Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
https://www.uniprot.org/uniprotkb/P00441/entry#sequences
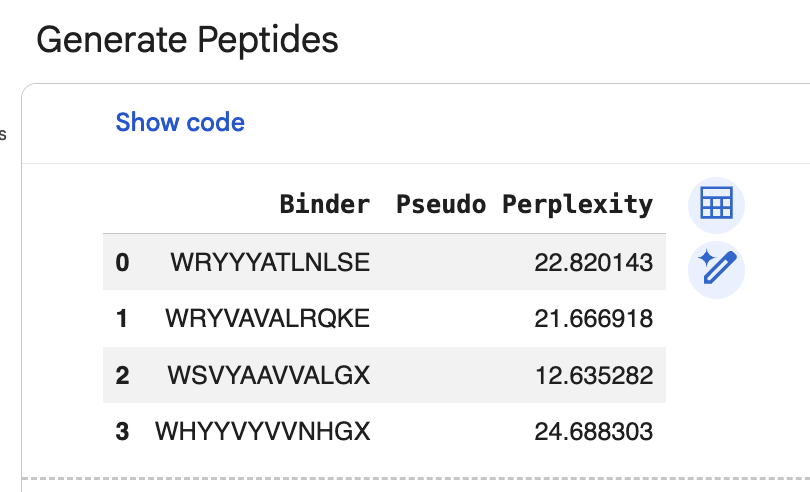
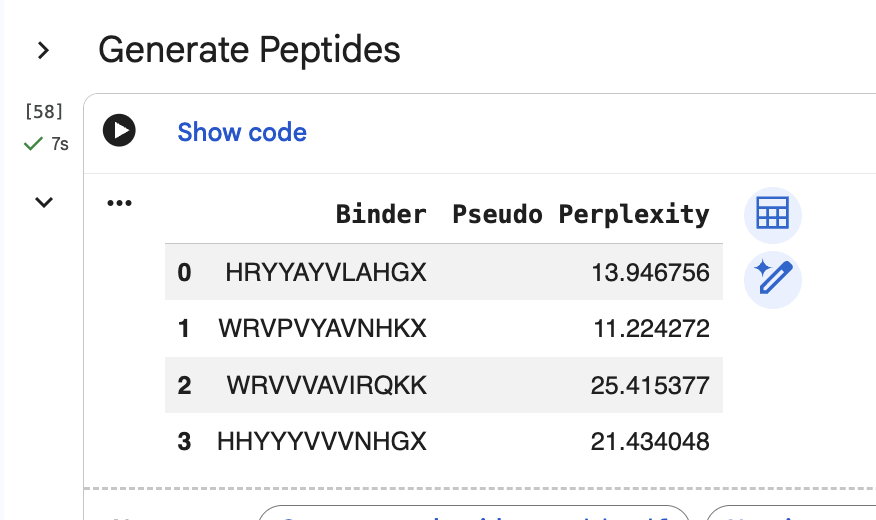
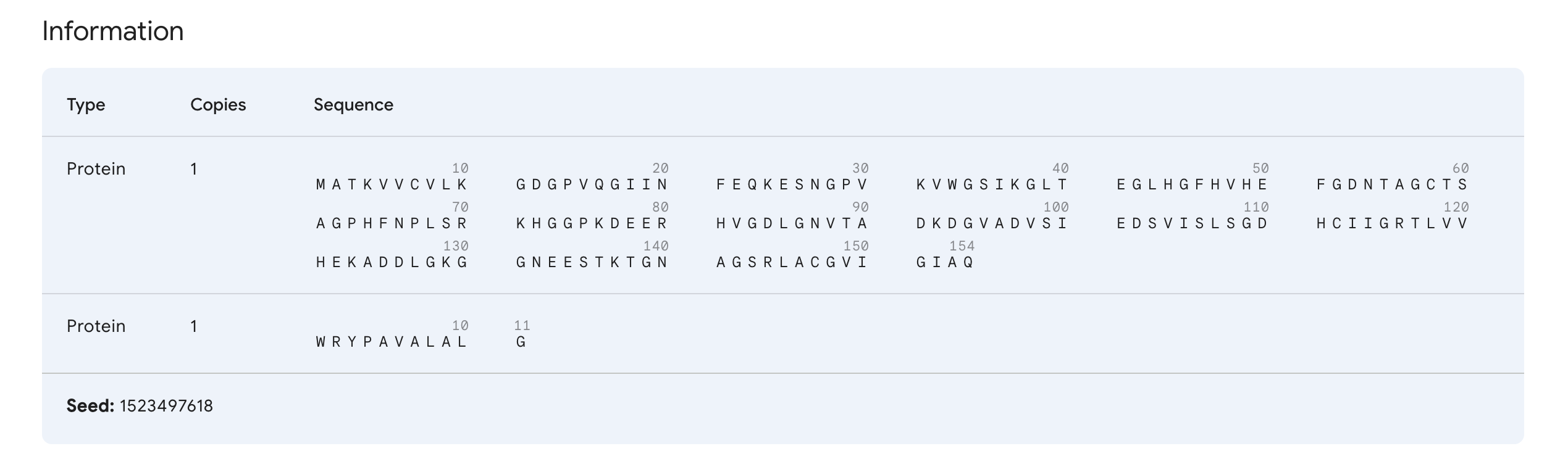
“Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.” I did this step several times. Here are two examples of my generating four peptides:
“To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.”
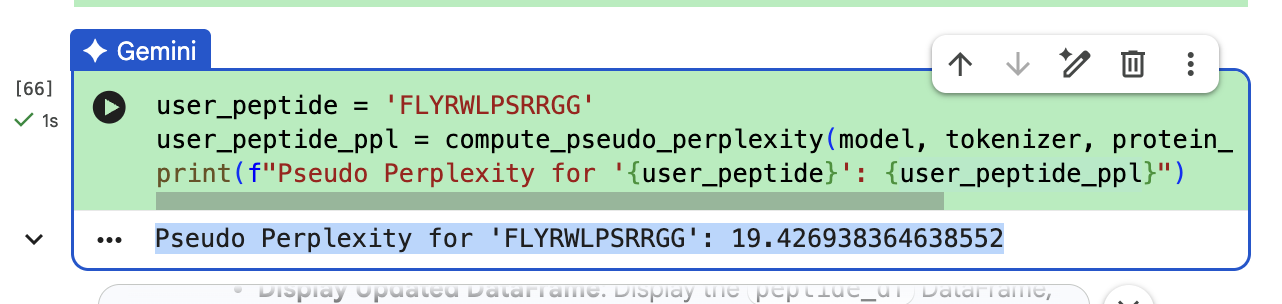
“Record the perplexity scores that indicate PepMLM’s confidence in the binders.” These can be seen in the above screenshiots, and I have also recorded them in an excel spreadsheet.
Part 2: Evaluate Binders with AlphaFold3
“Navigate to the AlphaFold Server: alphafoldserver.com For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.” This is one example:
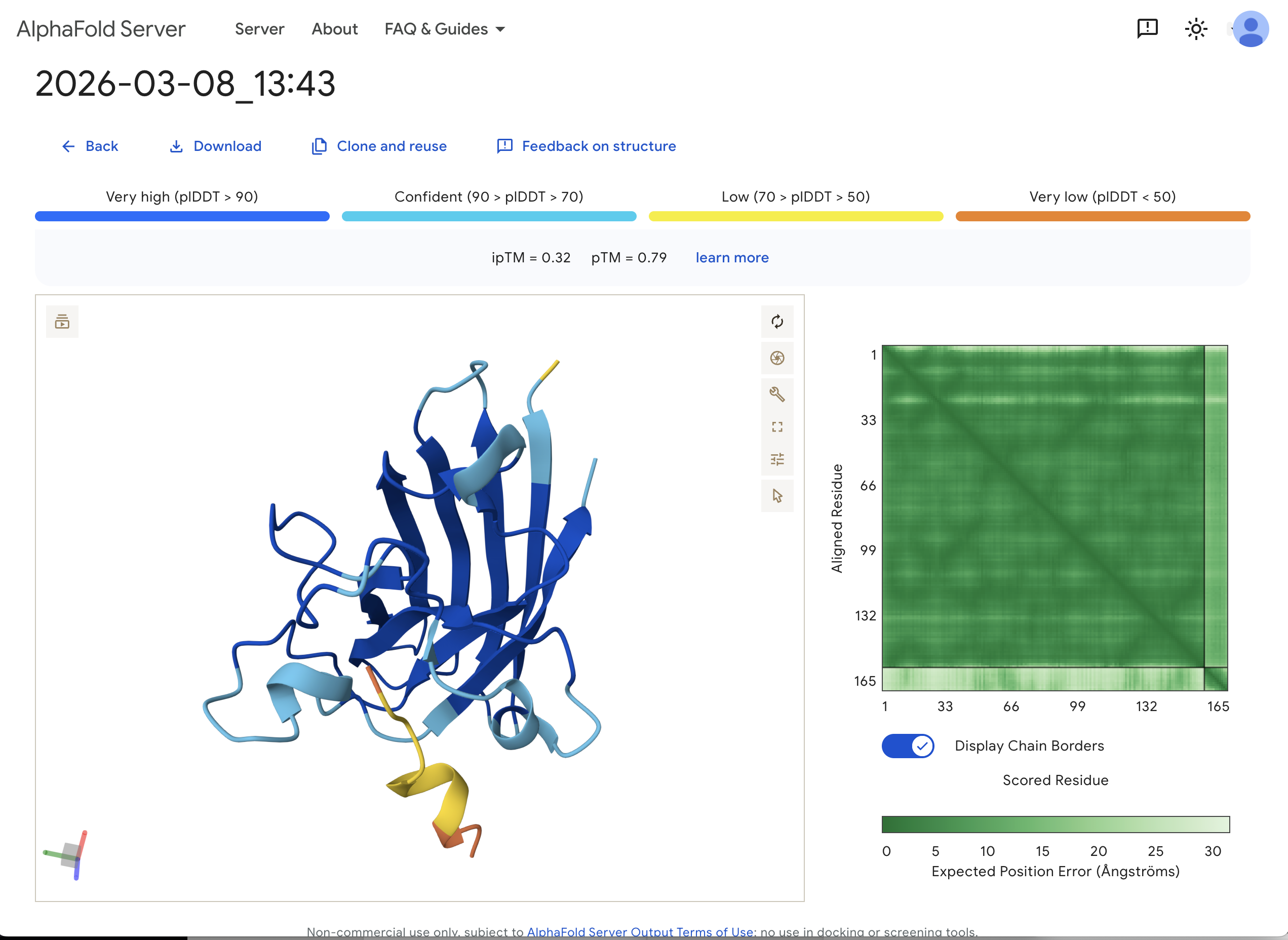
“Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?”
In teh one above, I’d say probably surface bound, and closer to the β-barrel region than to the N-terminus.
“In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.”
The ipTM value I got for the known binder was 0.31. The peptides I generated ranged from 0.28 to 0.43 in ipTM values. My understanding is that higher ipTM values show more confidence in the prediction. I hade a few that were slightly higher than the knonw binder, like WRYPAVALALGX at 0.38 and my highest at 0.43 was WHYYVYVVNHGX.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Below is a screenshot of part of my spreadsheet of results of my work in Peptiverse:
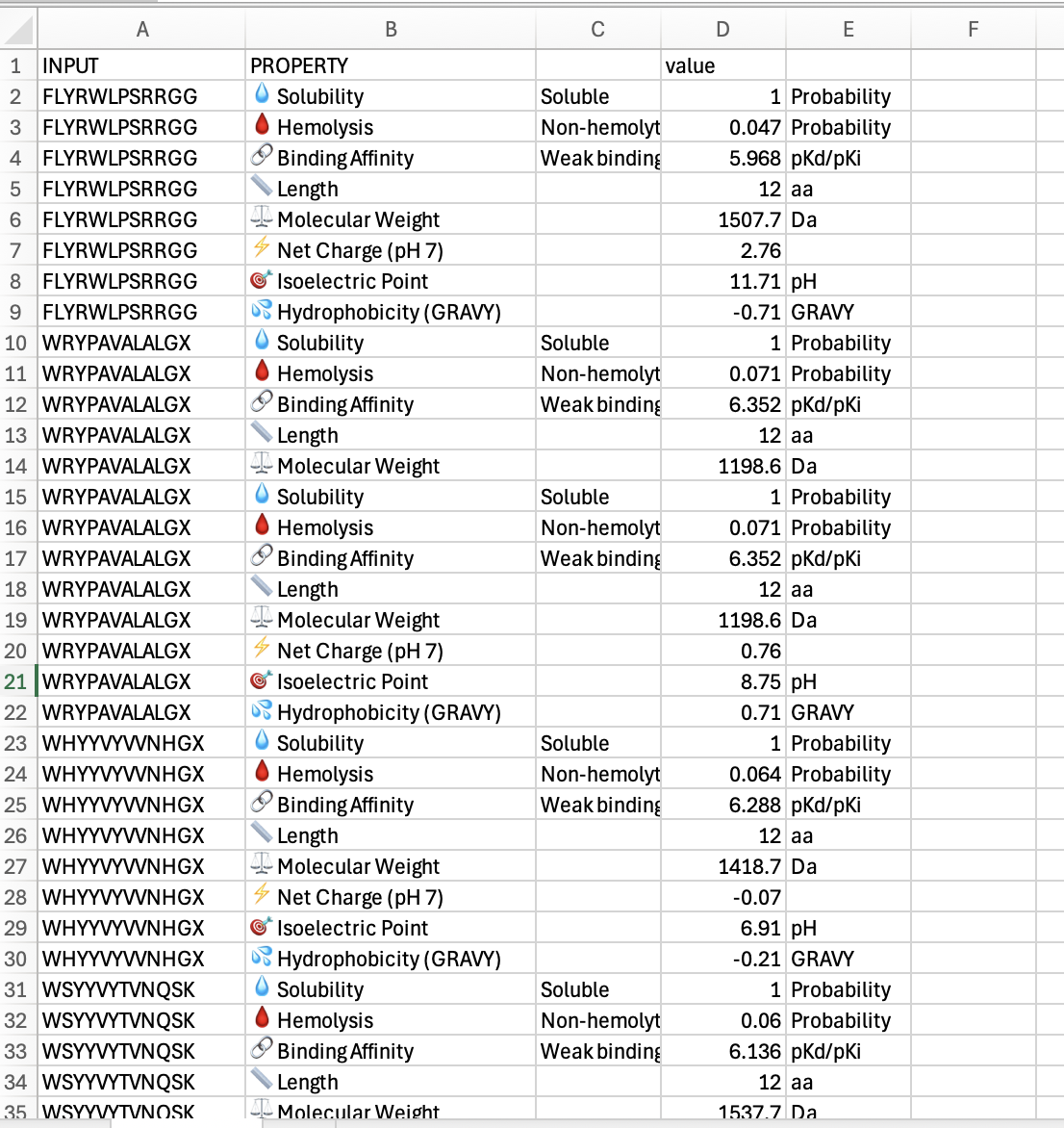
“Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties? Choose one peptide you would advance and justify your decision briefly.”
The few peptides I tested did not show much difference. They each were soluble, and each had weak binding affinity. The widest diferences I saw were in hydrophobicity, where i had some positives and some ngatives. The one that was closest to zero was at -0.21 which was WHYYVYWNHGX. Of these, that might eb the one that I would advance. I would probbaly go back and look for ones that had more binding affinity.
Part 4: Generate Optimized Peptides with moPPIt
I am rerunning this … ran on Sunday but did not save my results …
Some results of my rerun:
| Result | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| STKLHTKIKCQC | 0.9697153624147177 | 0.8333333134651184 | 6.467465877532959 | 0.7173478007316589 |
| SVTKKETQKRFA | 0.9688531029969454 | 0.75 | 5.76106595993042 | 0.7000954747200012 |
| GSAEMTCKKQRK | 0.9745583962649107 | 0.8333333134651184 | 6.006259441375732 | 0.6160933375358582 |
“briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?”
These seem to be getting different results by using different strategies. For advancing to trials, I would probably test a few of the best results found from each system.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
(optional, might get to later)
Part C: Final Project: L-Protein Mutants
“Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid”:
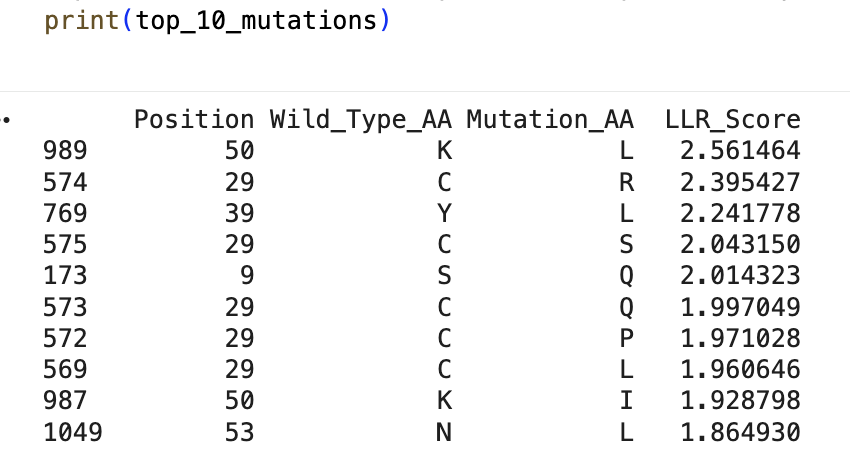
“does the experimental data correlate with the scores from the notebook”?
… I don’t think so … might’ve done something off … or maybe these are just different systems with different results …