Week 6 HW: genetic-circuits-part-i
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is a pre-mixed PCR solution containing several essential components for high-precision DNA amplification. Its main components and functions are:
- Phusion DNA Polymerase
This is a high-fidelity polymerase derived from Pyrococcus furiosus. Its function is to synthesize new DNA strands during PCR. It has 3’-> 5’ exonuclease proofreading activity, which reduces replication errors. - dNTPs (deoxynucleotide triphosphates)
These include dATP, dTTP, dCTP, and dGTP. They are the building blocks that the polymerase uses to form the new DNA strand. - Reaction Buffer (Phusion HF or GC buffer)
This maintains optimal chemical conditions for enzyme activity, such as appropriate pH and ionic stability. It also contains salts that improve PCR specificity. - Mg²⁺ (usually MgCl₂)
Magnesium is an essential cofactor for DNA polymerase. It allows the enzyme to catalyze the formation of phosphodiester bonds between nucleotides. - Stabilizers and detergents
Small molecules that protect the enzyme and improve reaction stability, helping to maintain activity during the thermal cycles of PCR. Together, these components allow for DNA amplification with high precision and a low error rate, which is especially important in applications such as site-directed mutagenesis or cloning, where even a single nucleotide error can alter the experimental outcome.
What are some factors that determine primer annealing temperature during PCR?
The annealing temperature in PCR depends primarily on the primer properties and reaction conditions. Some key factors are:
- Primer melting temperature (Tm): This is the most important factor. Tm depends on the primer sequence and represents the temperature at which 50% of the primer is bound to the template DNA. The annealing temperature is usually set 3–5 °C below Tm.
- GC content: G–C pairs form three hydrogen bonds, while A–T pairs form two. Therefore, a primer with a higher percentage of GC will have a higher Tm and require a higher annealing temperature.
- Primer length: Longer primers form more interactions with the template DNA, which increases hybrid stability and raises the annealing temperature.
- Complementarity with the template DNA: If mismatches exist between the primer and the target sequence, the binding is less stable and may require lower hybridization temperatures.
- Reaction conditions: Factors such as Mg²⁺ concentration, salts, and buffer additives influence the stability of DNA-DNA hybridization and can modify the optimal temperature.
- Primer secondary structures: The formation of primer dimers or hairpins can affect the primer’s availability to bind to the template DNA, altering the optimal hybridization temperature.
Together, these factors determine the temperature at which the primer binds specifically and efficiently to DNA during PCR, which is crucial for accurate amplification.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Two common methods used to generate linear DNA fragments are PCR amplification and restriction enzyme digestion, and they differ in both protocol and experimental applications.
PCR (Polymerase Chain Reaction) amplifies a specific DNA region using sequence-specific primers, a thermostable DNA polymerase, nucleotides, and thermal cycling. The process involves repeated cycles of denaturation, primer annealing, and extension, producing large amounts of a defined DNA fragment. PCR is particularly useful when the desired DNA segment must be selectively amplified from a template, when the starting DNA is present in small amounts, or when mutations, tags, or new restriction sites need to be introduced through primer design.
In contrast, restriction enzyme digestion uses enzymes that recognize specific DNA sequences and cut the DNA at those sites, generating defined fragments with blunt or sticky ends. The protocol is simpler: DNA is incubated with the appropriate restriction enzyme(s), buffer, and temperature conditions. This method is preferable when the DNA already contains the desired restriction sites, when one needs precise ends for cloning, or when verifying plasmids or isolating fragments from existing constructs.
In summary, PCR is best for generating or modifying specific DNA sequences and amplifying small amounts of DNA, while restriction enzyme digestion is ideal for cutting existing DNA molecules at known sites to produce fragments suitable for cloning or analysis. Both methods generate linear DNA but serve different experimental purposes.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure that DNA fragments generated by PCR or restriction digestion are suitable for Gibson Assembly, the key requirement is that all fragments share compatible overlapping ends.
- Design overlapping regions (most critical factor): Each fragment must have ~20–40 bp of sequence homology with the adjacent fragment.
• For PCR products, this is achieved by adding overlaps directly into the 5′ ends of primers.
• For restriction-digested fragments, you must verify that the cut sites preserve or expose the required overlaps, or add them via PCR if needed. - Ensure correct fragment sequence and orientation: The overlaps must correspond exactly to the adjacent fragment sequence and direction, otherwise assembly will fail or produce incorrect constructs.
- Use high-fidelity PCR: Amplify fragments with a high-accuracy enzyme (e.g., Phusion) to minimize mutations that could disrupt overlap regions or coding sequences.
- Generate clean, specific fragments:
• Confirm size by gel electrophoresis
• Purify fragments to remove primers, enzymes, and nonspecific products
• Contaminants can inhibit the Gibson reaction. - Avoid incompatible ends: Unlike traditional cloning, Gibson does not require restriction sites, but fragments must be linear and not have conflicting overhangs that interfere with exonuclease processing.
- Balance fragment concentrations: Use roughly equimolar amounts of each fragment to improve assembly efficiency.
How does the plasmid DNA enter the E. coli cells during transformation?
During transformation, plasmid DNA enters Escherichia coli cells only after the cells are made competent, meaning their membranes are temporarily altered to allow DNA uptake. There are two main mechanisms:
- Chemical transformation (CaCl₂ + heat shock): Cells are treated with calcium chloride, which helps neutralize the negative charges on both the DNA and the bacterial cell membrane. This allows the plasmid DNA to approach the cell surface. A brief heat shock (≈42 °C) then creates a thermal imbalance that induces transient pores or increases membrane permeability, enabling the plasmid DNA to enter the cytoplasm.
- Electroporation: Cells are exposed to a short, high-voltage electric pulse, which creates temporary pores in the membrane. The plasmid DNA is driven into the cell by the electric field and enters through these pores. After the pulse, the membrane reseals.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly is a DNA assembly technique that uses Type IIS restriction enzymes, such as BsaI, which cut outside of their recognition sequences. This allows researchers to design custom overhangs that determine how multiple DNA fragments will join together in a specific order. In a single reaction, the DNA fragments, restriction enzyme, and ligase are combined. The enzyme cuts the DNA to create compatible overhangs, and the ligase joins the fragments, resulting in a seamless assembly where the original restriction sites are removed. This method is highly efficient for assembling multiple DNA fragments simultaneously and in a defined orientation, making it ideal for modular cloning applications. It is particularly useful when working with standardized genetic parts, such as promoters, coding sequences, and terminators. However, it requires careful design to avoid internal restriction sites within the fragments and to ensure correct overhang compatibility. Overall, Golden Gate Assembly enables rapid, scarless, and high-throughput DNA construction.
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Diagram (simplified)
Step 1: Fragments with Type IIS sites
[Fragment A]– BsaI–> [Fragment B]–BsaI–> [Fragment C]
Step 2: Cutting creates custom overhangs
A: ATG |—-
B: | TAC
C: | GGA
Step 3: Complementary overhangs anneal
ATG —- TAC —- GGA
Step 4: Ligase seals fragments
[Fragment A][Fragment B][Fragment C] (final construct)
Assignment: Asimov Kernel
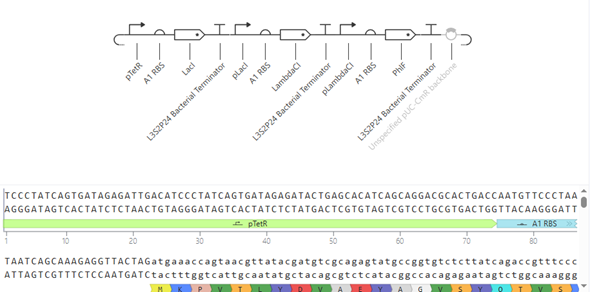
The construction consists of assembling three independent expression modules onto a single pUC-CmR vector: pTet–RBS A1–LacI–L3S2P24 terminator, pLac–RBS A1–λCI–L3S2P24, and pλ–RBS A1–PhlF–L3S2P24. Each module includes a specific promoter, a strong ribosomal binding site (A1), a repressor CDS, and a strong bacterial terminator to isolate transcription. The three cassettes are arranged in series to form a closed-loop cross-repression system.
The simulation was plausible; the design needs to be optimized, but I couldn’t do it due to lack of time.
Function
Each module should function as a “block” where the promoter activates the expression of its repressor, but that same repressor inhibits the next promoter in the chain; thus, pTet produces LacI which turns off pLac, pLac produces λCI which turns off pλ, and pλ produces PhlF which again turns off pTet, generating a cycle of cross-repression that, ideally, produces oscillations in the expression of each gene.
Explanation
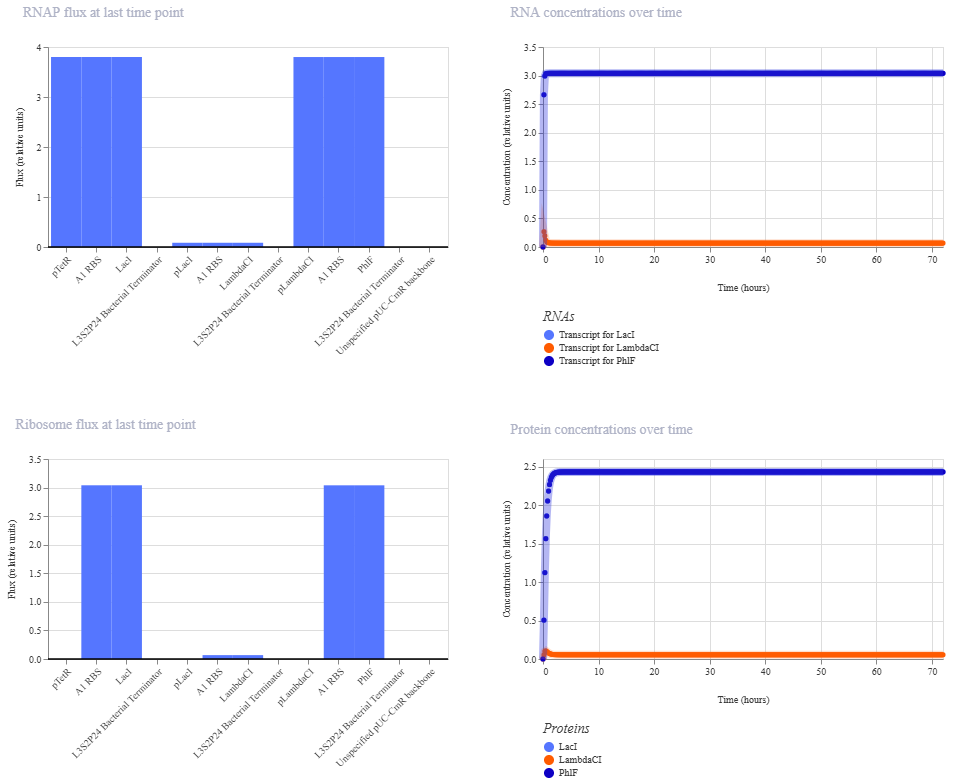
The simulation shows that the circuit is not generating repressilator-type oscillations, but rather rapidly converges to a steady state dominated by LacI and PhlF expression, while LambdaCI remains virtually repressed throughout the 72-hour simulation. Both the RNA and protein curves reach a plateau very early, approximately within the first few hours, and then remain constant until the end. This indicates that the system found a stable equilibrium rather than oscillatory dynamics.
RNAP and ribosome flow plots reinforce this interpretation. High transcriptional and translational activity is observed associated with the modules controlled by pTet and pLambda, while the pLac module exhibits almost no activity. Biologically, this suggests that LacI efficiently represses pLac, preventing significant LambdaCI production. Consequently, repression of pLambda is insufficient, allowing for the sustained accumulation of PhlF. The circuit is thus trapped in a stable, asymmetric state.
This behavior is consistent with what is expected for a repressilator without fine-tuning of kinetics. Although the logical architecture of the assembly is correct, the three modules appear too strong and too stable to produce spontaneous oscillations. The absence of rapid degradation labels (such as LVA/ssrA), the use of identical strong RBSs, and the high-copy-number pUC backbone likely favor excessive protein accumulation and dynamic damping. In real oscillatory systems, controlled degradation and a precise balance between synthesis and repression are critical to avoid steady states.
Furthermore, the fact that the system reaches stability so quickly despite a long simulation (72 h with 10-minute timesteps) indicates that the behavior does not correspond to slow, undersampled oscillations, but rather to a genuine convergence of the dynamic system. In other words, increasing the simulation duration would probably not substantially alter the result as long as the regulatory architecture remains the same.
Overall, the simulation demonstrates that the circuit is functional as a cross-repression system, but not as a robust oscillator under the current conditions. The design would likely require promoter/RBS tuning, protein stability reduction, or the introduction of active degradation to generate sustained oscillatory dynamics.









