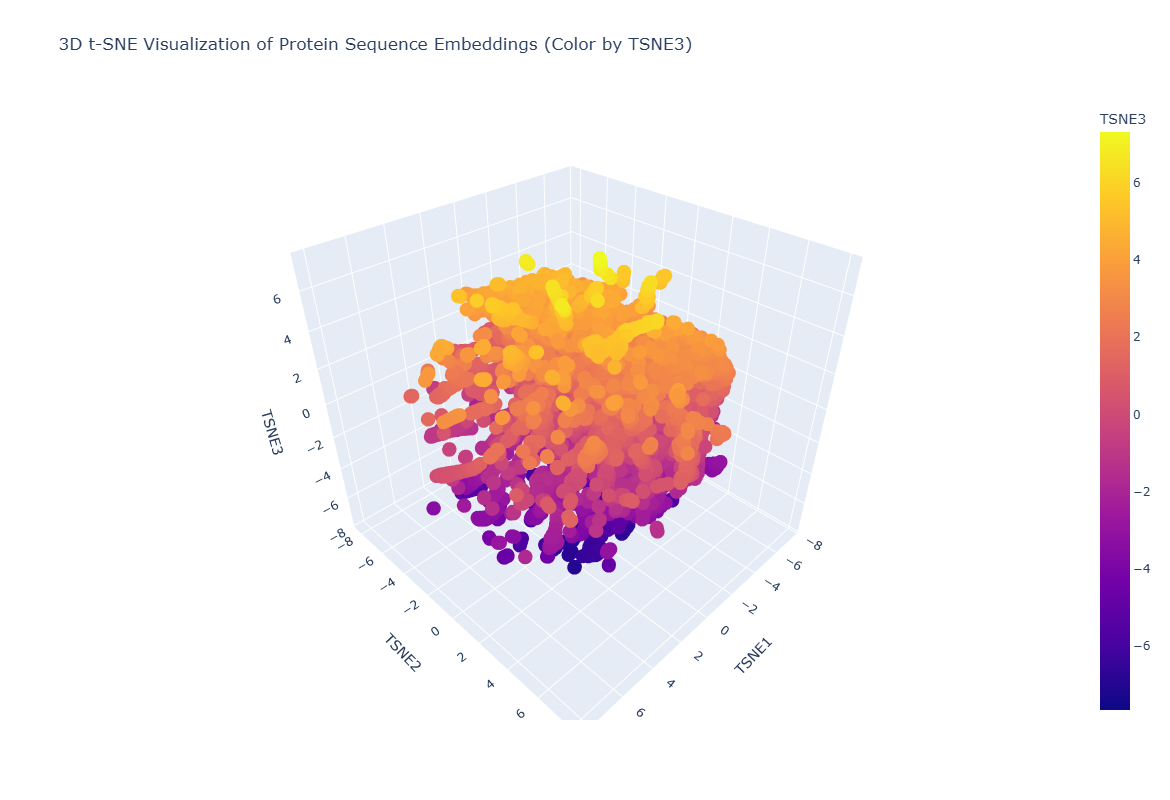
Figure 9. 3D t-SNE Visualitation of protein Sequence Embeddings.
C3. Protein Generation
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Due to the inability to run the code for the “Protein Folding with ESMFold” block, I chose a smaller protein hoping it would work by using less memory, but it wasn’t possible. I’ve included an image of the protein below, but the second part of the activity is missing.

Figure 9. 3D visualitation 1crn protein.