Week 5 HW: protein desing part 2
Homework — DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design (From Pranam)
Introduction
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M template card:
Generate four 12-amino-acid peptides conditioned to the mutant SOD1 sequence.
Add the known SOD1-binding peptide, FLYRWLPSRRGG, to the generated list for comparison.
Record the puzzle scores, which indicate PepMLM’s confidence in the ligands.

sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHE
FGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSI
EDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVI
GIAQ
Part 2: Evaluate Binders with AlphaFold3
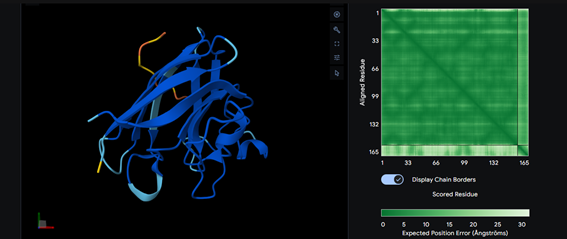
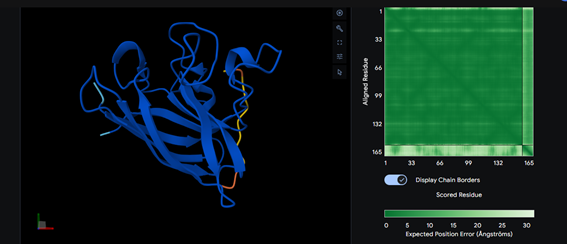
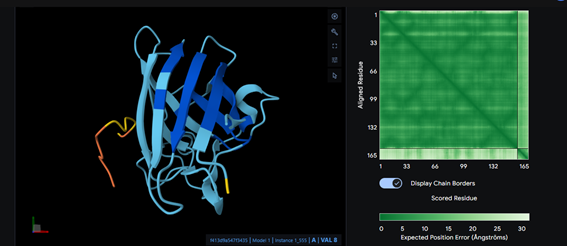
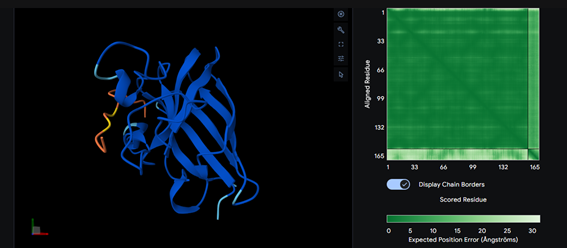
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
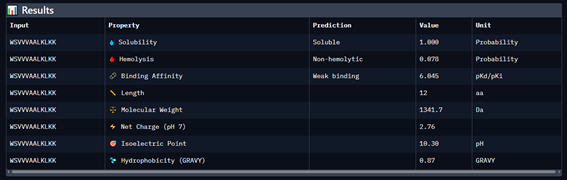
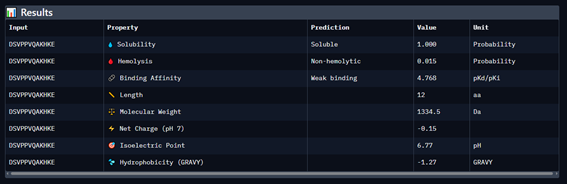
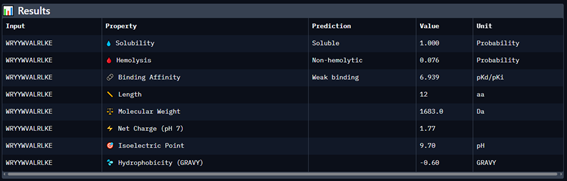
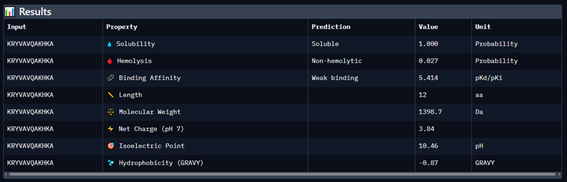
Part 4: Generate Optimized Peptides with moPPIt
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)

Part C: Final Project: L-Protein Mutants
The proposed mutations in the Bacteriophage MS2 lysis protein are generally biochemically consistent and plausible for improving stability without compromising function. The Q→E and S→T changes are conservative substitutions between polar residues that could favor electrostatic interactions or slightly increase local stability. The hydrophobic L→I and V→I mutations are particularly reasonable within the transmembrane domain, as they maintain the hydrophobicity necessary for membrane insertion and may improve helix packing. The F→Y substitution preserves aromaticity but introduces a hydroxyl group that could facilitate interactions at the membrane-water interface. Overall, these mutations do not significantly alter the length or overall hydrophobicity of the transmembrane segment, which is important because the L protein exerts its function by interfering with the MurJ lipid flippase II in Escherichia coli; therefore, the proposed changes are compatible with maintaining activity while potentially improving folding or structural stability.
Variants with more “aggressive” mutations (increasing hydrophobicity or propensity for helix formation in the transmembrane region):
- S -> L (increased hydrophobicity within the TM domain)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSLTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT - T -> L (reinforces the hydrophobic character of the propeller)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRLVTTLQQLLT - A -> L (greater transmembrane helix stability)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLLIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT - L -> F (introduces aromatic residue into TM)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFFSKFTNQLLLSLLEAVIRTVTTLQQLLT
These variants introduce less conservative changes that increase hydrophobicity, or the propensity to form α-helices, within the transmembrane domain of the Bacteriophage MS2 L protein. By replacing polar or small residues with more hydrophobic amino acids such as leucine or phenylalanine, more stable insertion into the bacterial membrane and more efficient packing of the transmembrane helix can be favored. This potentially increases the protein’s interaction with the lipid flippase II MurJ in Escherichia coli, the functional target of this lysis protein, which could intensify the inhibition of peptidoglycan precursor transport and accelerate the cell lysis process. Overall, mutations of this type aim to increase the structural stability and functional efficiency of the lysis mechanism.
Computational Analysis of Lysis Protein Mutations Using ESM Protein Language Models
Introduction
Protein language models (PLMs) such as ESM-1b and ESM2 enable the extraction of contextual evolutionary information directly from amino acid sequences. In this study, multiple computational approaches were applied to analyze mutational tolerance within a small lysis-associated membrane protein containing a predicted transmembrane (TM) domain. The objective was to evaluate whether PLM-derived representations correlate with experimentally relevant structural and functional constraints.
Methods
Mutational effects were evaluated using two complementary strategies:
- Log-Likelihood Ratio (LLR) Analysis using EsmForMaskedLM, where each residue was masked and alternative amino acids were scored according to evolutionary plausibility.
- Embedding-Based Representation Analysis, where hidden-state embeddings from ESM-1b/ESM2 were extracted and averaged to evaluate global perturbations caused by single-site mutations.
A heatmap representation of the embeddings was generated to visualize latent structural organization within the sequence.
 Image 17: ESM2 embedding heatmap representation of the protein sequence.
Image 17: ESM2 embedding heatmap representation of the protein sequence. Image 18: LLR ranking of top predicted mutations.
Image 18: LLR ranking of top predicted mutations. Image 19: Comparative mutational embedding analysis for positions 38–39.
Image 19: Comparative mutational embedding analysis for positions 38–39.Results
Table 1. Top Mutations Predicted by LLR Analysis
| Position | WT | Mutation | LLR Score | Structural Interpretation |
|---|---|---|---|---|
| 50 | K | L | 2.56 | Increased TM hydrophobicity |
| 39 | Y | L | 2.24 | TM helix stabilization |
| 52 | T | L | 1.81 | Reinforced membrane compatibility |
| 53 | N | L | 1.86 | Hydrophobic TM optimization |
| 45 | A | L | 1.54 | Helical stabilization |
Table 2. Rationally Proposed Mutations
| Mutation | Region | Expected Effect |
|---|---|---|
| S->L | TM | Increased membrane hydrophobicity |
| T->L | TM | Enhanced helix packing |
| A->L | TM | Improved TM stability |
| L->F | TM | Aromatic membrane interaction |
Embedding-based analyses at positions 38 and 39 produced highly similar effect_esm values (~0.00753–0.00759), indicating that single-point mutations do not substantially perturb the global latent representation of the protein. However, mutations preserving hydrophobic or aromatic character (Y->F, Y->L, Y->I, Y->W) appeared more compatible with the TM environment.
Discussion
The results demonstrate that protein language models effectively capture structural and evolutionary constraints associated with transmembrane domains. High-scoring mutations consistently increased hydrophobicity, reinforcing the idea that PLMs encode membrane-associated biophysical principles. However, structural compatibility predicted by ESM does not necessarily imply preserved lytic activity. Experimental observations previously indicated that some functionally essential residues, such as K50, lose activity despite appearing structurally favorable when mutated.
Thus, PLMs appear to model sequence plausibility and structural coherence more strongly than specific biological function. Consequently, these methods are highly informative for identifying conserved structural regions and designing rational variants, but experimental validation remains essential for assessing functional activity.