Eric Millikin is an American artist based in Baltimore, Maryland, (previously in Detroit, Michigan, and Richmond, Virginia) with over 30 years of experience creating computer, internet, video, biological, and artificial intelligence artwork. Millikin comes from a working-class family, growing up in a mobile home in the woods of rural Michigan. He is a first-generation college student and National Merit Scholar who earned his BFA from Michigan State University where he created AI-generated postmodern poetry and interactive video installation art, and his MFA from Virginia Commonwealth University where he created live-AI-generated computer art installations and political protest art. His artwork has been featured by WIRED, USA Today, Ripley’s Believe It or Not!, and The New York Times Sunday Arts section. His work has been included in recent exhibitions at the Royal Scottish Academy in Edinburgh, Charles University in Prague, and the Festival and Congress Centre in Varna, Bulgaria. His artwork has won well over 50 international, national and regional visual journalism awards, which includes interactive data visualizations for Pulitzer Prize-winning investigations, motion graphics for Emmy-winning documentaries, and a Society for News Design medal for newspaper front page illustration. Millikin has recently been awarded international residencies at Cow House Studios in County Wexford, Ireland, the Ayatana Biophilium Artists’ Research Program “Symbiosis,” hosted from Ottawa, Canada, and the virtual reality residency “Artist is Absent” hosted by Nazar Voitovich Art Residence (NVAIR) in Travneve, Ukraine as the Russian military invaded. He is an Assistant Professor of Visual Arts, Animation and Interactive Media at the University of Maryland, Baltimore County.
SCOBY DNA Steganography A biological engineering application or tool that I want to develop is a system for biomaterials for artist books that contains the book contents in its DNA. Above is an example of a a previous project of mine, where I was making masks out of SCOBYs, Symbiotic Cultures Of Bacteria and Yeast. A next level project might be to use SCOBY to create pages of artist books, where the the content of the pages (illustrations, text, relief sculptures, etc.) is also written into the DNA of the yeasts and/or bacteria that makes up the SCOBY. The reasons I am interested in this, and believe others will be interested as well, is primarily twofold: 1) I am interested in this as an unusual form of artwork that “grows on you” on multiple levels (such as genetic level, personal level, cultural(!) level, etc.) and 2) I am interested in this as a form of storytelling and story distrubution that can replicate itself, where the DNA creates new copies as the yeast reproduces, then thise new copies are used to create new copies of books.
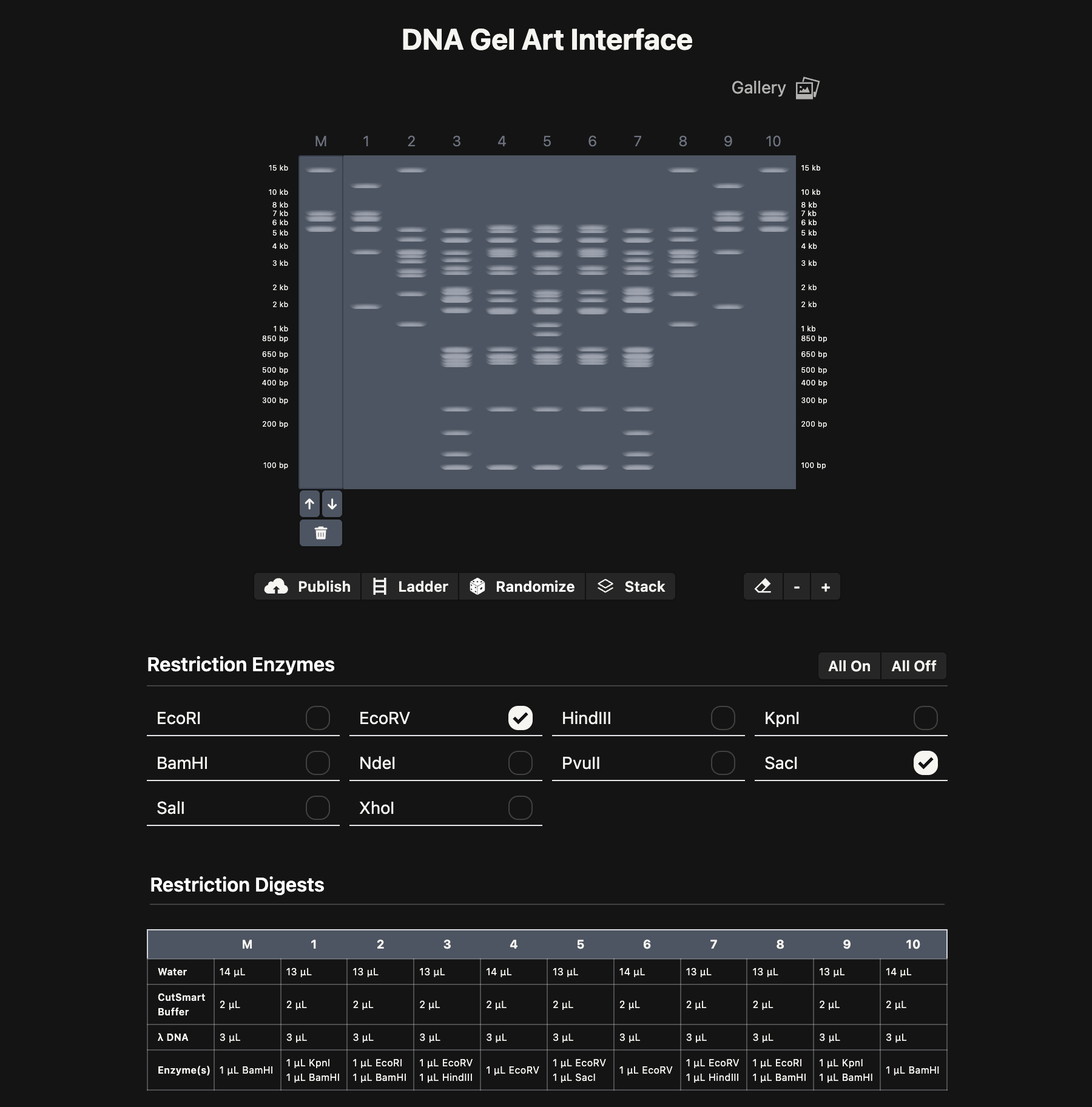
Part 1: Benchling & In-silico Gel Art Above is maybe my favorite Gel Electrophoresis design I created. I was trying to make monster faces, so hopefully this looks sort of like a skull with horns! I made this with the “DNA Gel Art Interface” website created by Ronan at https://rcdonovan.com/gel-art
Part 1: Generate an artistic design NOTE: Some of my newer and hopefully maybe better images are toward the end!
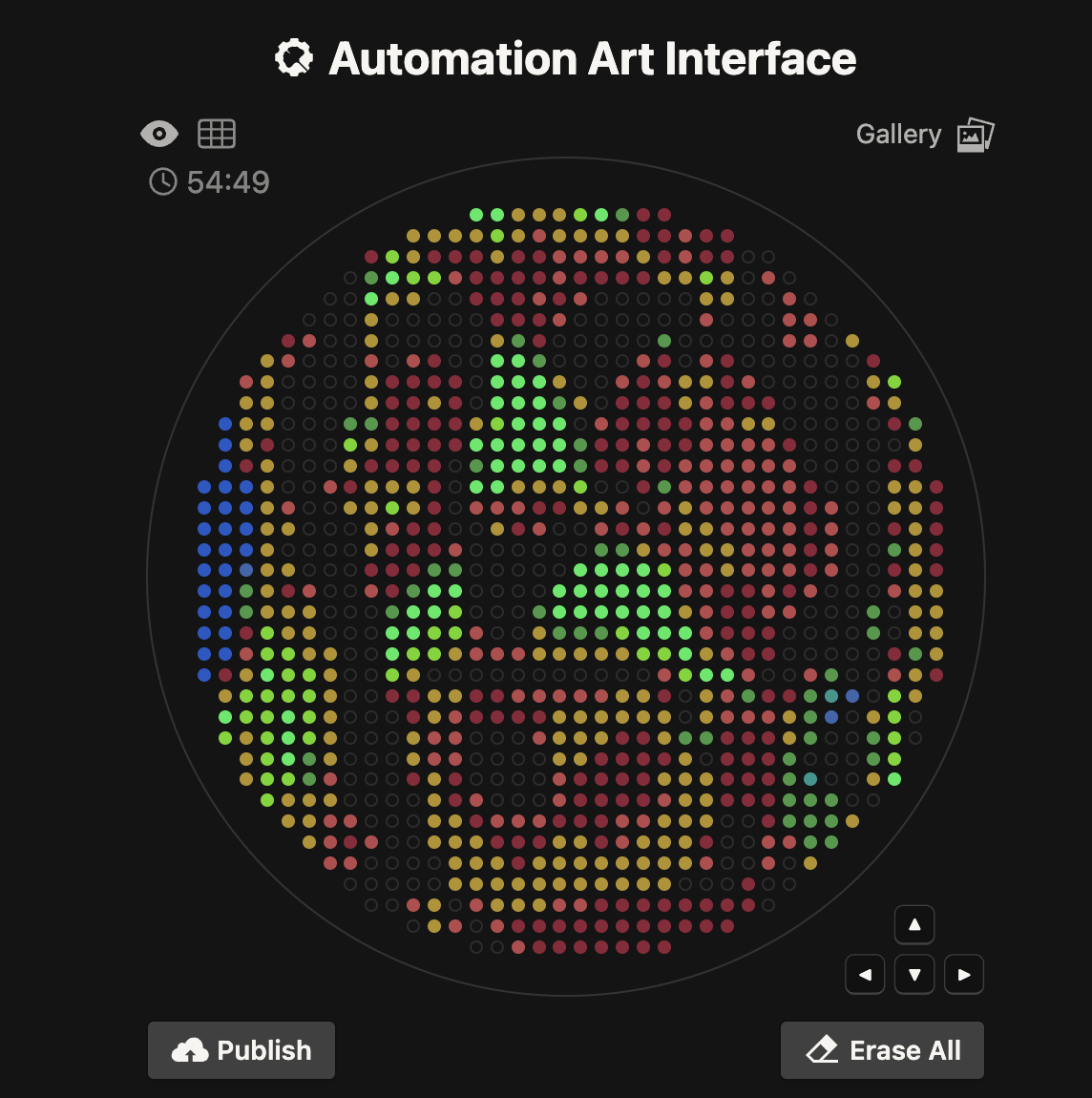
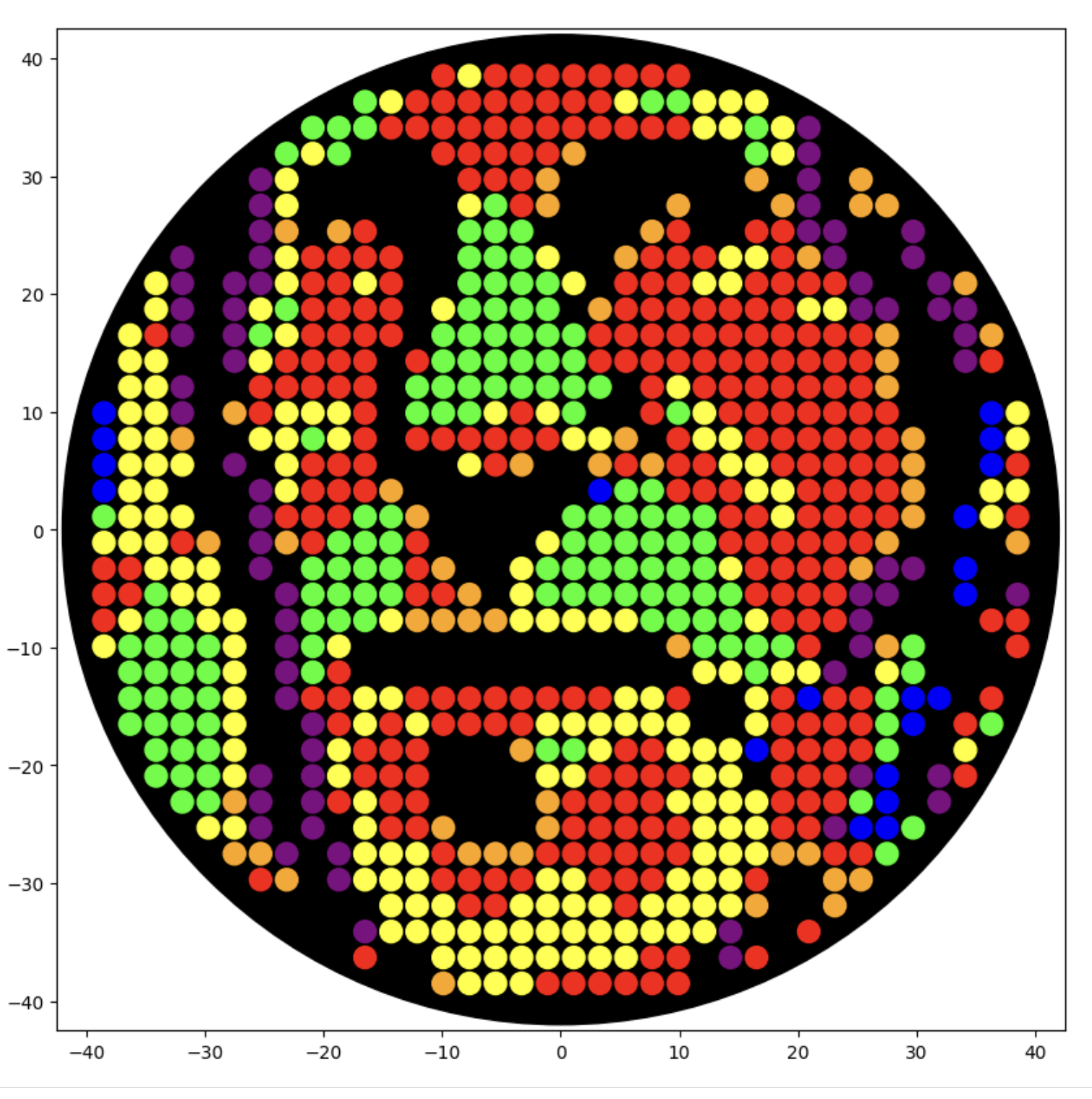
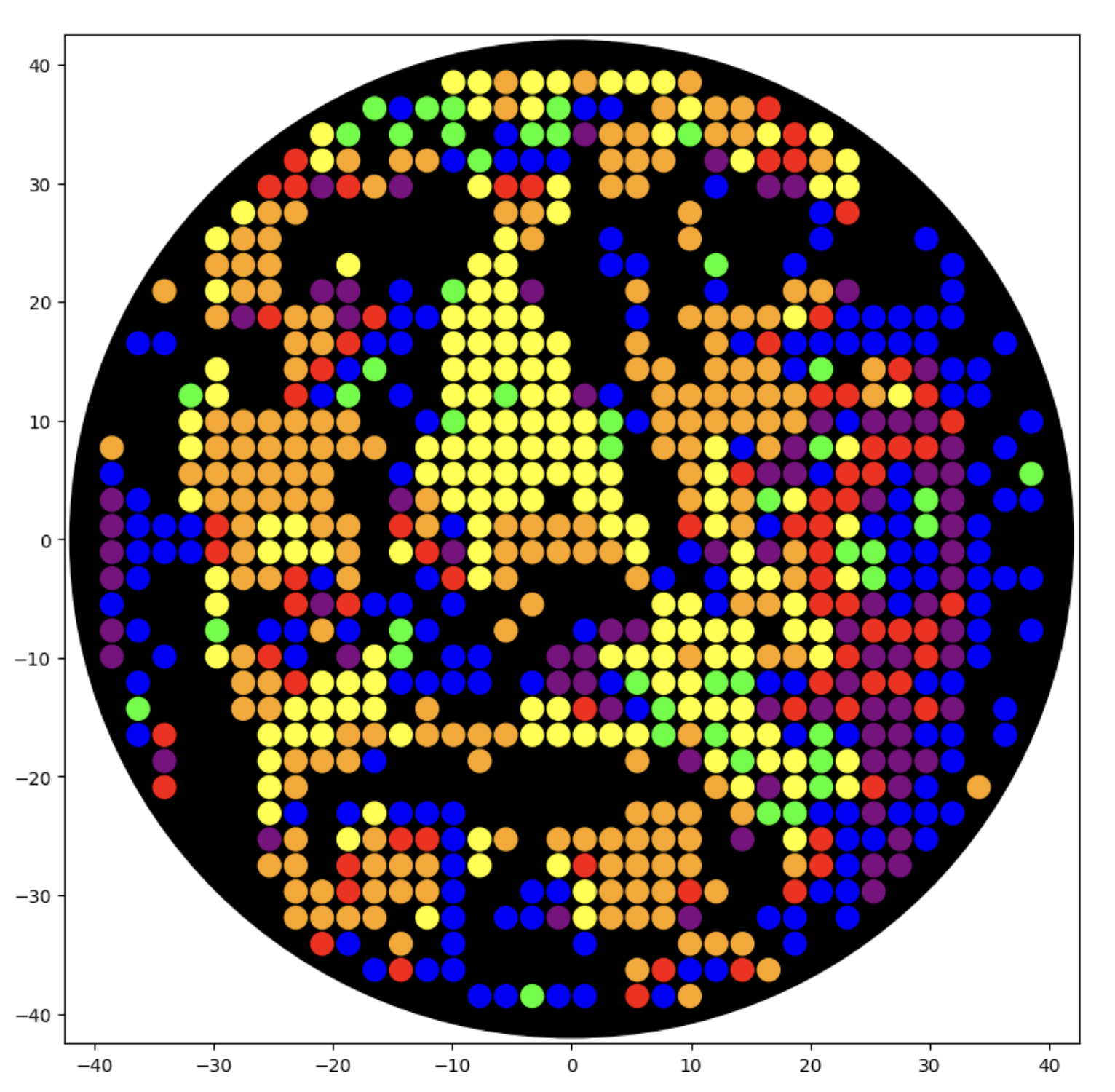
I generated the above artistic design – a self portrait – using the GUI at opentrons-art.rcdonovan.com – NOTE: BUGSS Lab has colors: orange, green, yellow, purple, red, blue
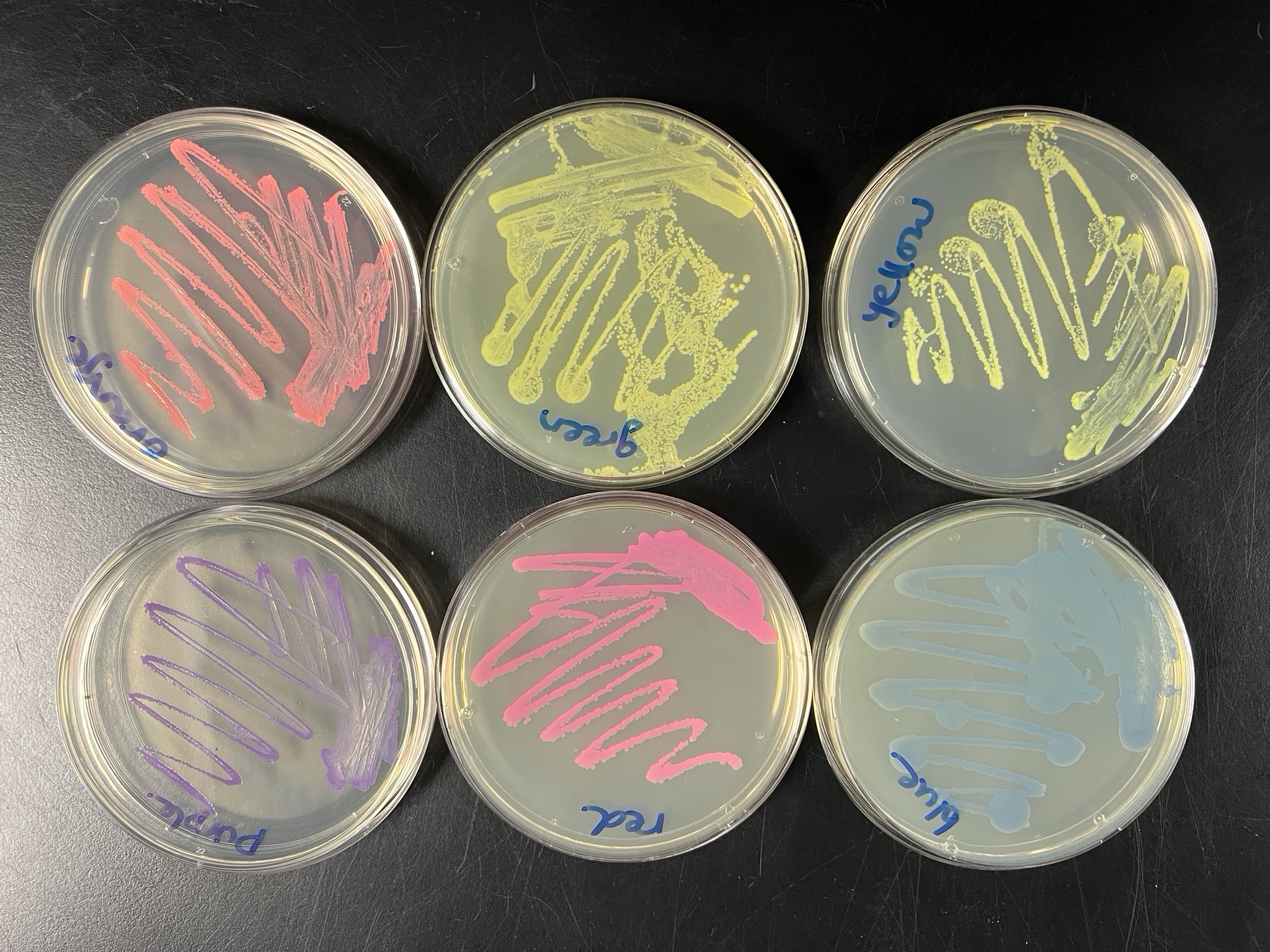
A below is me working on simulating that, redrawing and recoloring little bit in the Google Colab notebok with the six colors at BUGSS Lab. Thanks to Amanda and everyone for helping hack up that code.
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is about 20% protein, so that is 100 g of protein. There are 6.022e+23 Daltons per gram, so for 100 grams that is 6.022e+25 Daltons. Then if there are 100 Daltons in an average amino acid, we’re back to 6.022e+23 molecules of amino acids.
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM https://www.uniprot.org/uniprotkb/P00441/entry#sequences
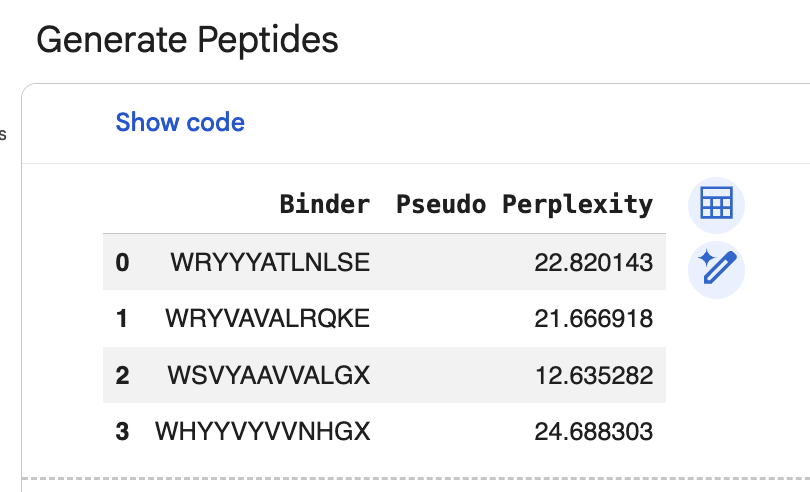
“Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.” I did this step several times. Here are two examples of my generating four peptides:
Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? From New England Biolabs “Phusion High-Fidelity PCR Master Mix with HF Buffer is a 2X master mix consisting of Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2. All that is required is the addition of template, primers and water.”
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits are binary digital on/off. IANNs can be more analog, with greater ranges, which is shown in the blue gradient diagrams in the neuromorphic wizard.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
#General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
From “A User’s Guide to Cell-Free Protein Synthesis,” https://pmc.ncbi.nlm.nih.gov/articles/PMC6481089/
Two advantages would be 1) “elimination of reliance on living cells,”" so we don’t hve to wait for the living organism’s growth cycle an maintain their living conditions, and 2) “the ability to focus all system energy on production of the protein of interest.” Two cases where cell-free systems have benfits over in vivo methods include 1) “The production of functional antibodies and antibody fragments in vitro using CFPS has the potential to allow for simplification of the antibody production process for more rapid manufacturing,” and 2) “Metalloproteins … are difficult to produce in vivo … have the potential to enable renewable hydrogen fuel and other important biotechnological advancements.”
Homework: Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I contributed a few early pixels to the HTGAA global artwork experiment at https://rcdonovan.com/1536
I will say that they were largely decorative, incremental changes because I was working when the wait time between each person’s changes was one minute, so I wasn’t able to make any really drastic changes. I was mostly just trying to add some additional variety and visual interest to what other collaborators had already drawn.
Subsections of Homework
Week 1 HW: Principles and Practices
SCOBY DNA Steganography
A biological engineering application or tool that I want to develop is a system for biomaterials for artist books that contains the book contents in its DNA. Above is an example of a a previous project of mine, where I was making masks out of SCOBYs, Symbiotic Cultures Of Bacteria and Yeast. A next level project might be to use SCOBY to create pages of artist books, where the the content of the pages (illustrations, text, relief sculptures, etc.) is also written into the DNA of the yeasts and/or bacteria that makes up the SCOBY. The reasons I am interested in this, and believe others will be interested as well, is primarily twofold: 1) I am interested in this as an unusual form of artwork that “grows on you” on multiple levels (such as genetic level, personal level, cultural(!) level, etc.) and 2) I am interested in this as a form of storytelling and story distrubution that can replicate itself, where the DNA creates new copies as the yeast reproduces, then thise new copies are used to create new copies of books.
The chose the mask example above because these might be sort of like Lovecraftian grimoires, sell books with monstrous faces on them, as I am thinking of suffucuenty advanced technologies being indistinguishable from magic, computer coded and genetic codes as forms of magic, etc. This is also based on “steganography,” the practice of embedding hidden coded information within another object. The term “steganography” dates back to the “Steganographia” from 1499, whcih is a book of cryptography disguised as a book of magic.
I could also possibly drink the brewed kombucha to let the DNA into my own body’s systems, and possibly sell the drink to others during art events, where people coul ddrink the kombucha that makes the SCOBYs that make the pages of the books …
Governance goals and actions
My main governance/policy goals are to make sure that this biomaterial system is safe for artists like myself when they are creating the materials, and also to make sure they are safe for viewers and collectors of the artist books. I will name these goals as the standard goals provided in class, 1) Enhance Biosecurity, 2) Foster Lab Safety, 3) Protect the environment, and 4) Other considerations.
My three different potential governance “actions” are 1) Develop guide for safe SCOBY DNA Steganography creation for artists, 2) Provide screening and training of potential collecting institutions so they can safely handle and preserve the books, and 3) Register any kombucha drink manufacturing facility with the FDA, and adhere to FDA regulations related to creating and selling drinks.
Scoring from 1-3 with 1 as the best, 3 as the worst:
Does the option:
Develop guide for safe SCOBY DNA Steganography creation for artists
Provide screening and training of potential collecting institutions so they can safely handle and preserve the books
Register any kombucha drink manufacturing facility with FDA, adhere to FDA regulations
Enhance Biosecurity
• By preventing incidents
1
2
1
• By helping respond
2
2
1
Foster Lab Safety
• By preventing incident
1
3
1
• By helping respond
2
3
1
Protect the environment
• By preventing incidents
1
1
1
• By helping respond
2
2
1
Other considerations
• Minimizing costs and burdens to stakeholders
3
1
2
• Feasibility?
1
3
2
• Not impede research
1
2
2
• Promote constructive applications
1
3
2
Based upon this scoring, I think the governance option I would prioritize is “Develop guide for safe SCOBY DNA Steganography creation for artists.” This is not only an effective option for creating lab safety for artists, and protecting the environment and others in it, it is also a governance option that I know would be highly feasible as I would be th eprimary artist to use it. If this project scales up and includes othwrs drinking the kombucha used to make the SCOBYS, then FDA registration and following FDA regualtiosn woudl be required and highly beneficial.
Refelctning on this project proposal and new ethical concerns that I am thinking about, I would say that the combination of this as a lab safety project, an art preservation project, and a potential food safety project. I would say that those are areas that I have previously thought of individually, and am now thinking of them all together on the same project for the first time. I am also now thinking about whether this also introduces other governance/policy, perhaps related to shipping, storage, etc.
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
The error rate of polymerase is 1:106. The human genome length is roughly 3 x 109 base pairs. The error rate is lower because of MutS Repair System in Error Correcting Gene Synthesis (slides 14 and 15).
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Google AI Overview says: “An average human protein (~500 amino acids) can be encoded by a practically astronomical number of different DNA sequences, potentially exceeding (10^{100}) combinations, due to codon redundancy where 61 triplets encode 20 amino acids. However, only a few of these codes are functionally efficient or viable in vivo due to factors like codon usage bias, mRNA stability, and proper folding.”
What’s the most commonly used method for oligo synthesis currently?
Solid phase synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Google AI Overview says: “Making oligonucleotides (oligos) longer than 200 nucleotides (nt) via direct chemical synthesis is difficult primarily because of exponentially decreasing yields caused by imperfect coupling efficiency and the accumulation of errors. "
Why can’t you make a 2000bp gene via direct oligo synthesis?
Google AI Overview says: “A 2000bp gene cannot be produced via direct (single-pass) chemical oligonucleotide synthesis because the efficiency of the coupling reaction drops significantly, leading to low yields of full-length product and high rates of sequence errors (insertions/deletions). "
Using Google & Prof. Church’s slide #4 What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Google AI Overview says: “The 10 essential amino acids that all animals must obtain from their diet (as they cannot synthesize them in sufficient quantities) are phenylalanine, valine, threonine, tryptophan, isoleucine, methionine, histidine, arginine, leucine, and lysine. This reality makes the “Lysine Contingency” from Jurassic Park scientifically flawed, as all vertebrates, including engineered dinosaurs, would already be unable to synthesize lysine, rendering the engineered deficiency redundant.”
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
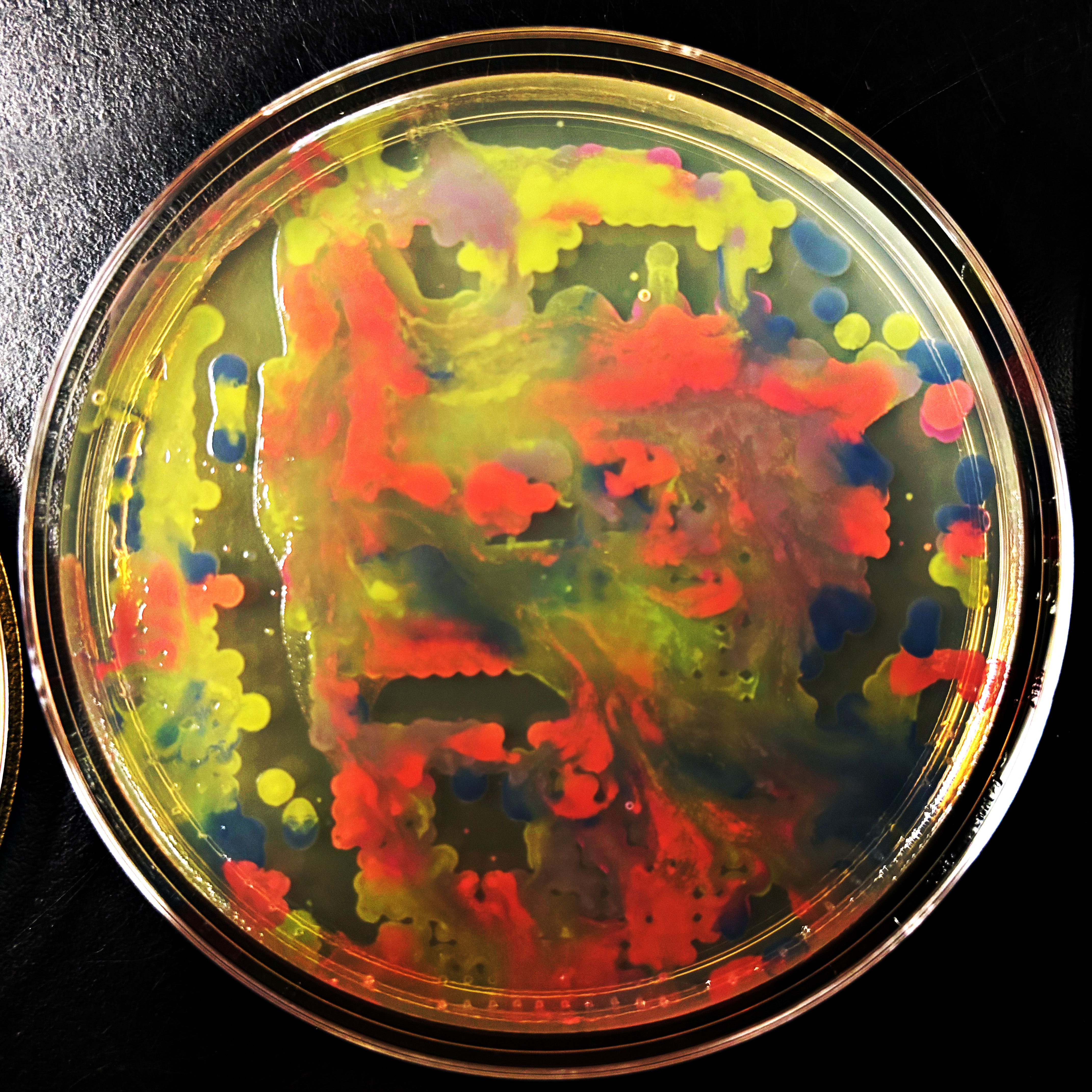
Above is maybe my favorite Gel Electrophoresis design I created. I was trying to make monster faces, so hopefully this looks sort of like a skull with horns! I made this with the “DNA Gel Art Interface” website created by Ronan at https://rcdonovan.com/gel-art
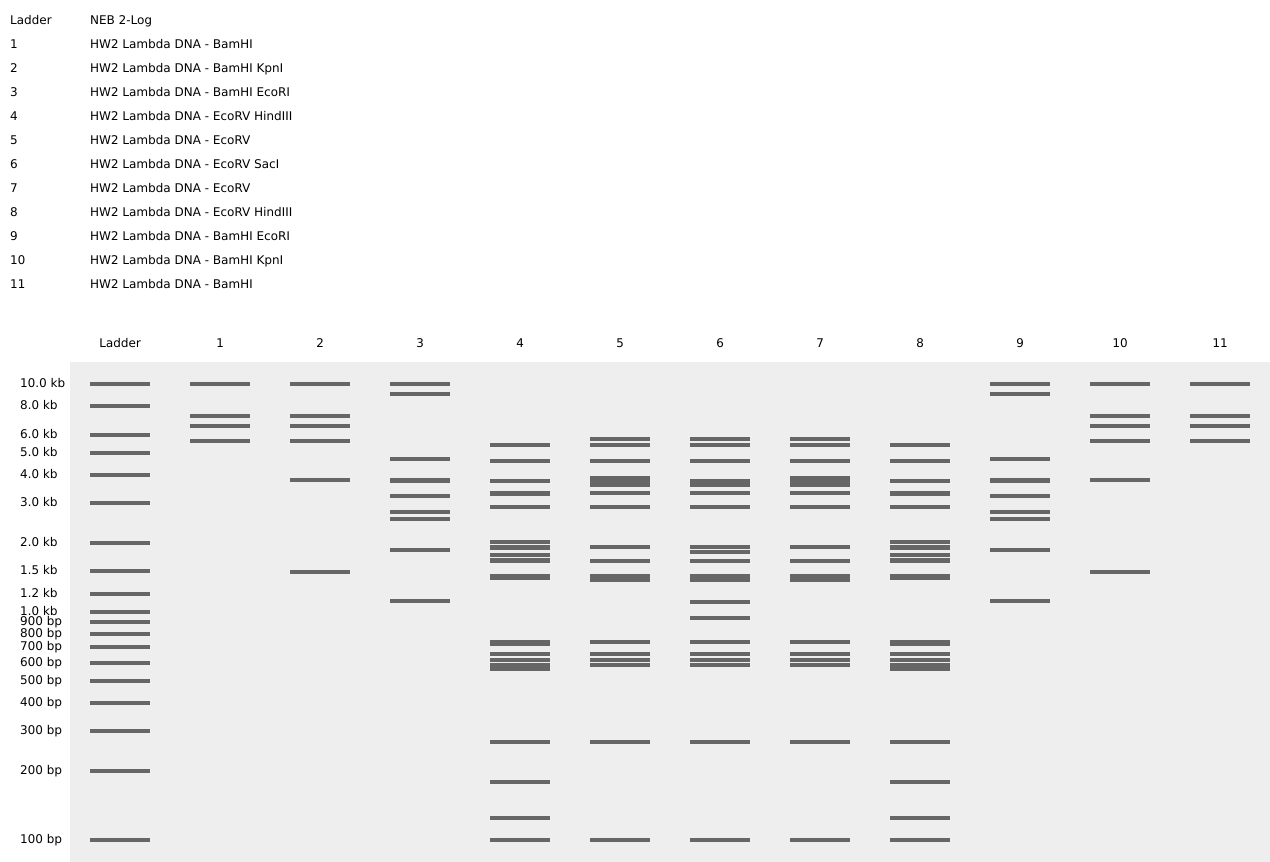
And below is a screenshot of some of my work in Benchling with the Lambda DNA from https://raw.githubusercontent.com/htgaa/htgaa2023/main/02_gel-art/Lambda_NEB.fasta and restriction enzymes, with the “NEB 2-log” ladder selected in the Virtual Digest tab, and with multiple Digests appearing in the same Virtual Digest, which I reorderdd by dragging the tabs around. These are the same restriction enzymes used in the horned skull drawing at the top of the page.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
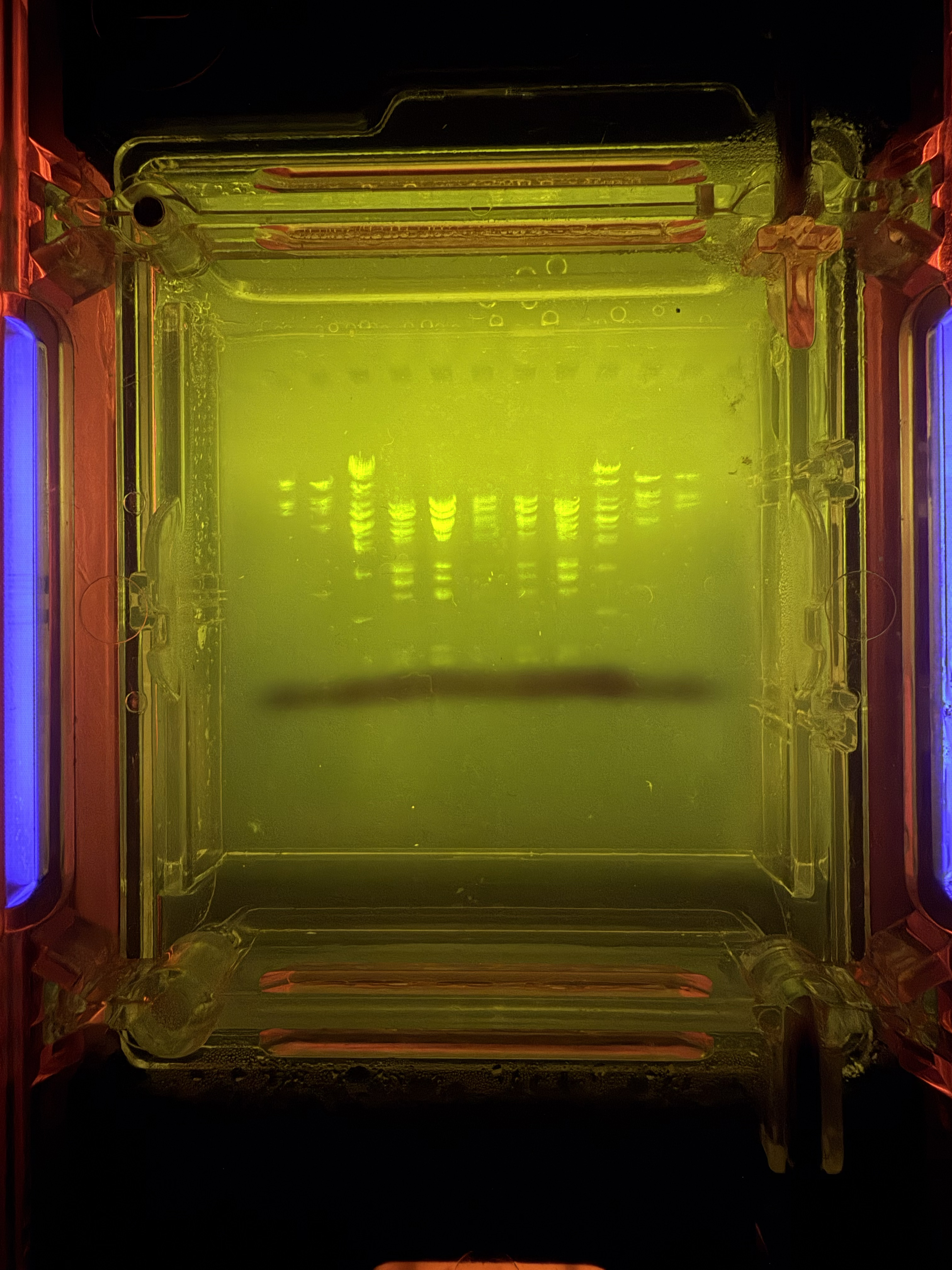
Below is my DNA gel electrophoresis art looking pretty good after about 45 minutes. This was done at the BUGSS Lab (Baltimore UnderGround Sceince Space) on Sunday, Feb 15. Thanks to Amanda and Joel and everyone else!
Part 3: DNA Design Challenge
Will do in lab on Sunday? Or on my own on Monday?
Part 4: Prepare a Twist DNA Synthesis Order
I created my account, still need to prepare my order …
Week 3 HW: Lab Automation
Part 1: Generate an artistic design
NOTE: Some of my newer and hopefully maybe better images are toward the end!
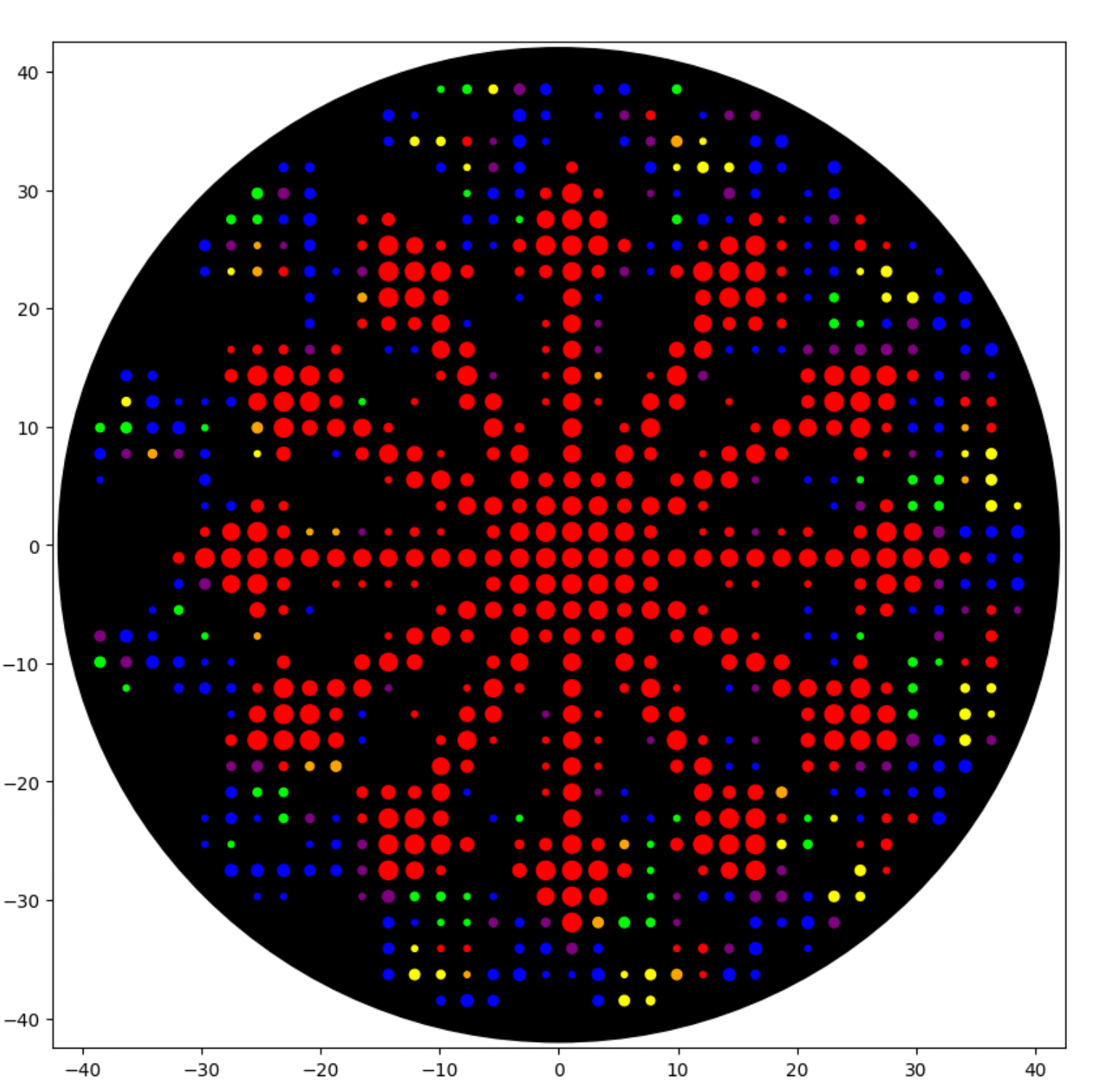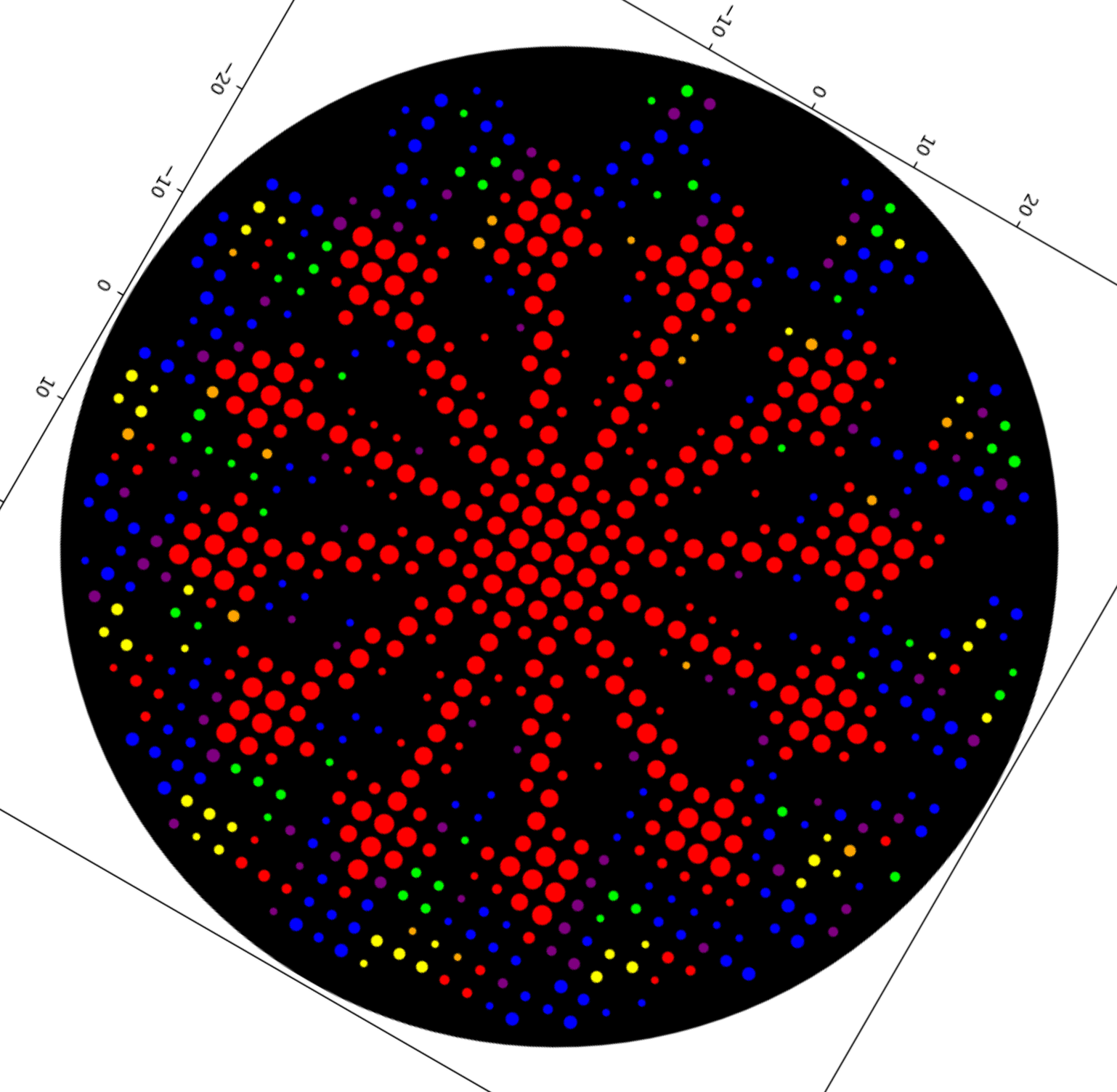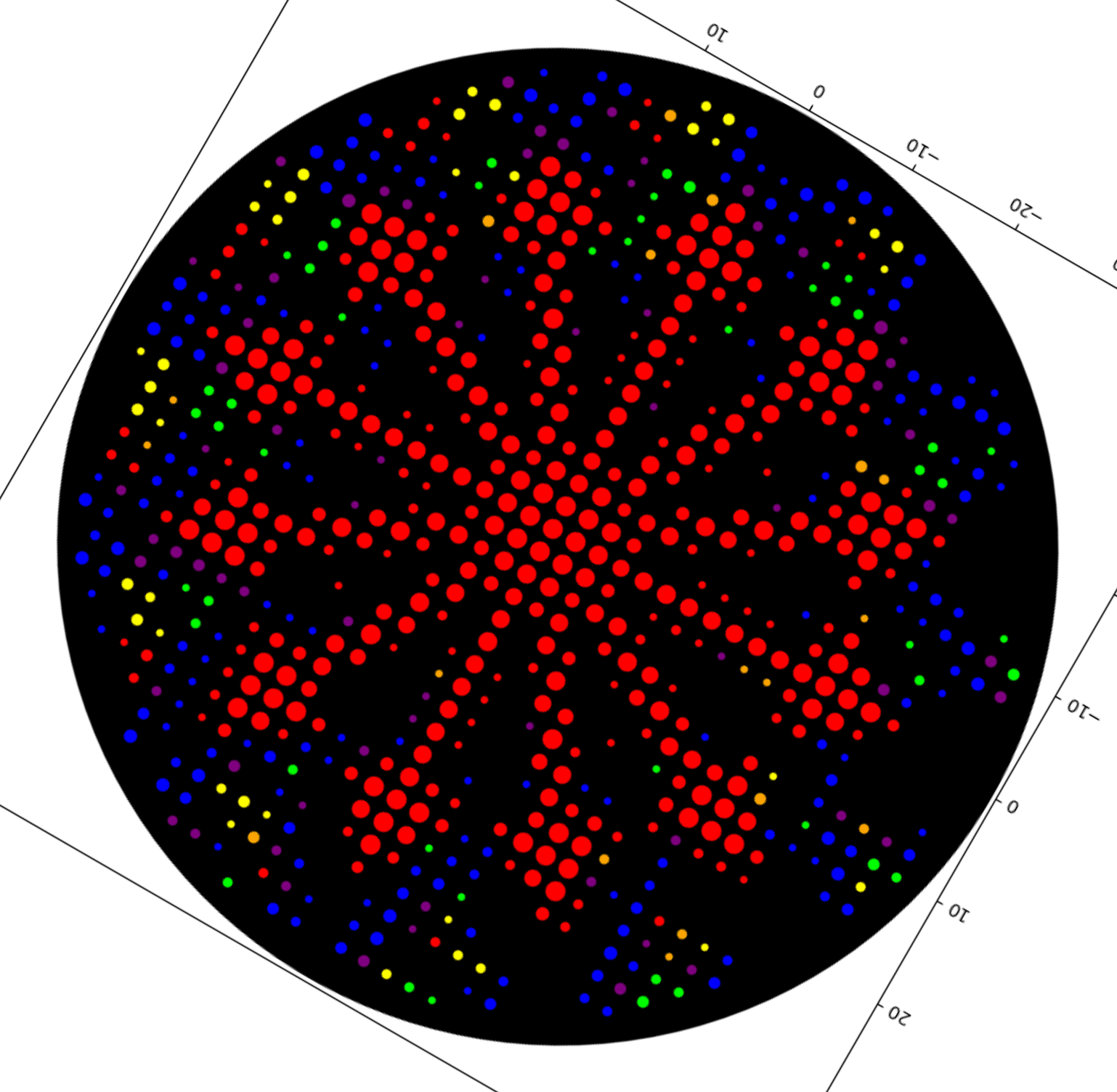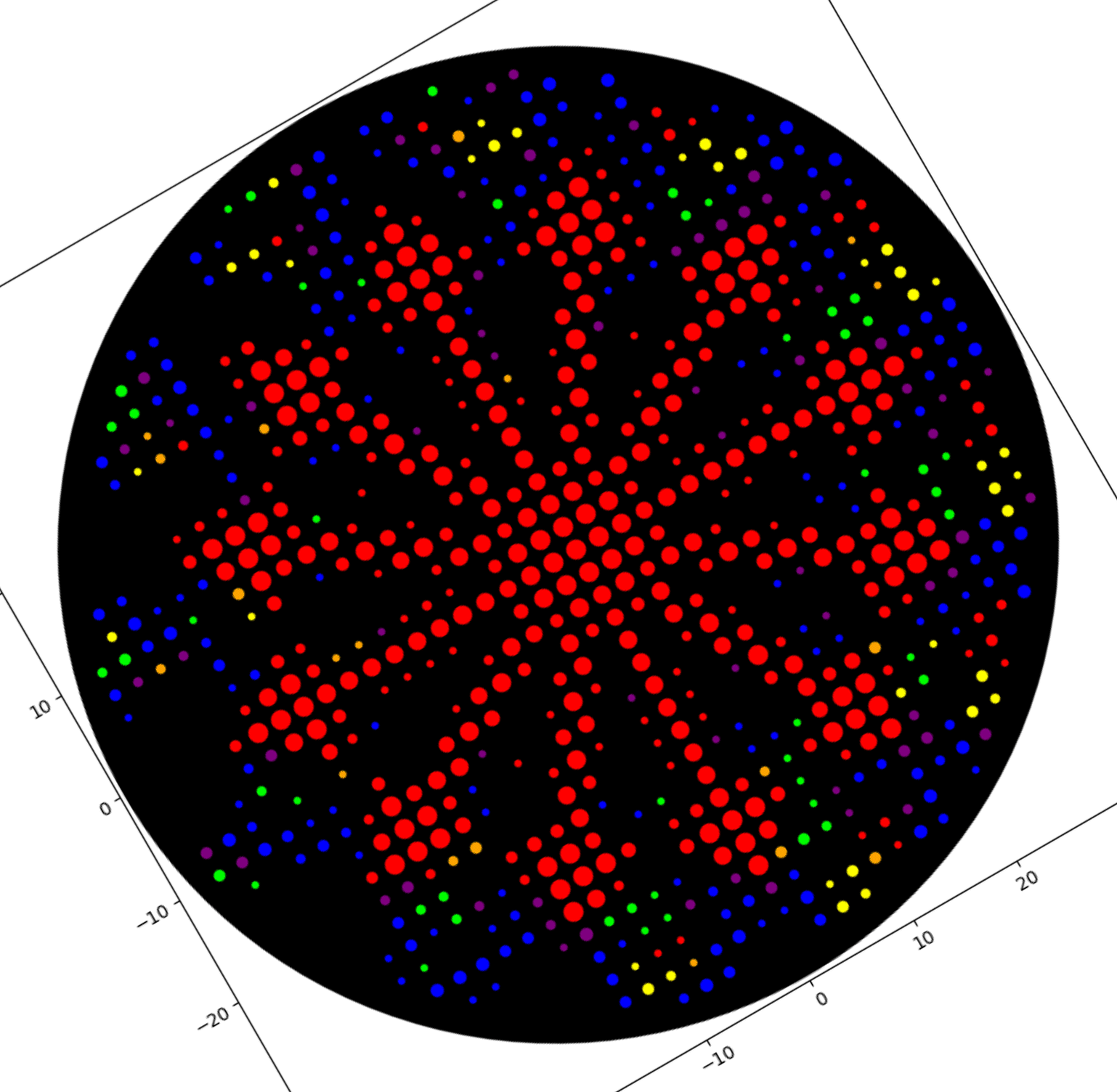
I generated the above artistic design – a self portrait – using the GUI at opentrons-art.rcdonovan.com – NOTE: BUGSS Lab has colors: orange, green, yellow, purple, red, blue
A below is me working on simulating that, redrawing and recoloring little bit in the Google Colab notebok with the six colors at BUGSS Lab. Thanks to Amanda and everyone for helping hack up that code.
And below this is the photo of the six colors at BUGSS Lab:
And then below this is my self-portrait trying to get closer to those colors:
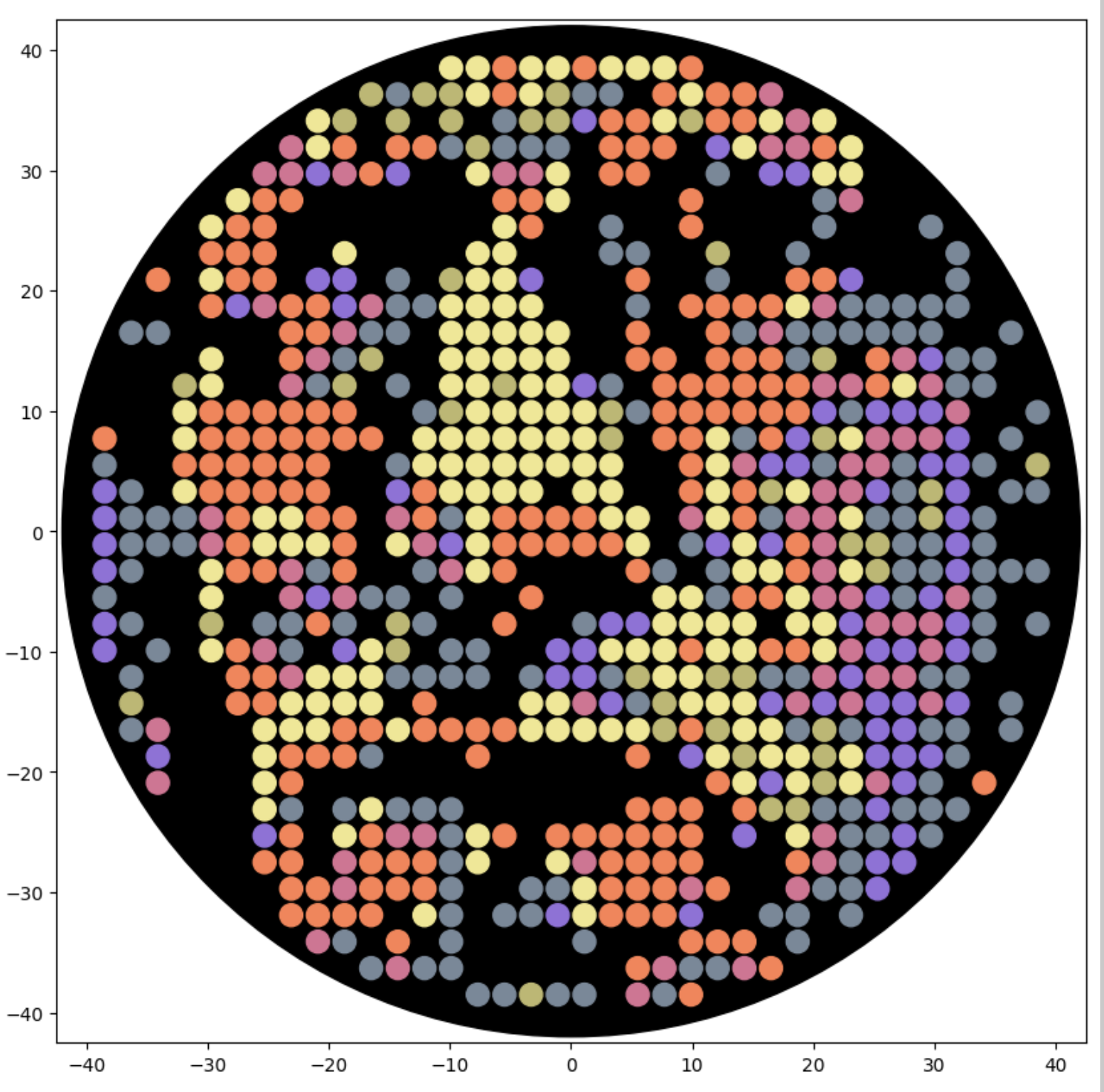
And below this is me working in the google colab notebook to make symmetrical multicolor designs. This is me drawing directly with google colab python code, rather than importing code from Ronan’s website.
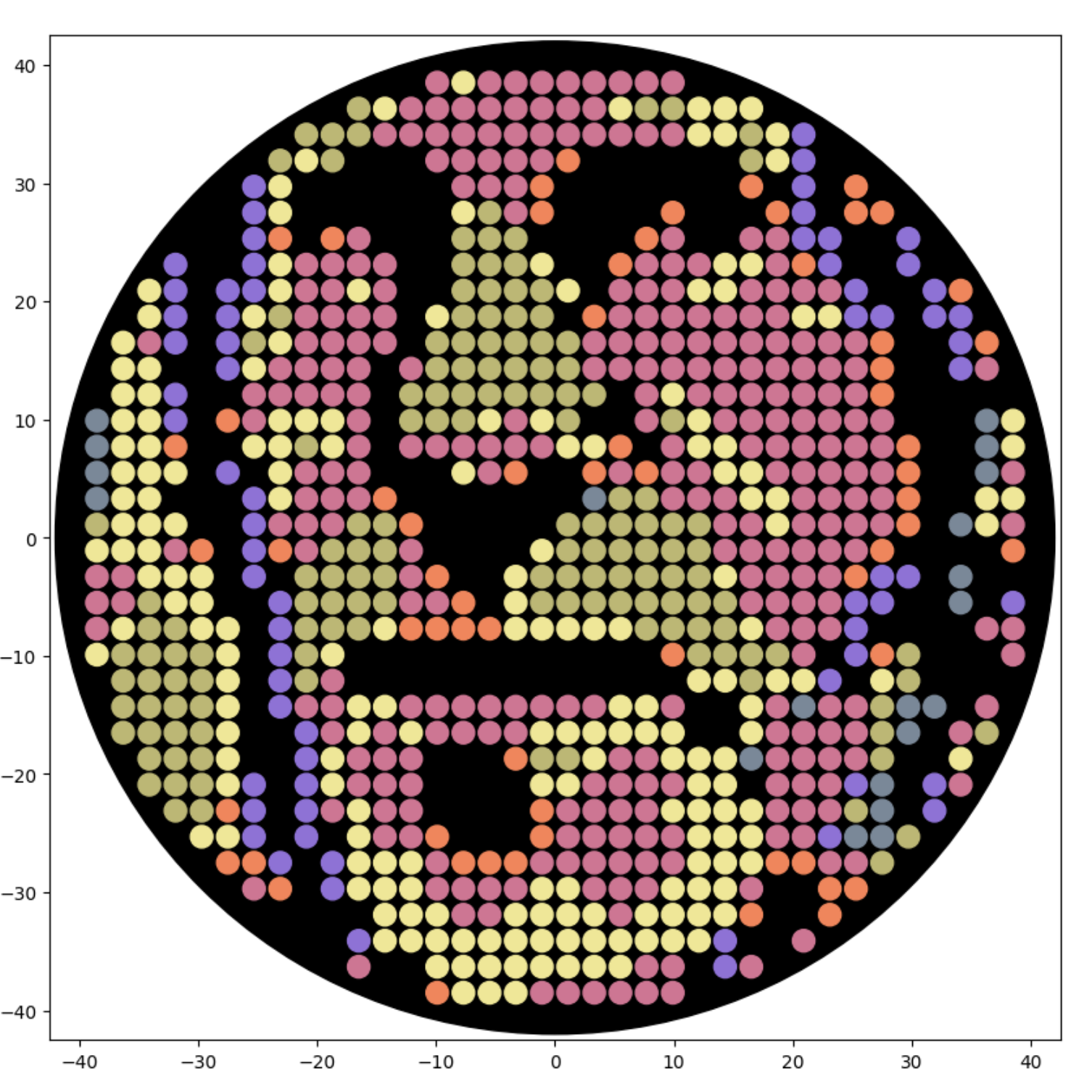
Updated Feb 21: Here is a new self-portrait, closer up, with less flat areas of color, in a screenshot from the Google Colab notebok:
Also updated Feb 21: Here is that same new self-portrait, trying to simulate the six colors at BUGSS Lab:
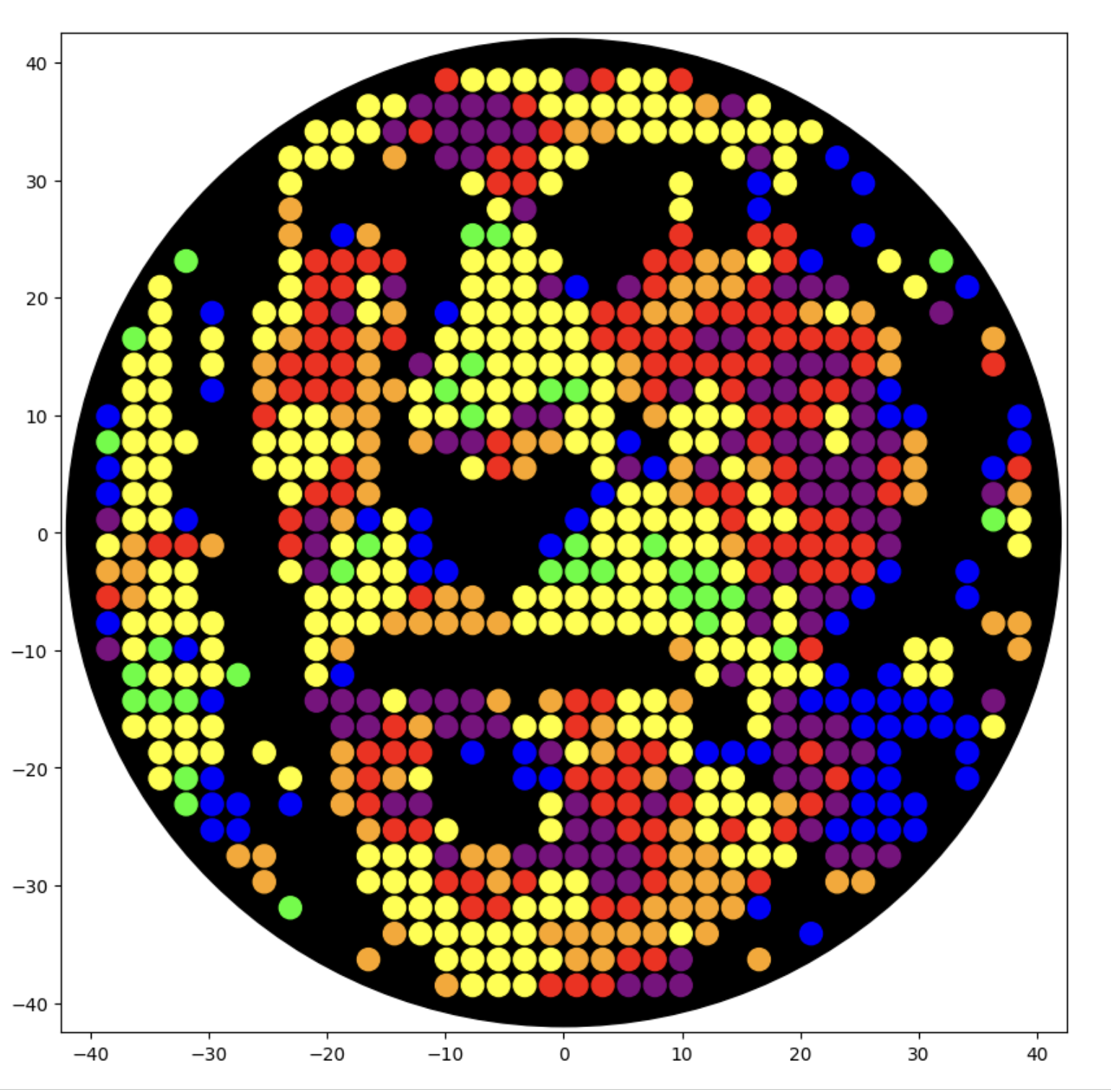
OK! Last set of uopdates before lab! Here is myabe my best, trying to hit the sweet spot of everything I did so far. Her eon Ronan’s site:
That with saturated colors in the google colab:
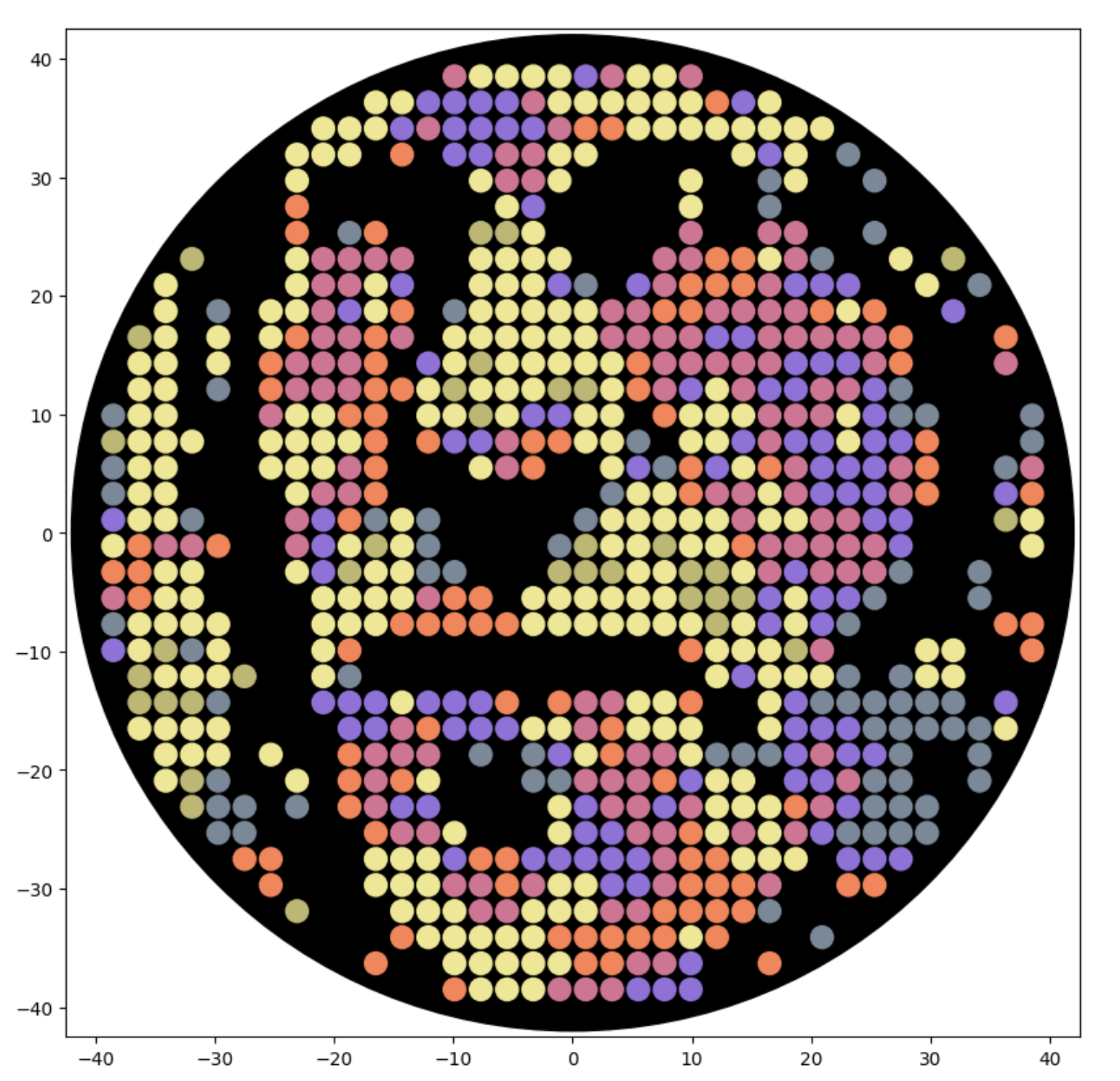
And finally that trying to simulate colors in the photo from BUGSS Lab:
And, update March 9! Here is a photo our awesome TA Joel Tyson took, who said “messing with the opentrons a little. Here are some runs I tried with Eric’s design w 0.2ul drops. I also put a pause between drops to see if it helps dry it.” Thanks, Joel!
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is about 20% protein, so that is 100 g of protein. There are 6.022e+23 Daltons per gram, so for 100 grams that is 6.022e+25 Daltons. Then if there are 100 Daltons in an average amino acid, we’re back to 6.022e+23 molecules of amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because or food prepartion and digestive systems have broken the cow (and cow DNA), the cow DNA does not mix with human DNA to produce new cells, and our immune systems fight DNA other than our own.
To be continued … ??? … Will finish later …
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I chose the protein “Oxidoreductase cns1” from Cordyceps militaris (strain CM01) (Caterpillar fungus). (https://www.uniprot.org/uniprotkb/G3JF08/entry) I chose it because I am interested in parastic fungi and because it is “part of the gene cluster that mediates the biosynthesis of cordycepin (COR)” and “Cordycepin has antitumor, antibacterial, antifungal, antivirus, and immune regulation properties.”
2. Identify the amino acid sequence of your protein.
XP_006669647.1 oxidoreductase domain-containing protein [Cordyceps militaris CM01]
MAMNENAYPTTFPSFERENHRDALRQPFDPAFRRTWSNGVALRQLVDFARPTVANHTMSYALIEYCLSRL
PMQHLERLGQLKIPVELHAAPFQYLQKHHRACGFDWVERFVWRTHDLHKPYNFLRPELLLAQESGSQRIV
ALLTIMPGEDYIRHYASILEVAQHDGAISSHHGPIRCVLYPHLTQSMMAWTGLTELSLSVEPGDILILGF
VAELLPRFASLVPTARVIGRQDAQYYGLVRLELRPGLVFSLIGAKYSYWGNLGGRVVRELAARRPRAICY
IAKQGTLLSPGDIHRTIYSPTRYCVFDKGQACWHGDDHSALPINPLSSRFPTFDRGLHVSTPTIVEQDVD
FRTQVEAHGASSVDNELAQMARALTDVHEENPSMERVQLLPLMFITDYLRRPEELGMTVPFDLTSRNETV
HRNKELFLARSAHLVLEAFNVIERPKAIIVGTGYGVKTILPALQRRGVEVVGLCGGRDRAKTEAAGNKHG
IPCIDVSLAEVQATHGANLLFVASPHDKHAALVQEALDLGGFDIVCEKPLALDMATMRHFANQSQGSSQL
RLMNHPLRFYPPLIQLKAASKEPSNILAIDIQYLTRRLSKLTHWSAGFSKAAGGGMMLAMATHFLDLIEW
LTSSSLTPASVQDMSTSNSIGPLPTEDAGATKTPDVESAFQMNGCCGLSTKYSVDCDGAADTELFSVTLR
LDNEHELRFIQRKGSPVLLEQRLPGREWLPLKVHWEQRVREGSPWQISFQYFAEELVEAICMGTRSAFAD
KATGFSDYARQVGVFGSKVGIA
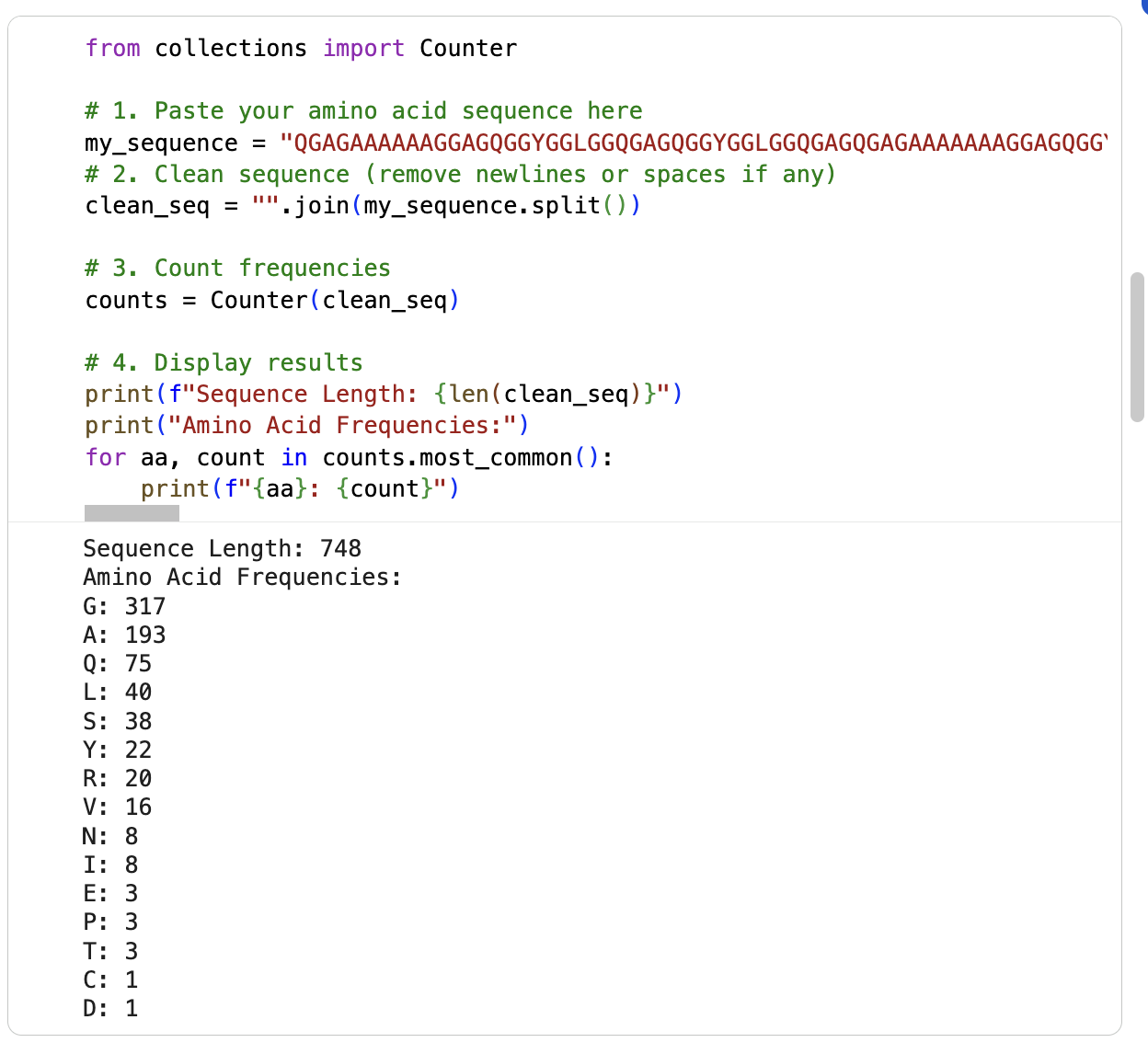
I am running the google collab notebook to count the frequency of amino acids (had some errors) … it says the sequence length is 748 and the most frequent amino acid is glycine (317) followd by alanine (193). See below:
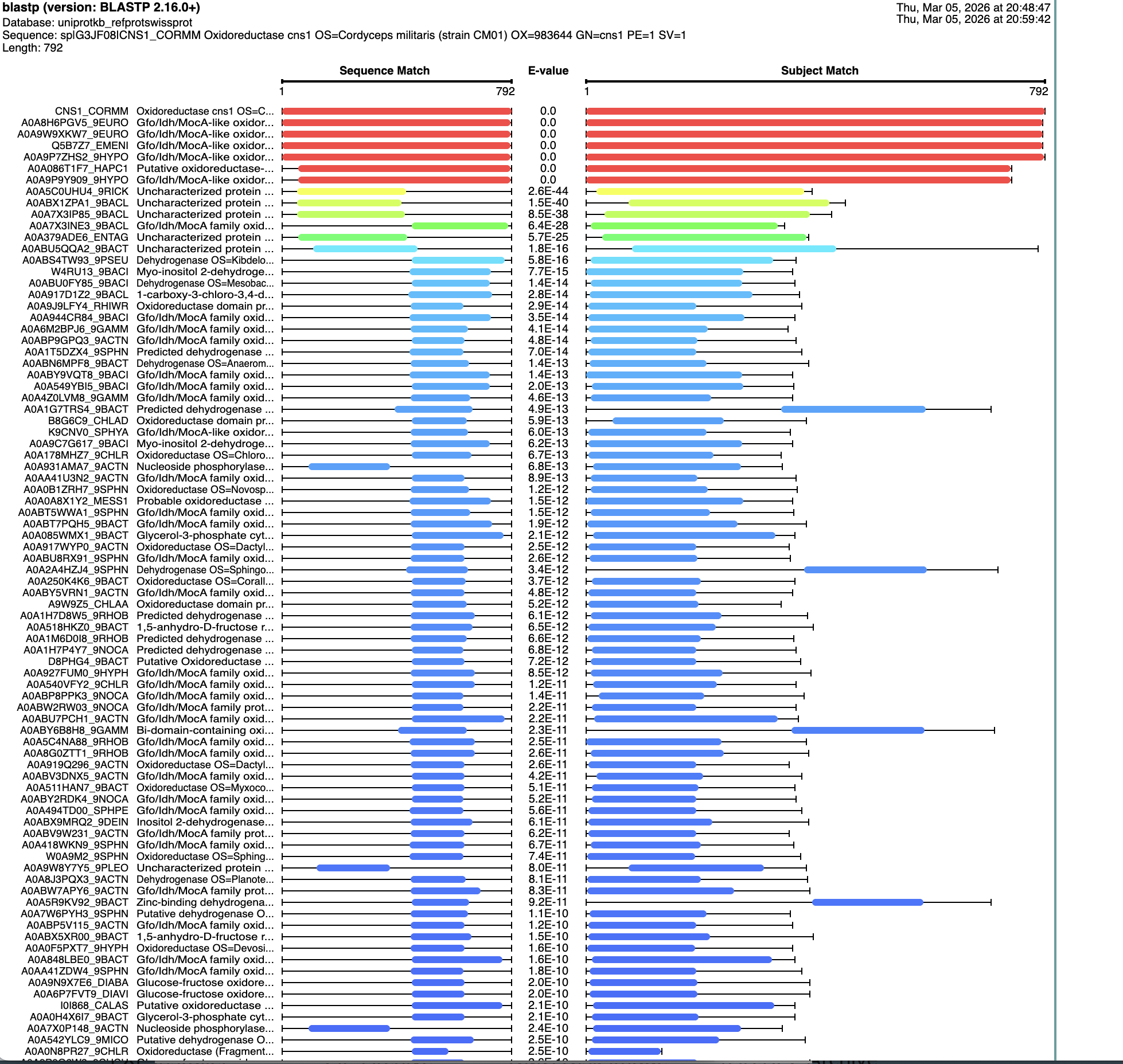
I ran a search for homologs on BLAST. There are six that are in the red, that have an E value of about 0. See below:
3. Identify the structure page of your protein in RCSB.
I dod not find the “Oxidoreductase cns1” from Cordyceps militaris (strain CM01) (Caterpillar fungus)” in RCSB. I did find “Crystal Structure of endo-beta-N-acetylglucosaminidase from Cordyceps militaris D154N/E156Q mutant in complex with fucosyl-N-acetylglucosamine” at https://www.rcsb.org/structure/6KPN
I don’t see a solved date. It was Deposited: 2019-08-15. I don’t see it listed as part of any structure classification family at https://www.ebi.ac.uk/pdbe/scop/
4. Open the structure of your protein in any 3D molecule visualization software
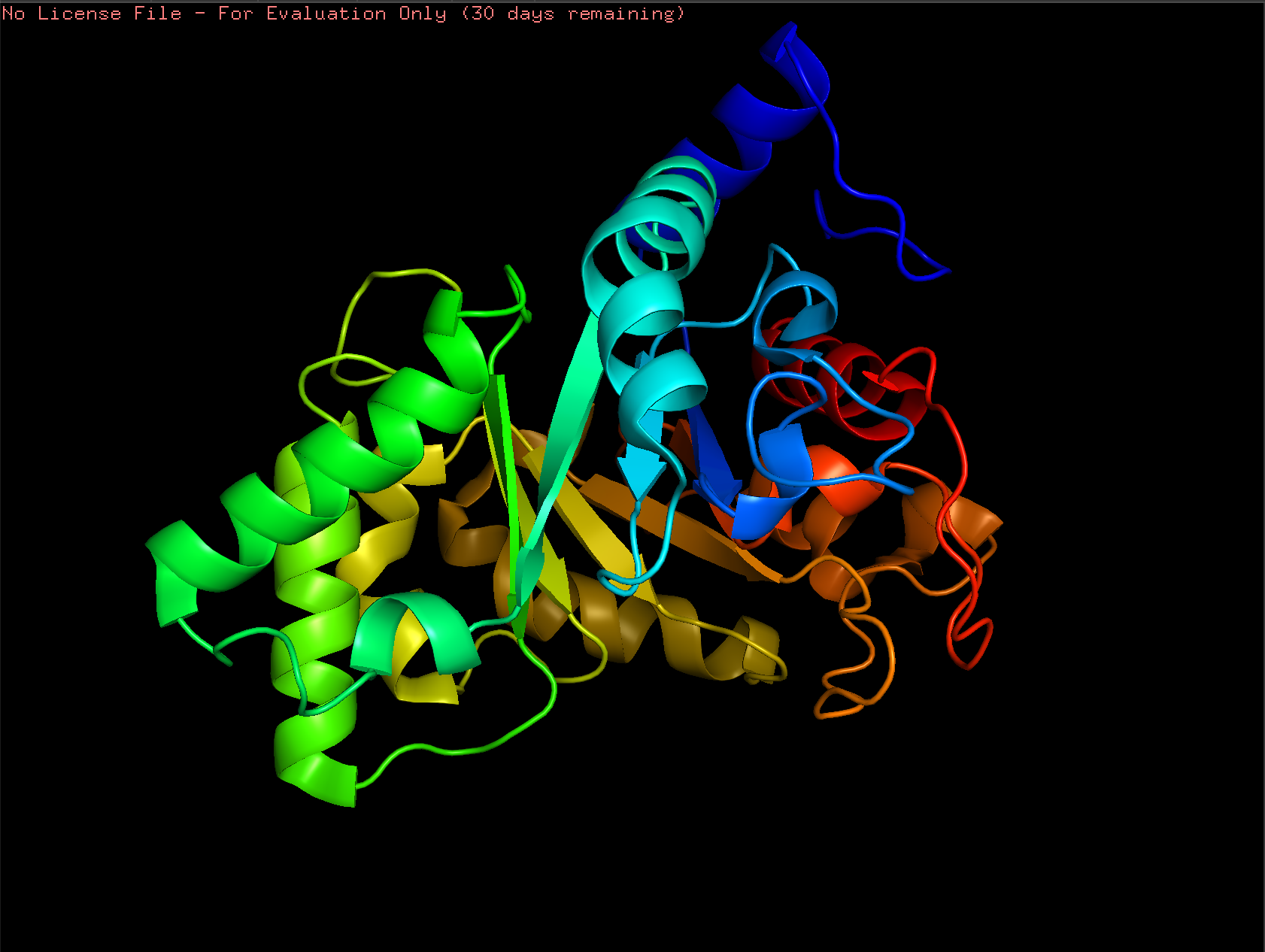
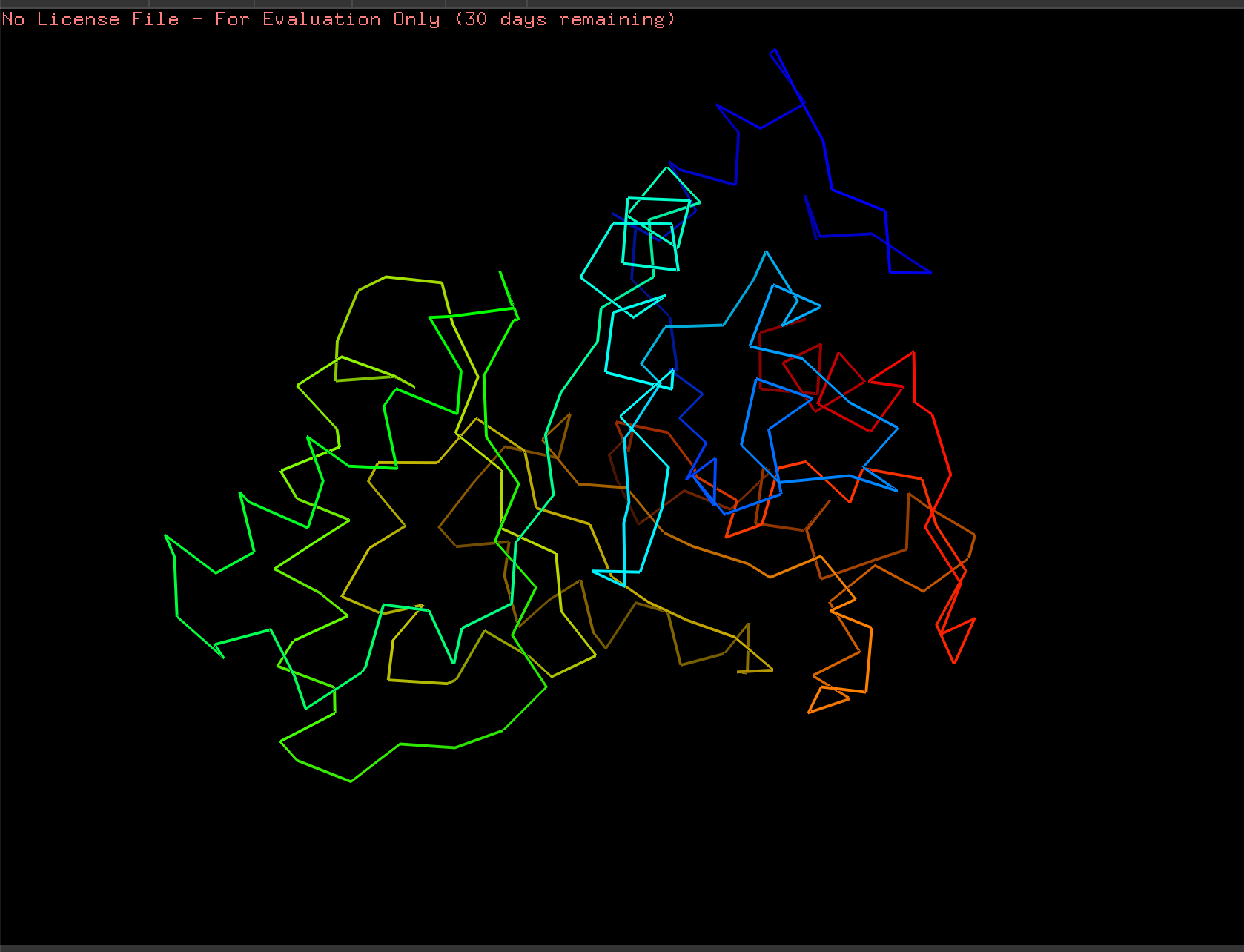
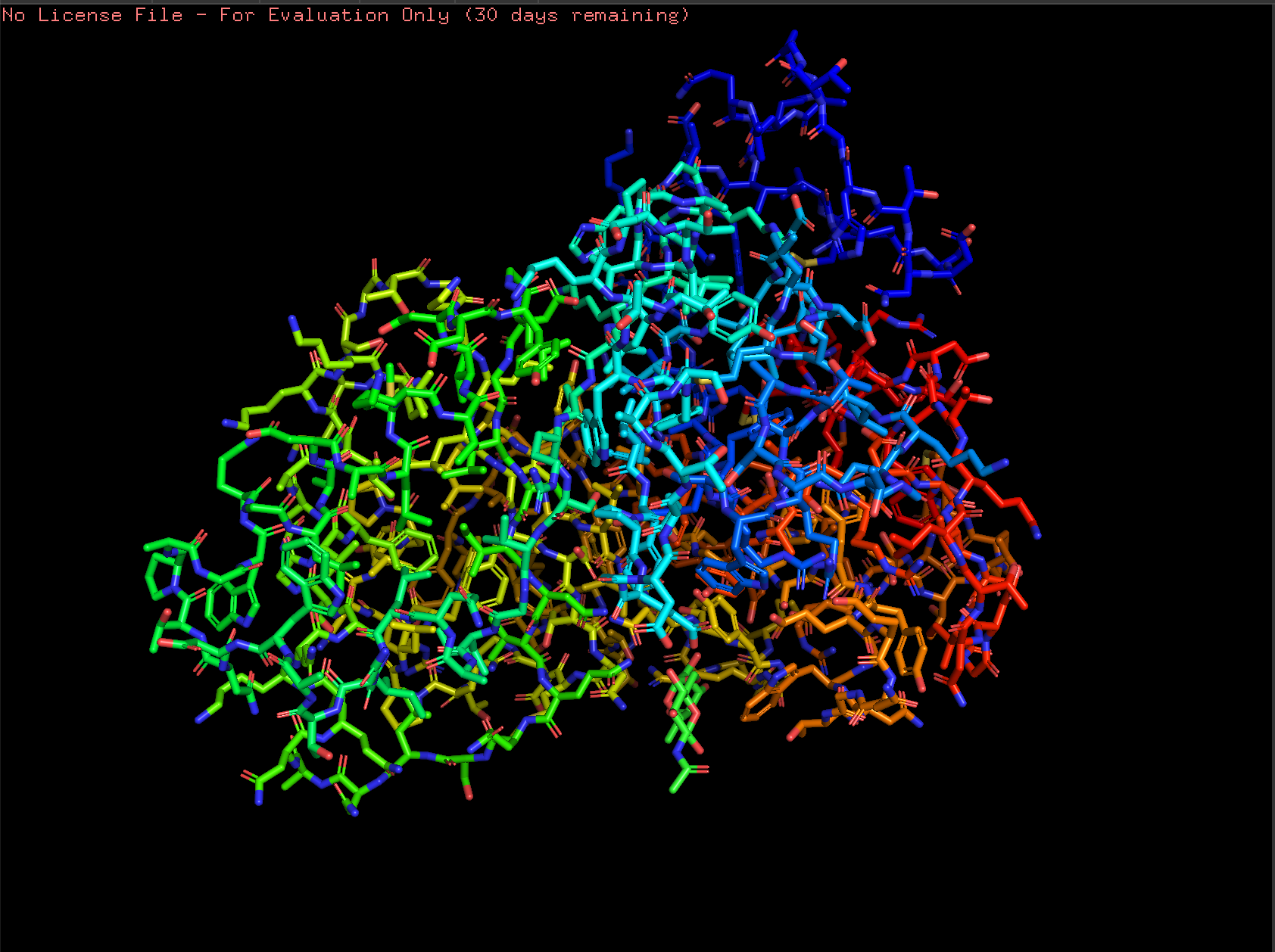
Cartoon:
Ribbon:
Ball and Stick:
To Do:
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C1. Using ML-Based Protein Design Tools: Protein Language Modeling
To do: Can you explain any particular pattern? (choose a residue and a mutation that stands out) and (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
2. Latent Space Analysis
To do: Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbors.
Part C2. Using ML-Based Protein Design Tools: Protein Folding
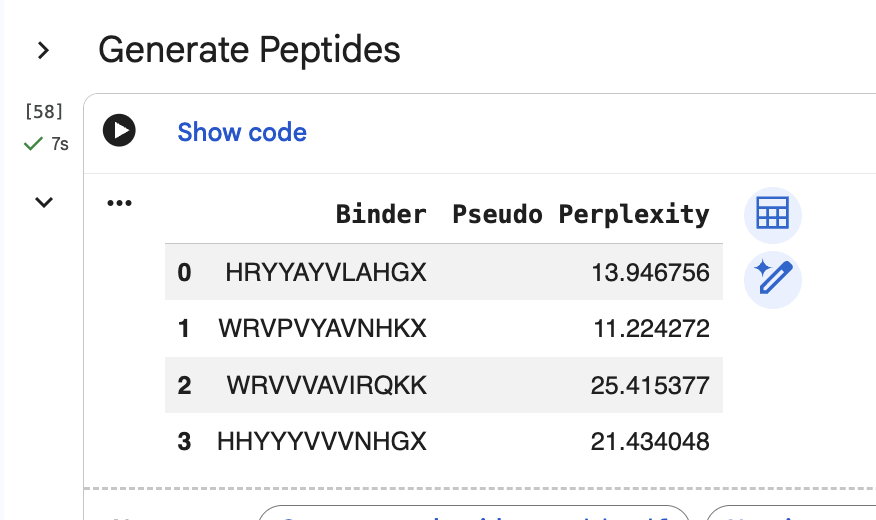
“Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.” I did this step several times. Here are two examples of my generating four peptides:
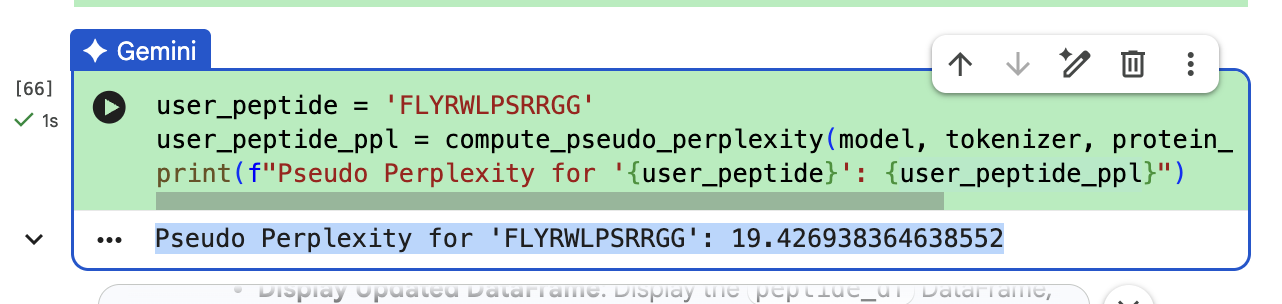
“To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.”
“Record the perplexity scores that indicate PepMLM’s confidence in the binders.” These can be seen in the above screenshiots, and I have also recorded them in an excel spreadsheet.
Part 2: Evaluate Binders with AlphaFold3
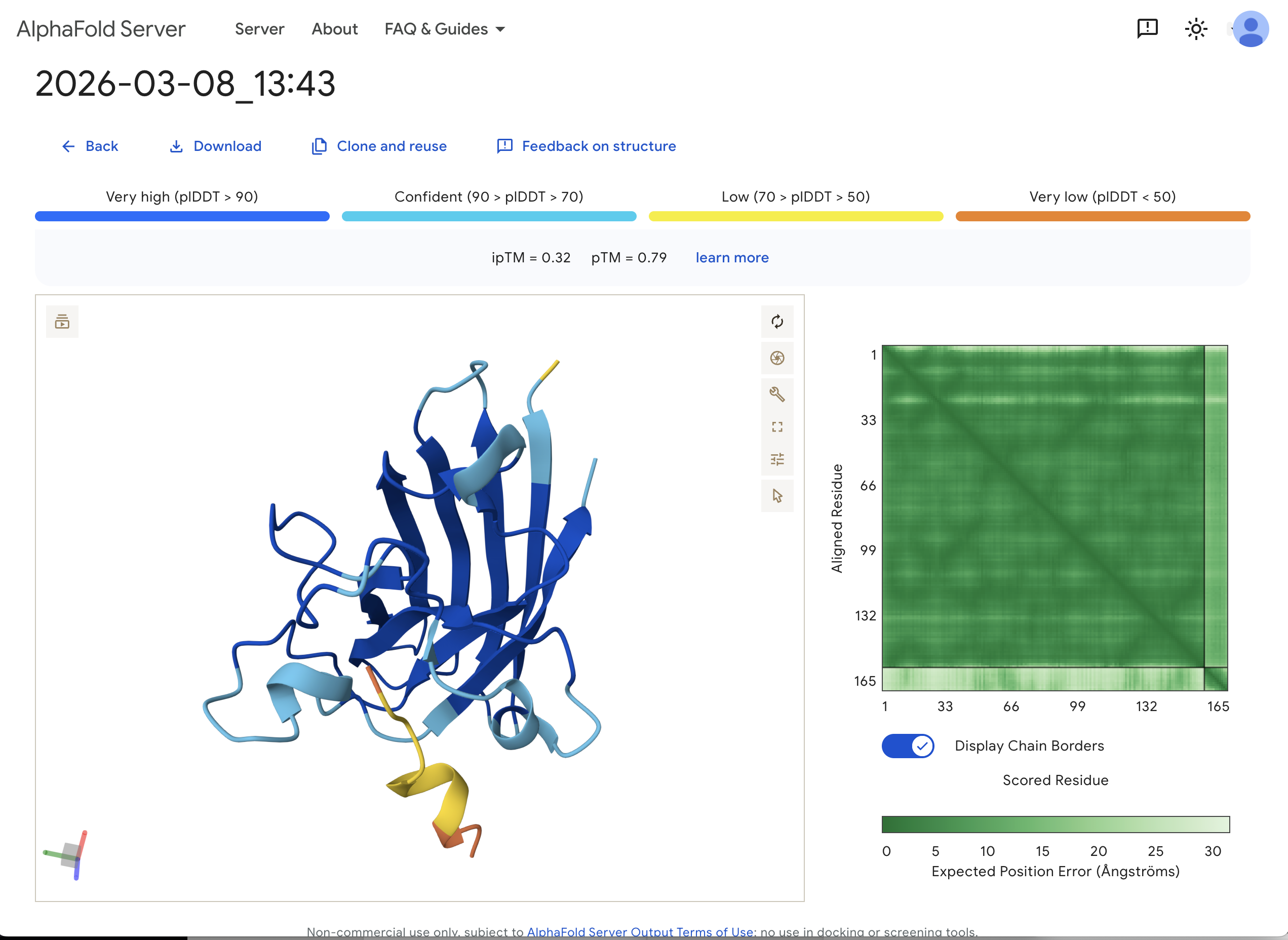
“Navigate to the AlphaFold Server: alphafoldserver.com For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.” This is one example:
“Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?”
In teh one above, I’d say probably surface bound, and closer to the β-barrel region than to the N-terminus.
“In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.”
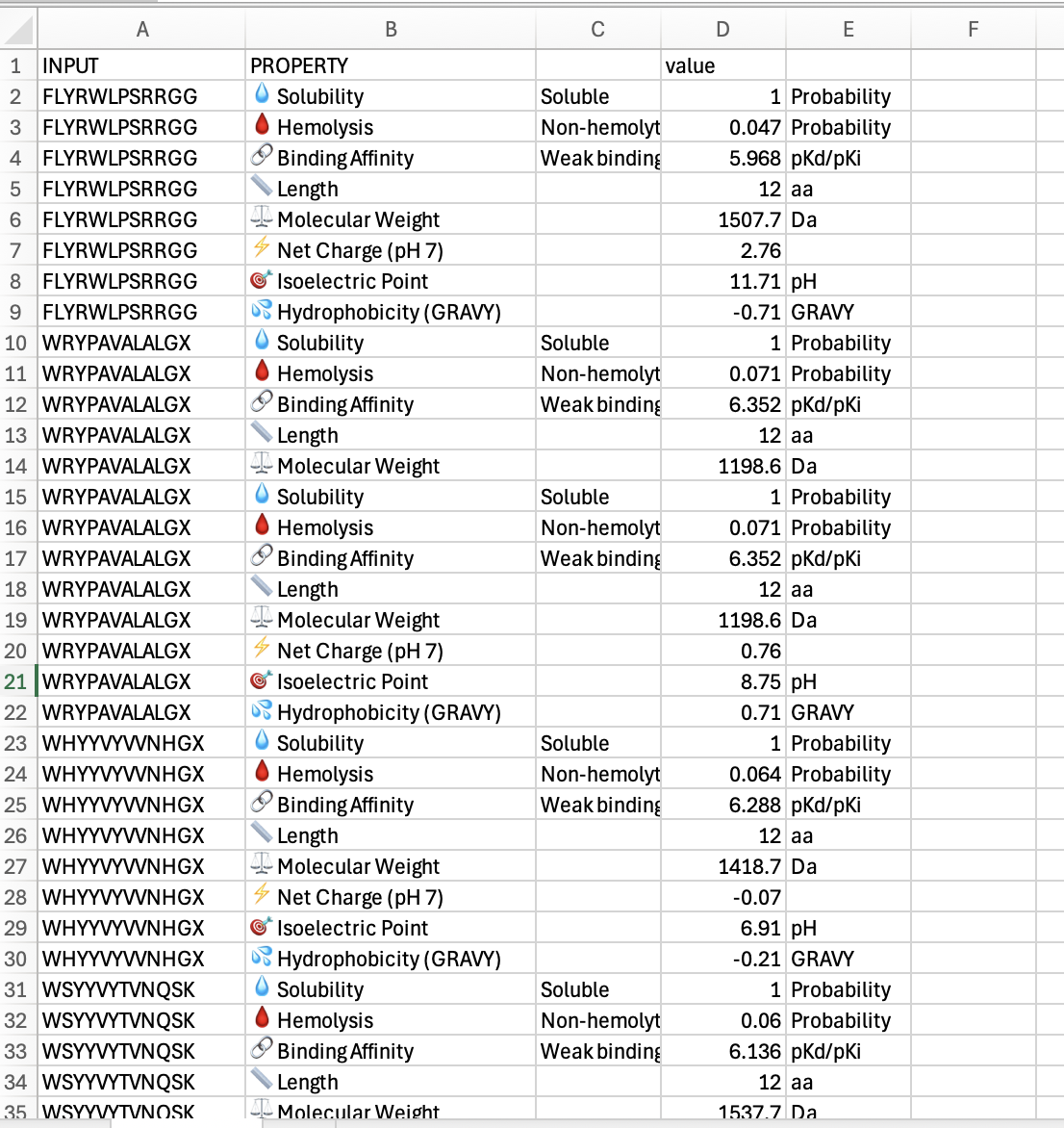
The ipTM value I got for the known binder was 0.31. The peptides I generated ranged from 0.28 to 0.43 in ipTM values. My understanding is that higher ipTM values show more confidence in the prediction. I hade a few that were slightly higher than the knonw binder, like WRYPAVALALGX at 0.38 and my highest at 0.43 was WHYYVYVVNHGX.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Below is a screenshot of part of my spreadsheet of results of my work in Peptiverse:
“Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties? Choose one peptide you would advance and justify your decision briefly.”
The few peptides I tested did not show much difference. They each were soluble, and each had weak binding affinity. The widest diferences I saw were in hydrophobicity, where i had some positives and some ngatives. The one that was closest to zero was at -0.21 which was WHYYVYWNHGX. Of these, that might eb the one that I would advance. I would probbaly go back and look for ones that had more binding affinity.
Part 4: Generate Optimized Peptides with moPPIt
I am rerunning this … ran on Sunday but did not save my results …
Some results of my rerun:
Result
Hemolysis
Solubility
Affinity
Motif
STKLHTKIKCQC
0.9697153624147177
0.8333333134651184
6.467465877532959
0.7173478007316589
SVTKKETQKRFA
0.9688531029969454
0.75
5.76106595993042
0.7000954747200012
GSAEMTCKKQRK
0.9745583962649107
0.8333333134651184
6.006259441375732
0.6160933375358582
“briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?”
These seem to be getting different results by using different strategies. For advancing to trials, I would probably test a few of the best results found from each system.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
(optional, might get to later)
Part C: Final Project: L-Protein Mutants
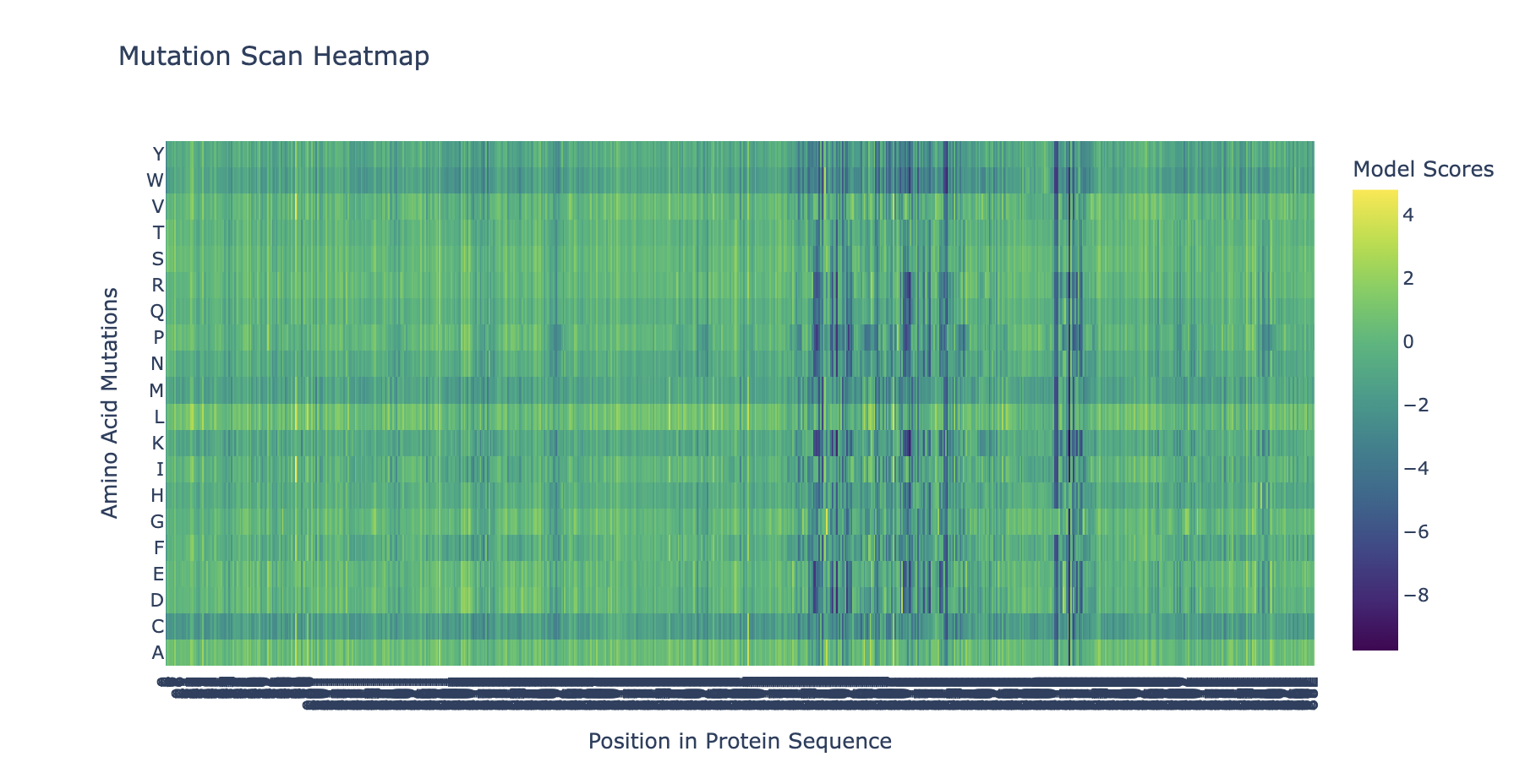
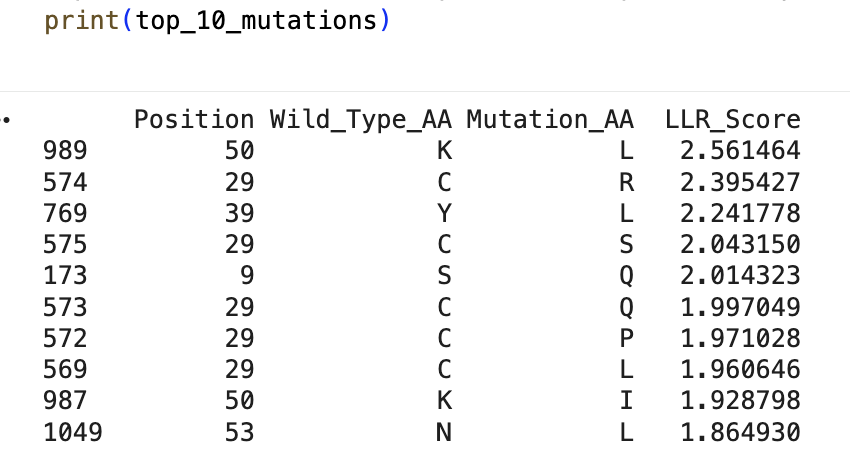
“Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid”:
“does the experimental data correlate with the scores from the notebook”?
… I don’t think so … might’ve done something off … or maybe these are just different systems with different results …
Week 6 HW: Genetic Circuits Part I
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
From New England Biolabs “Phusion High-Fidelity PCR Master Mix with HF Buffer is a 2X master mix consisting of Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2. All that is required is the addition of template, primers and water.”
2. What are some factors that determine primer annealing temperature during PCR?
From ThermoFisher Scientific “The annealing temperature is determined by calculating the melting temperature (Tm) of the selected primers for PCR amplification. A general rule of thumb is to begin with an annealing temperature 3–5°C lower than the lowest Tm of the primers … One important consideration in Tm calculation is the use of PCR additives, co-solvents, and modified nucleotides. The presence of these reagents lowers the Tm of the primer-template complex.”
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR is used to make many copies of a section of DNA. Restriction enzyme digests cut the DNA at specific points, and do not make multiple copies.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
5. How does the plasmid DNA enter the E. coli cells during transformation?
6. Describe another assembly method in detail (such as Golden Gate Assembly)
7. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
8. Model this assembly method with Benchling or Asimov Kernel!
Assignment: Asimov Kernel
1. Create a Repository for your work
I created a repository called “Millikin Constructs HTGAA BUGSS HW06”
2. Create a blank Notebook entry to document the homework and save it to that Repository
I created a notebook called “HW06 Documentation Notebook”
3. Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
In the “Bacterial demos” repository, the “info” for the Reprissilator lists “Simulation parameters: Chassis: E. coli Duration: 24 hours Timestep: 10 minutes Transfection: Transient transfection.” I clicked the “Sim” button on the right, entered those parameters (only had to change duration) and then simulated at 10:14 AM 3/31/26. These are the graphs of my results:
4. Create a blank Construct and save it to your Repository
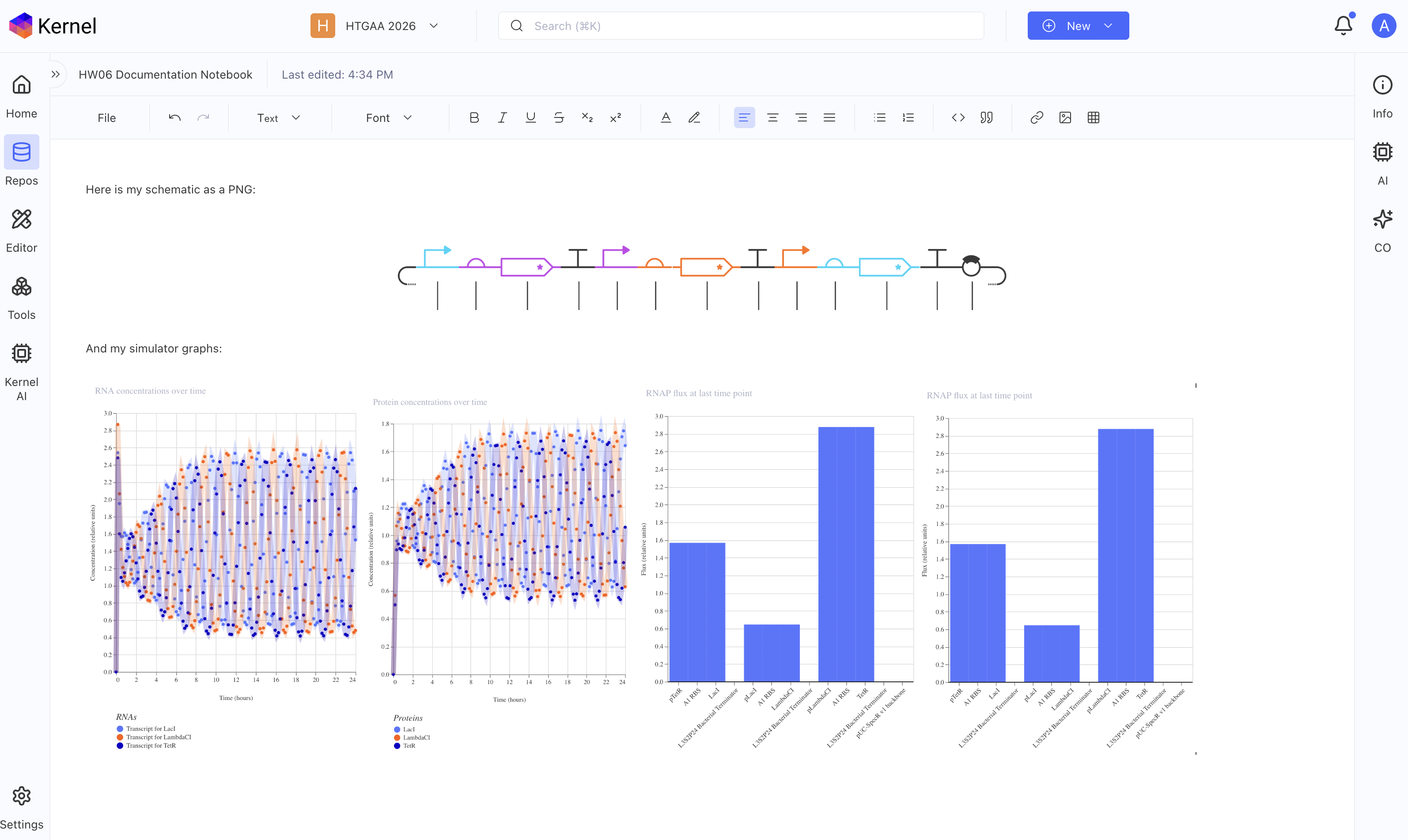
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository. Search the parts using the Search function in the right menu.Drag and drop the parts into the Construct. Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository. Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook*
Yes! I did all of the above, below should be a screenshot of my notebook with my glyph image and simulator graphs from my recreated repressilator.
5. Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
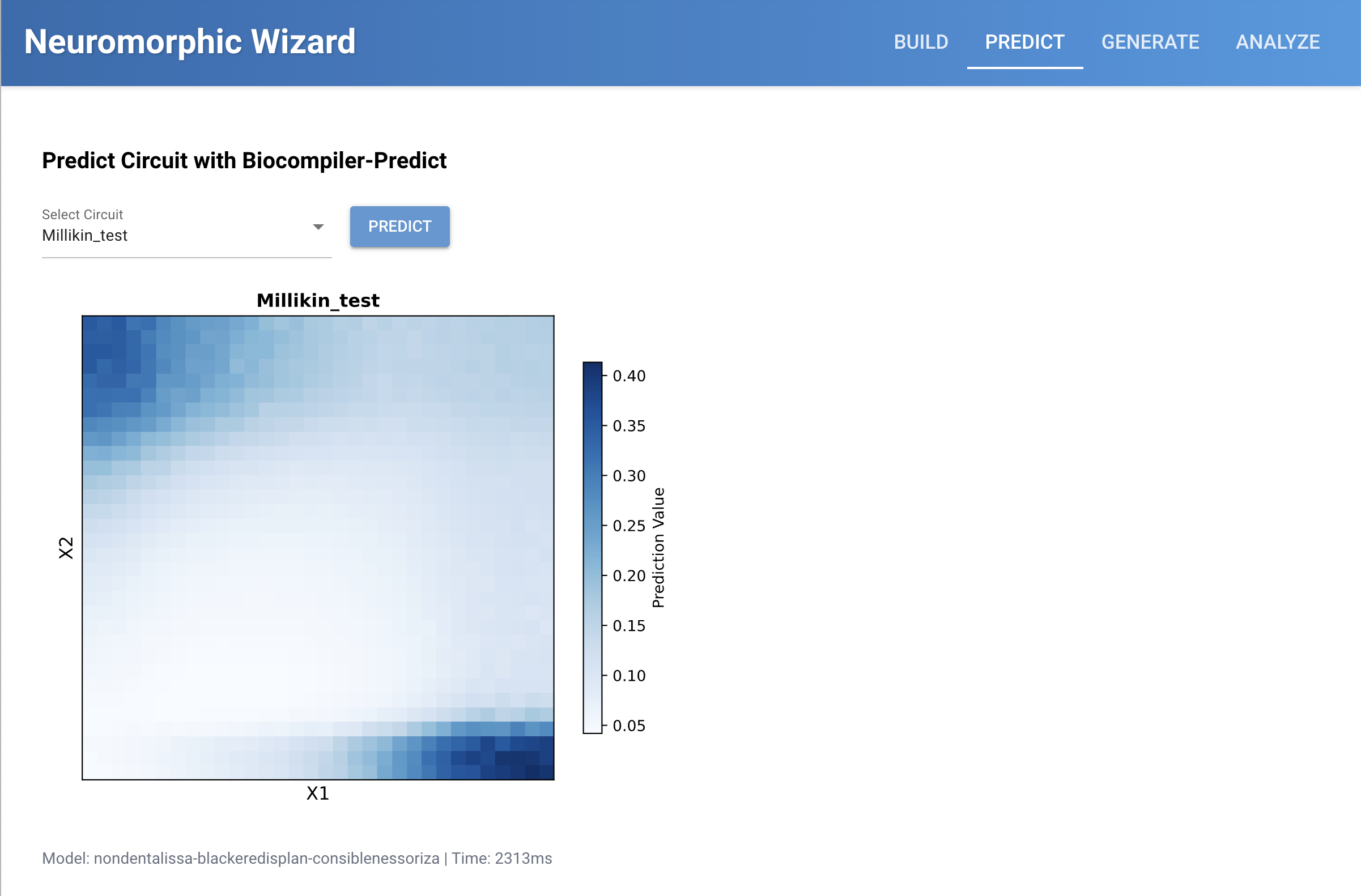
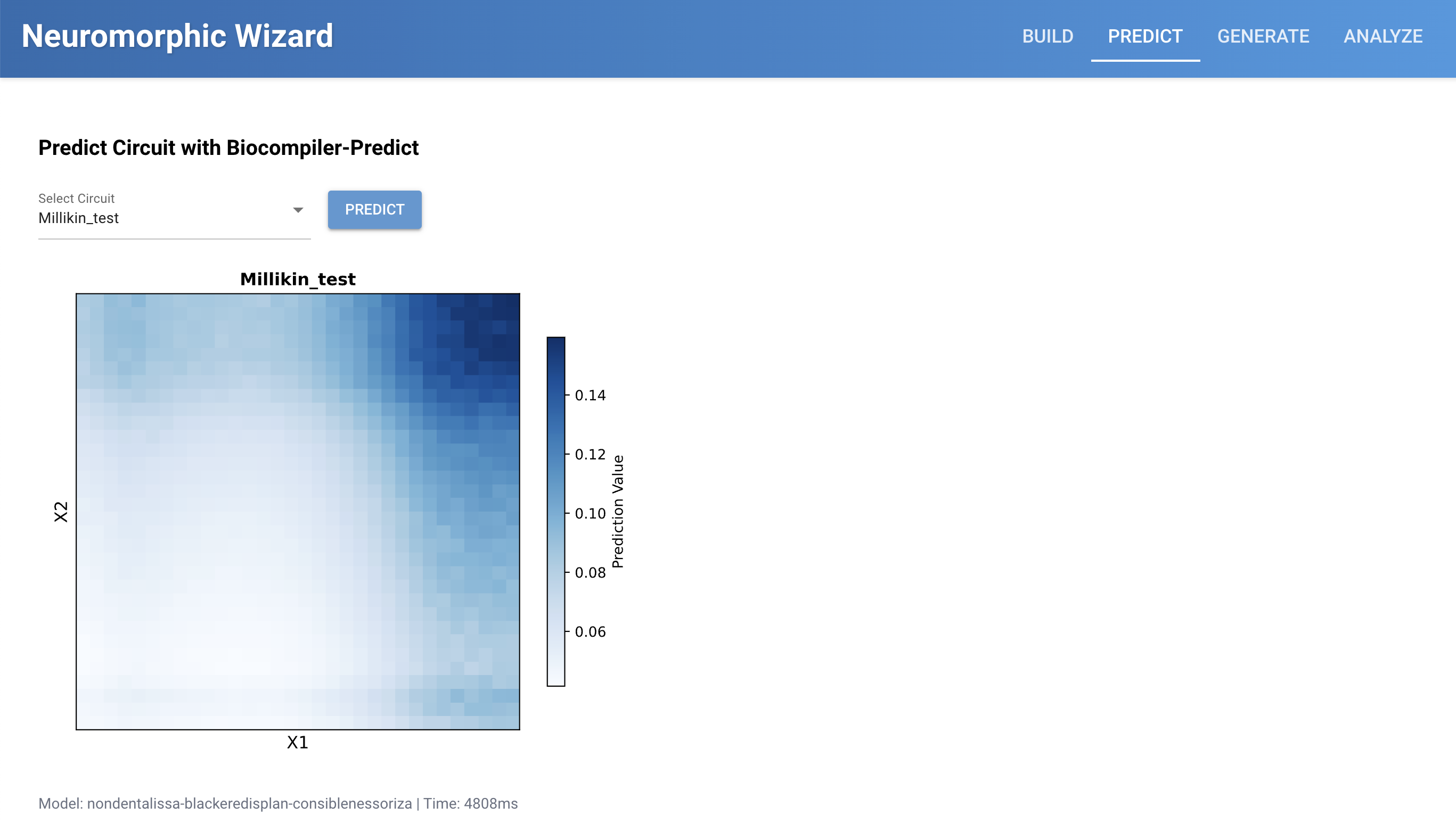
Traditional genetic circuits are binary digital on/off. IANNs can be more analog, with greater ranges, which is shown in the blue gradient diagrams in the neuromorphic wizard.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
My memory of examples described in class involved cancer fighting Intracellular Artificial Neural Networks, which could take multiple inputs of different biological signs of cancer, give them different weights, and then could output either just a biosignal that suspected cancer has been detected, or maybe as outpur release a biological chemotherapy. Current limitations include that these are currently fairly difficult to design and test, which is why systes like the neuromorphic wizard are being developed to model them in simulation and predict their behavior.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
One example of fungal materials is objects made from mycelium, which can be grown into pretty much any shape you can make a form for, much like tradiitional casting. We saw several examples of mycelium projects including acoustic tile, packing material, insulation, and bricks. Yeast is also a fungus, so materials made from SCOBY (Symbiotic Culture Of Bacteria and Yeast) are also in part fungal materials. We saw images of several SCOBY fashion projects in recitation.
I have used fungal mycelium and SCOBYs in previous projects. Below is an image showing, on the left, a sculptural mask made of mycelium and mushrooms, and on the right is a sheet of SCOBY leather with embedded venus flytrap leaves.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
For biomaterials, genetically engineered fungi could maybe grow mycelium faster, or maybe spend less energy on fruiting and spore production and more energy on creating mycelium. A natural or agricultural fungus that has been bred and selected for creating large fruit and for reproducing well might actually be less useful for biomaterials than a fungus that has been genetically engineered for faster and stronger mycelium.
Assignment Part 3: First DNA Twist Order
Thinking about this …
Week 9 HW: Cell Free Systems
#General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Two advantages would be 1) “elimination of reliance on living cells,”" so we don’t hve to wait for the living organism’s growth cycle an maintain their living conditions, and 2) “the ability to focus all system energy on production of the protein of interest.” Two cases where cell-free systems have benfits over in vivo methods include 1) “The production of functional antibodies and antibody fragments in vitro using CFPS has the potential to allow for simplification of the antibody production process for more rapid manufacturing,” and 2) “Metalloproteins … are difficult to produce in vivo … have the potential to enable renewable hydrogen fuel and other important biotechnological advancements.”
Describe the main components of a cell-free expression system and explain the role of each component.
“In CFPS, a solution containing all the cellular machinery needed to direct protein synthesis (e.g., ribosomes, tRNAs, enzymes, cofactors, amino acids, etc.) is used to transcribe and translate a supplied nucleic acid template (e.g., plasmid DNA, linear DNA or mRNA).”
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
“Supplying energy for cell-free protein synthesis reactions is one of the biggest challenges to the success of these systems. Oftentimes, short reaction duration is attributed to an unstable energy source. "
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Week 10 HW: Imaging and Measurement
Homework: Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Homework: Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Note (from meeting) that GFP loses weight (a water, and two hydrogen?) when it folds, so you have to subtract 20 daltons from what expasy reports …
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I will say that they were largely decorative, incremental changes because I was working when the wait time between each person’s changes was one minute, so I wasn’t able to make any really drastic changes. I was mostly just trying to add some additional variety and visual interest to what other collaborators had already drawn.
The final artwork has changed so much since my early contributions, which is of course fine and cool, but maybe a future experimental version of this project could make it so there was always at least one or more pixels from each participant.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Looking at each omponent’s role is in the cell-free reaction:
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): has a T7 RNA polymerase gene, which means that it can express proteins with the T7 promoter upstream in its genes
Salts/Buffer
Potassium Glutamate: helps recreate the cell’s native ionic environment by supplying potassium ions that stabilize ribosomes and enhance efficient protein synthesis
HEPES-KOH pH 7.5: maintains a stable pH, ensuring optimal activity of enzymes involved in transcription and translation in the cell-free reaction.
Magnesium Glutamate: Supplies Mg²⁺ ions, which are essential cofactors for ribosomes, RNA polymerase, and other enzymes, directly enabling transcription and translation.
Potassium phosphate monobasic: Acts as part of a phosphate buffer system and contributes to maintaining pH and ionic strength in the reaction.
Potassium phosphate dibasic: Pairs with the monobasic form to establish the buffering equilibrium that stabilizes the reaction pH.
Energy / Nucleotide System
Ribose: Serves as a precursor for nucleotide regeneration pathways, supporting sustained synthesis of ATP and other nucleotides in the reaction.
Glucose: Acts as an energy source that can be metabolized (in extract-based systems) to regenerate ATP and maintain reaction longevity.
AMP: Functions as a nucleotide precursor that can be phosphorylated to regenerate ATP, helping sustain the reaction’s energy supply.
CMP: Serves as a precursor for CTP synthesis, supporting continued RNA transcription.
GMP: Acts as a precursor for GTP, which is required for both RNA synthesis and translation processes.
UMP: Serves as a precursor for UTP, enabling ongoing RNA transcription.
Guanine: Functions as a nucleobase precursor that can be converted into GMP and subsequently GTP for transcription and translation.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Provides the bulk of the amino acids required for protein synthesis during translation, excluding those supplied separately for stability or solubility reasons.
Tyrosine: Supplies tyrosine as a protein building block, often added separately due to its limited solubility in concentrated amino acid mixes.
Cysteine: Provides cysteine for protein synthesis and proper folding, typically added separately because of its reactivity and instability in solution.
Additives
Nicotinamide: Acts as a precursor to NAD⁺/NADH, supporting metabolic and redox reactions that help sustain energy regeneration in extract-based systems.
Backfill
Nuclease Free Water: Serves as a solvent to dissolve components and maintain reaction volume while preventing degradation of nucleic acids.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
For the 6 fluorescent proteins we used for our collaborative painting, here are biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Such as maturation time, acid sensitivity, folding, oxygen dependence, etc)
sfGFP: Superfolder GFP is engineered for robust folding, allowing efficient fluorescence even under suboptimal conditions in cell-free systems.
mRFP1: This red fluorescent protein has relatively slow maturation, which can delay the appearance of fluorescence after protein synthesis.
mKO2: mKO2 has fast maturation but is somewhat sensitive to acidic conditions, which can reduce fluorescence intensity if pH is not well controlled.
mTurquoise2: This cyan fluorescent protein has high quantum yield and brightness but requires proper folding to achieve full fluorescence efficiency.
mScarlet-I: mScarlet-I combines high brightness with rapid maturation, improving signal output in cell-free systems compared to older red fluorescent proteins.
Electra2: Electra2 is a flavin-binding fluorescent protein that depends on oxygen-independent chromophore formation but requires flavin cofactors, which can influence fluorescence based on cofactor availability.
My hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation is:
For mScarlet-I, I think increasing potassium glutamate and magnesium glutamate will enhance fluorescence by improving folding and translation. This might allow for better/faster maturation of the fluorescent protein. And maybe more Ribose or Glucose just for more energy.
On the 1536 artwork at https://rcdonovan.com/1536 I was able to “customize the cell-free reagent composition of wells of your choosing … up to 8 wells”
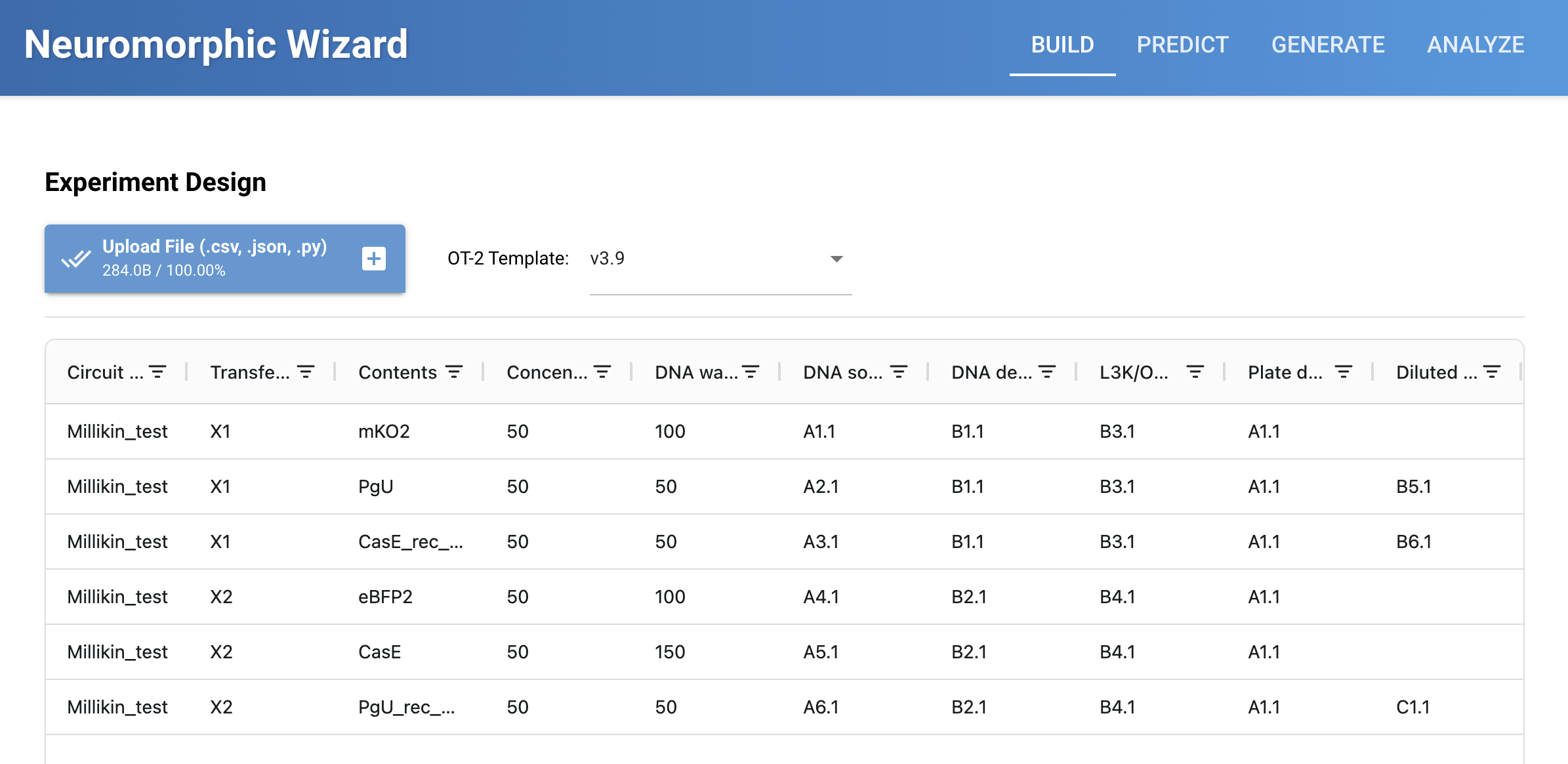
OK, I wanted to do one of each color. And only change two reagents for each one, so as not to be adjusting too many variables at once. So this si what I did:
sfGFP: I raised Magnesium Glutamate for same reasons above. And, I raised Glucose for more energy. This was in Q4-P24, lower right, last pixel of the whole thimg!
mRFP1: I raised Magnesium Glutamate for same reasons above. And, I raised Glucose for more energy. This was in Q3-O22, end of the A in HTGAA
mKO2: I raised Magnesium Glutamate for same reasons above. And, I raised Glucose for more energy. This was in Q1-A1, upper left, first pixel of the whole thing!
mTurquoise2: I raised Magnesium Glutamate for same reasons above. And, I raised Glucose for more energy. This was in Q4-C4, the first crossing of the DNA double helix.
mScarlet-I: I raised Magnesium Glutamate for reasons above. And, I raised Glucose for more energy. This was in Q3-B6, in the heart in “LOVE”
Electra2: I raised Magnesium Glutamate for reasons above. Let’s mix things up: I raised Nicotinamide to hopefully help with flavin metabolism. This was in Q1-D23, the second peak of the M in the Meida Lab logo.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
I used the simulation tool at https://racs.rcdonovan.com/ to create the cloud lab pictured below out of the Ginkgo Reconfigurable Automation Carts. It is pretty rough! Just testing things out.
Week 6 Lab: Gibson Assembly Prelab, use a codon table and convert the following figure to a table of colors of DNA sequences.
Variant Bases Original TGTCAG Orange GTTGGA ? Pink GCATGT ? Some photos from lab …
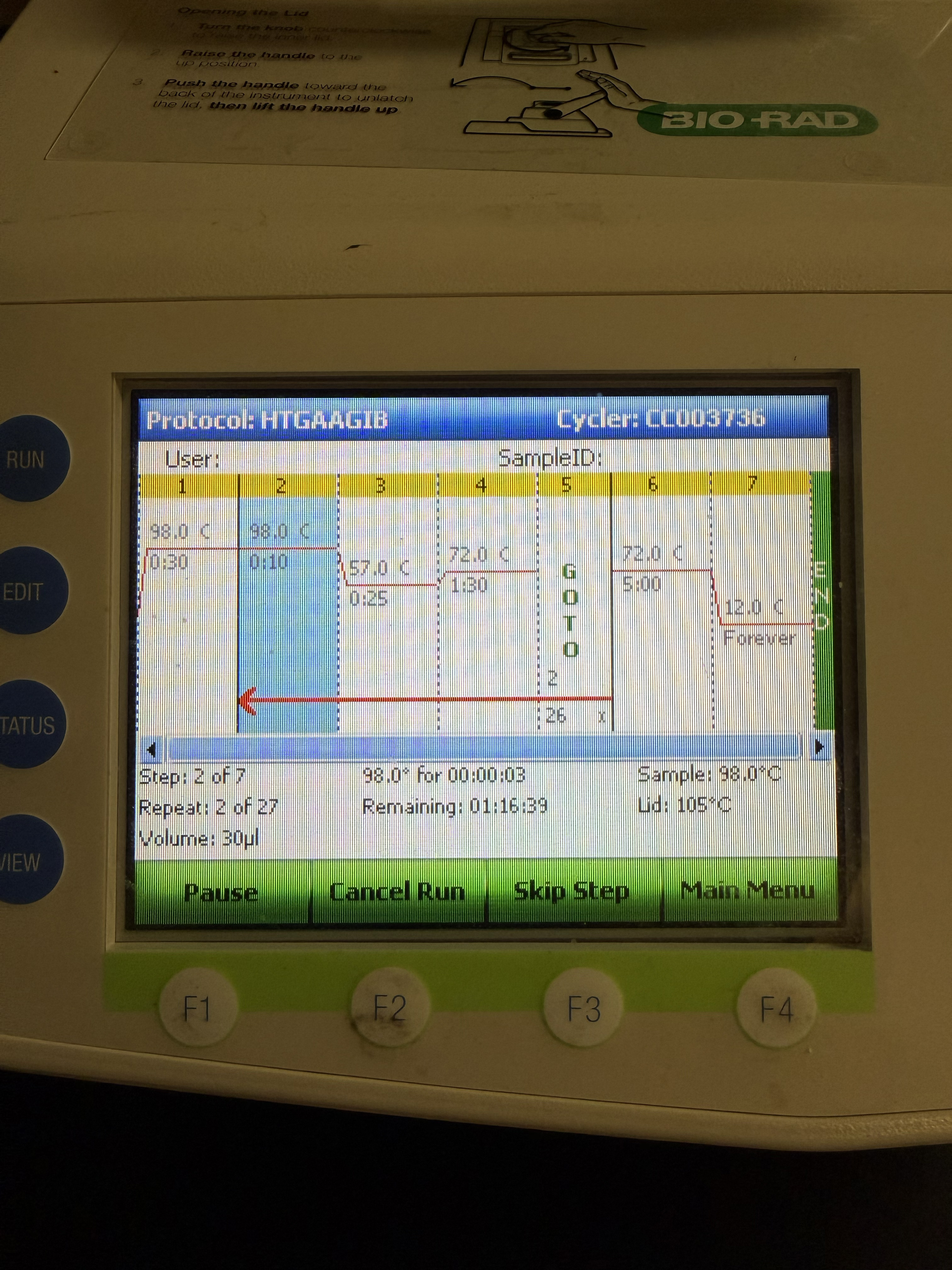
Polymerase Chain Reaction (PCR) Above is a photo of our Bio-Rad C1000 PCR Thermal cycler, set for the Backbone Fragment PCR, with Initial Denature: 98 C, 30 seconds, then for 26 Cycles: Denature: 98 C, 10 seconds; Anneal: 57 C, 25 seconds; Extend: 72 C, 1.5 minutes, hen afteer the 26 Cycle: Final Extension: 72 C, 5 minutes; Hold: 12 C, Forever.
Day 1: Write instructions for the OT-2 to execute. I made a few of these, working on getting different results. I tested over 30 of these! I had a few that returned errors!
Another design test:
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production. Describe the main components of a cell-free expression system and explain the role of each component.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Questions etc. are here: https://2026a.htgaa.org/2026a/course-pages/weeks/week-12/lab/index.html
Post Lab Questions | Mandatory for All Students
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively? Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene? What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures? Generally describe what “OD600” measures and how it can be interpreted in this experiment. OD600 is optical density at 600 nanometer wavelngth … this helps measure how cloudy the liquid is, to determine cell growth.
Subsections of Labs
Week 1 Lab: Pipetting
Week 6 Lab: Gibson Assembly
Week 6 Lab: Gibson Assembly
Prelab, use a codon table and convert the following figure to a table of colors of DNA sequences.
Variant
Bases
Original
TGTCAG
Orange
GTTGGA ?
Pink
GCATGT ?
Some photos from lab …
Polymerase Chain Reaction (PCR)
Above is a photo of our Bio-Rad C1000 PCR Thermal cycler, set for the Backbone Fragment PCR, with Initial Denature: 98 C, 30 seconds, then for 26 Cycles: Denature: 98 C, 10 seconds; Anneal: 57 C, 25 seconds; Extend: 72 C, 1.5 minutes, hen afteer the 26 Cycle: Final Extension: 72 C, 5 minutes; Hold: 12 C, Forever.
Day 1: Write instructions for the OT-2 to execute.
I made a few of these, working on getting different results. I tested over 30 of these! I had a few that returned errors!
Another design test:
Week 9 Lab: Cell-Free Systems
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production.
Describe the main components of a cell-free expression system and explain the role of each component.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
what outcomes might we see at different temperatures and – if we change growht conditiosn we will see difference in gorowth and pigment productionn
OD600 – optical density at 600 wavelngth helps easure how cloudy the liquid is, to determine cell growth.
other experimental setups use acetone to separate … can do protein percipitate analysis … could also use ethanol or isopropyl alcohol to separate diferent pieces from one another …
why might we want to engineer e coli to produce pigments … because … e coli grows fast and because it does fewer cellular functions
biosynthetic pathway knowckout experiments … knock out one gene at a time to see
which should you modify e coli to sacrromycese – maybe stick with what uis easiest to modify – also how large is your gene insert – sometimes have to put into e coli before getting into yeast – e coli grows fast and is cheaper … sacrromycese is slower and constructs may have to simplified … yeast might be better if maybe trying to modify another fungus …
chose one enzyme and outline – main peices we need when making a construct – promoter (ex. T7, conducive, on all the time), RBS (Rhibozome binding site), your coding sequence, terminator, plasmid origin of replication (tells cell to allow plasmid to copy into host), and antibiotic resistance marker
Week 12 Lab: Bioproduction of Beta-Carotene and Lycopene
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively?
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene?
What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures?
Generally describe what “OD600” measures and how it can be interpreted in this experiment.
OD600 is optical density at 600 nanometer wavelngth … this helps measure how cloudy the liquid is, to determine cell growth.
What are other experimental setups where we may be able to use acetone to separate cellular matter from a compound we intend to measure?
We could also use acetone to separate out proteins …
Why might we want to engineer E. coli to produce lycopene and beta-carotene pigments when Erwinia herbicola naturally produces them?
Post Lab Questions | For Committed Listeners Only
Let’s get in touch with our metabolic pathway!
What are the enzymes of the carotene pathway?
Within this pathway, which is the rate determining step (the step that takes the longest)? Which enzyme is responsible for this step?
Notes for design of a DNA construct for bioproduction
The first thing to do is to decide what organism you are going to use for this (E. coli or S. cerevisiae) for production. Which would you choose and why (emphases on production differences)?
Now choose one of the enzymes and lets outline the parts of the construct for expression.
For E. coli lets create a expression vector that works as a plasmid you choose E. coli let’s create a expression vector that works as a plasmids
Eric Millikin, May 2026
SECTION 1: ABSTRACT: Large-format Animated Opentrons Art
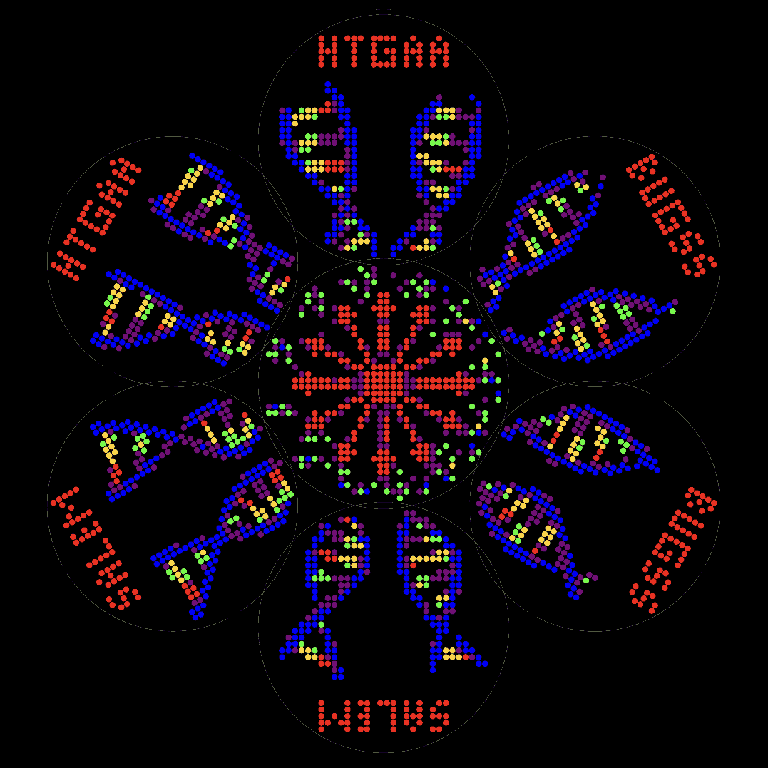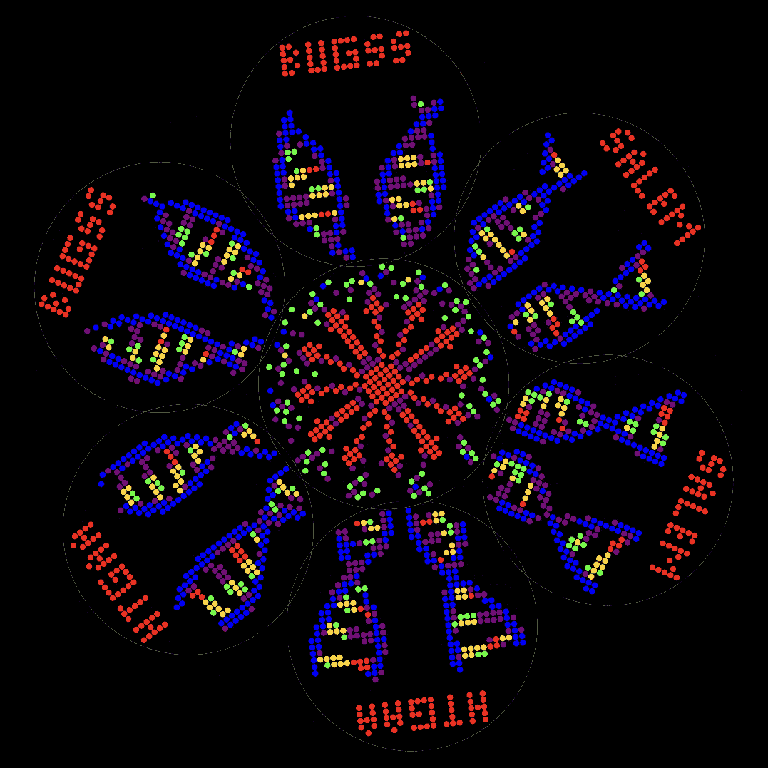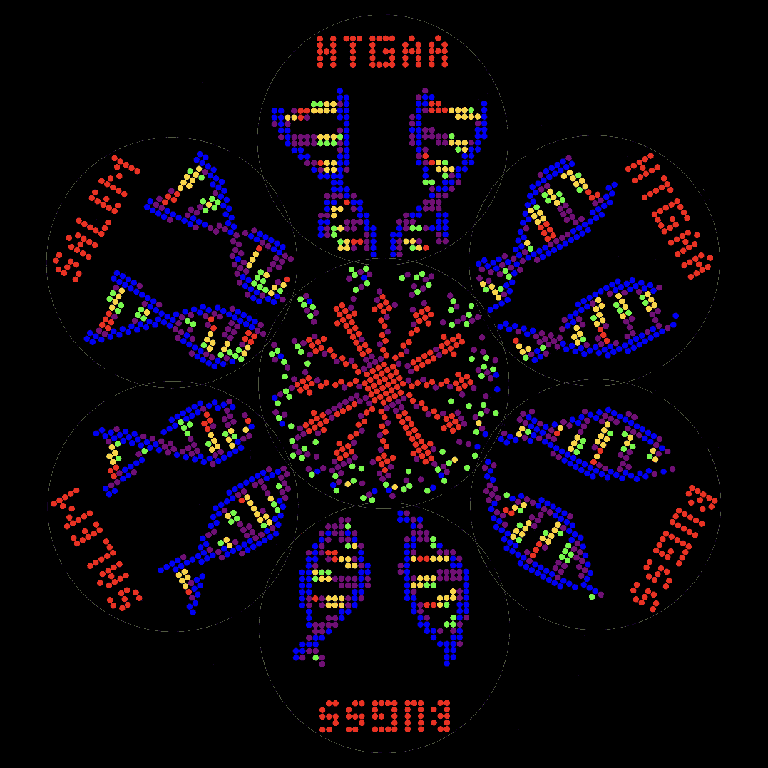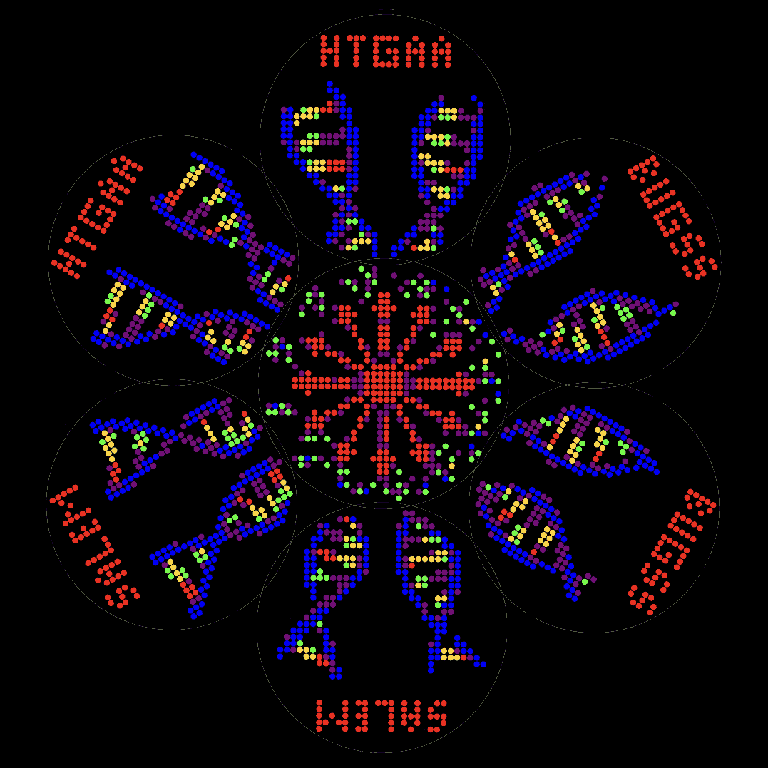
The idea here is to create large-format and animated Opentrons art mosaics, by developing my lab automation and fluorescent bacteria skills to the point where I can create larger format artwork from multiple petri dishes. Shown above is my design featuring rotating DNA strands in seven petri dishes of Opentrons agar art. My art/design here emphasizes the colors that show up best in fluorescent bacteria art; for example, green, red, and blue. My main synthetic biology challenges are 1) Can I create new custom colors biodesigned by me, based on existing mScarlet? and 2) Can I design fluorescent proteins with hidden DNA messages from my ancestors?
Individual Final Project: SALEM II: Synthetic Animated Luminescent Engineered Microbes
Eric Millikin, May 2026
SECTION 1: ABSTRACT:
Large-format Animated Opentrons Art
The idea here is to create large-format and animated Opentrons art mosaics, by developing my lab automation and fluorescent bacteria skills to the point where I can create larger format artwork from multiple petri dishes. Shown above is my design featuring rotating DNA strands in seven petri dishes of Opentrons agar art. My art/design here emphasizes the colors that show up best in fluorescent bacteria art; for example, green, red, and blue. My main synthetic biology challenges are 1) Can I create new custom colors biodesigned by me, based on existing mScarlet? and 2) Can I design fluorescent proteins with hidden DNA messages from my ancestors?
I am descended from women who were executed during the Salem Witch Trials, so I carry part of their DNA with me. I am calling this project “SALEM: Synthetic Animated Luminescent Engineered Microbes.” (My original working title was “Something Something Sacred Sigil System.”)
I aim to engineer fluorescent protein variant(s) based on my ancestor Mary Eastey who was executed at the Salem Witch Trials. These will be mutants based on mScarlet, will include a quote from Mary Eastey encoded into their DNA, may be spectrally shifted toward or into invisible-to-humans infrared, and may be fused with NanoLuciferase for bioluminescence. (This represents several aims of this project.)
The quote I will embed into the DNA is from Mary Eastey’s final petition to the court, “I petition to your honours not for my own life for I know I must die and my appointed time is set … if it be possible no more Innocent blood may be shed.” From the University of Virginia’s Salem Witch Trials Documentary Archive https://salem.lib.virginia.edu/n45.html#n45.22
I will create custom designs for fluorescent bacteria colors and Opentrons python code based on the “Fluorescent Pixel Art” tools created by HTGAA TA Ronan Donavan. See https://opentrons-art.rcdonovan.com/ I am building upon these by creating a series of UNIX shell scripts that I run in Mac Terminal, that largely duplicate Ronan Donavan’s system of converting pixel color positions into Opentrons coordinates. My scripts add a few other features helpful to my project, such as the ability to process all seven images at once.
As shown below, the design of the petri dish setup – six circles around a single center circle – is based on what is described as the “Egg of Life” of sacred geometry. The original chart on the left below is from https://pardesco.com/blogs/news/sacred-geometry-art-symbols-meanings
The animation system where a still image becomes animated when it is rotated is based on the “phenakistoscope,” an early pre-film motion picture device created in the 1830s. The phenakistoscope is sort of like a flip book, but instead of flipping through pages to create the illusion of motion, you spin a disc with images. You can see more info at https://publicdomainreview.org/collection/phenakistoscopes-1833/
The phenakistoscope is a precursor to the more well-known “zoetrope,” where the motion picture frames are arranged on the inside of a cylinder rather than on a disc. “Zoetrope” is based on the Greek words for “wheel of life.”
So, the basic idea here is to create a sacred geometry “egg of life,” that animates like a zoetropic “wheel of life,” based on DNA, the “blueprint of life,” containing convicted “witch” Mary Eastey’s quote “I petition to your honours not for my own life …”
Engineer and test fluorescent protein variant(s) based on my ancestor Mary Eastey who was executed at the Salem Witch trials. My initial “aim one” is to create DNA containing a quote from Mary Eastey inside a genetic construct, which will also contain a red fluorescent protein based on mScarlet.
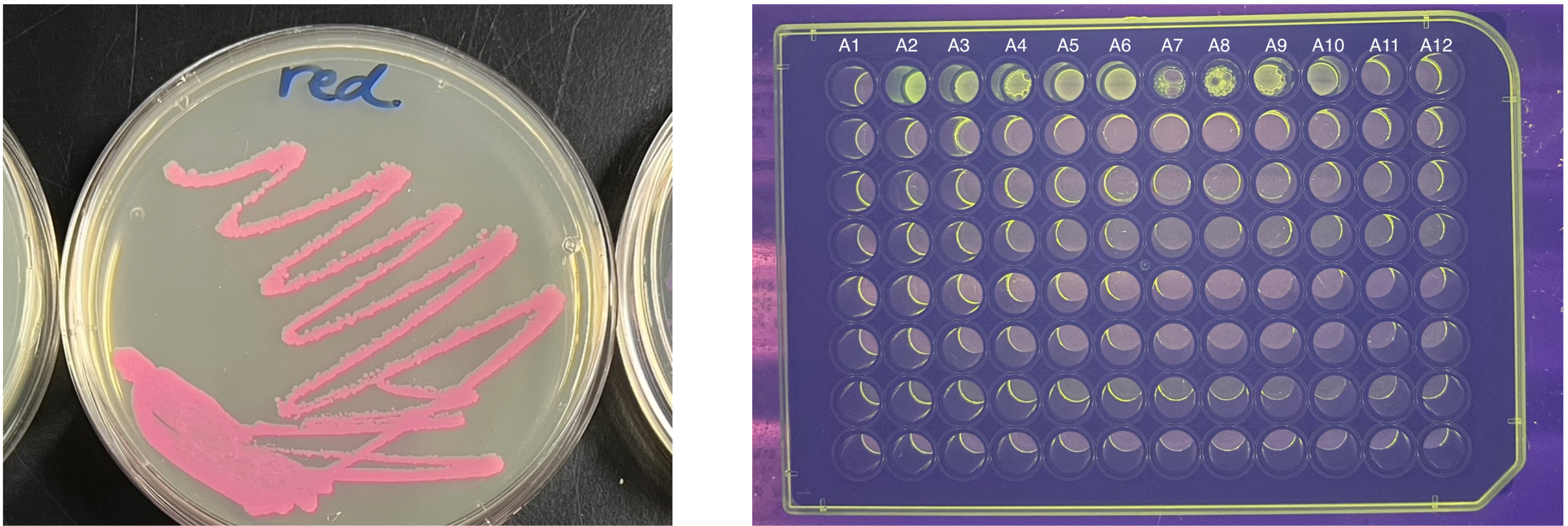
I will design these synthetic biological experimental colors in Asimov Kernel and Benchling, and I will order them from Twist Bioscience. I will initially test the color(s) by hand by streaking on an agar plate with an inoculating loop in the BUGSS Lab. I can also measure the fluorescent proteins with fluorometry and ImageJ.
Aim 2: Development Aim:
Possible further development aims include:
Create the ultimate design itself
Create the Opentrons liquid handling robot code
Design and create devices for live rotating display, with correct speed and lighting, rather than digital rotation
Design possible systems for time lapse animation, during the printing process and/or growing process.
Maybe phosphorescent bacteria that eat other phosphorescent bacteria over time.
Longer form animation, with multiple sets of petri dishes that form longer video(s).
New mutant colors spectrally shifted toward or into invisible-to-humans infrared
New mutant colors fused with NanoLuciferase for bioluminescence.
Thanks to HTGAA BUGSS crew Amanda, Joel, Juhi, Mantis, Violeta, and Marian with whom I discussed these aims (some of these are their ideas!)
Aim 3: Visionary Aim:
Ultimately this project is headed towards live performances featuring Opentrons art. Below should be an embedded video clip from a previous performance with live-coded video and sound art, with my friend, mentor and collaborator Wes Taylor, professor at Wayne State University, on the right and myself on the left. A next-level aim for this project would be for somewhat similar live-coded phenakistoscope performances, featuring large-format animated video projection Opentrons art.
SECTION 3: BACKGROUND:
Briefly summarize two peer-reviewed research citations relevant to your research.
Gadella, T.W.J., van Weeren, L., Stouthamer, J. et al. “mScarlet3: a brilliant and fast-maturing red fluorescent protein.” Nature Methods 20, 541–545 (2023). https://doi.org/10.1038/s41592-023-01809-y
From the abstract: “We report the evolution of mScarlet3, a cysteine-free monomeric red fluorescent protein with fast and complete maturation, as well as record brightness, quantum yield (75%) and fluorescence lifetime (4.0 ns).” This paper describes the process of creating multiple mutations of mScarlet to maximize growth rate as well as fluorescence. mScarlet-I3-NCwt, the protein I am using in this project, is one of the mutants they created.
Ryan, John Charles. “Biological Processes as Writerly?: An Ecological Critique of DNA-based Poetry.” Environmental Humanities 9 (1), 129–148 (2017). https://doi.org/10.1215/22011919-3829163
From the abstract: “This article examines the DNA-based biopoetry of Christian Bök in relation to its antecedents in the art-science experiments of Joe Davis, Pak Chung Wong, and Eduardo Kac. In particular, I develop an ecocritical analysis of the process of encipherment at the center of their works.” This paper is of interest to me because it is looking at other artists use of text encoded within DNA, and it is looking at it from a humanities perspective.
Explain how your project is novel or innovative.
My project uses Opentrons liquid handling robots to create agar art in new and unusual ways. I have successfully explored larger format (multiple petri dish) designs. I have also successfully explored animated agar art using early pre-film motion picture phenakistoscope techniques. Also, my DNA design with text encoded adds another innovative layer to agar art.
Explain why your project matters and what impact it could have. (Minimum 5 sentences.)
One of the things that matters most about this project is that it provides a system for unique visual art to help lead conversations about synthetic biology concepts among audiences that do not typically work with it or discuss it. This could provide an accessible way to expand public engagement with synthetic biology. My goal is to ultimately create a series of animations that can be combined into a short film. Screening this film at film festivals and art galleries should provide new venues for experiencing and considering synthetic biology. The project also demonstrates new ways of using laboratory equipment and laboratory automation for visual, literary, and ultimately performance, art.
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility).
While the strains of E. coli used in agar art are nonpathogenic, I still have a responsibility to follow biosafety standards, regulations, containment, sterilization, etc. Also, since this is intended as part of an art series meant for viewing by the general public, communication about safety issues is also important. This also ties into beneficence, as exhibiting this work can contribute positively to society by helping people engage more thoughtfully with synthetic biology.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take).
I need to convert the quote from Mary Eastey into a DNA sequence.
One way would be to first convert the sentence into letters available as IUPAC amino acid codes – https://www.bioinformatics.org/sms/iupac.html – which only has 20 letters, so I changed every B to a P, every O to a Q, every U to a V, and removed spaces:
I need to get the fluorescent protein to combine this with
I am thinking mScarlet-I3-NCwt – https://www.fpbase.org/protein/1VSM7/ – since it is 1) Scarlet and there are many occult scarlet references, 2) I3 is almost the number 13, which is the typical number of witches in a coven, etc.; and 3) NCwt is almost “newt” as in “eye of newt” from the witches’ potion chant of Shakespeare’s “Macbeth. “In the caldron boil and bake; Eye of newt and toe of frog, Wool of bat and tongue of dog … For a charm of powerful trouble, Like a hell-broth boil and bubble.”
I need to design this, preferably in Asimov Kernel …
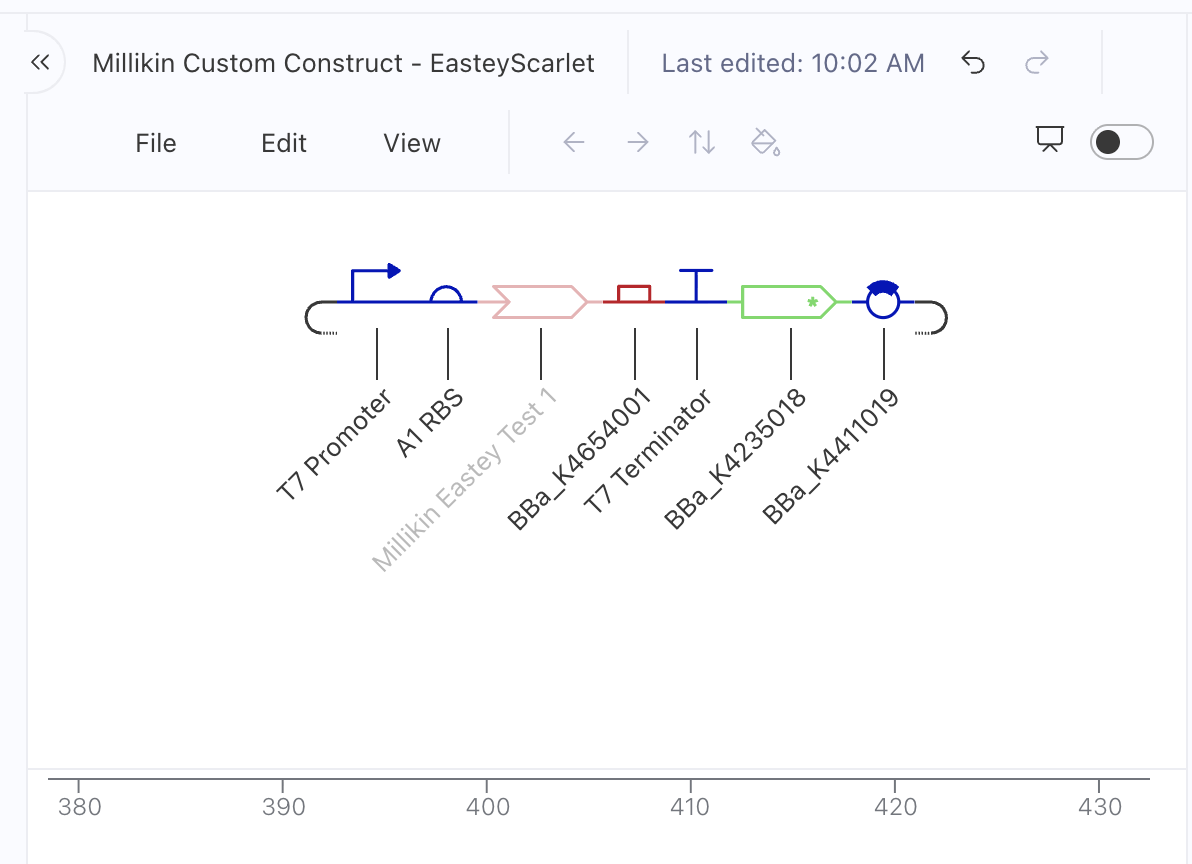
Here was my construct design in Asimov Kernel as of April 15, 2026, this is now out of date! I am keeping it here for documentation and explanation purposes:
From left to right, this early version had:
T7 promoter
A1 RBS
Millikin Eastey Test 1 (my code insert)
BBa_K4654001 (mScarlet-I3 red fluorescent reporter; I can switch this to mScarlet-I3-NCwt)
T7 Terminator
BBa_K4235018 (Ampicillin Resistance Gene)
BBa_K4411019 (pET28a-backbone)
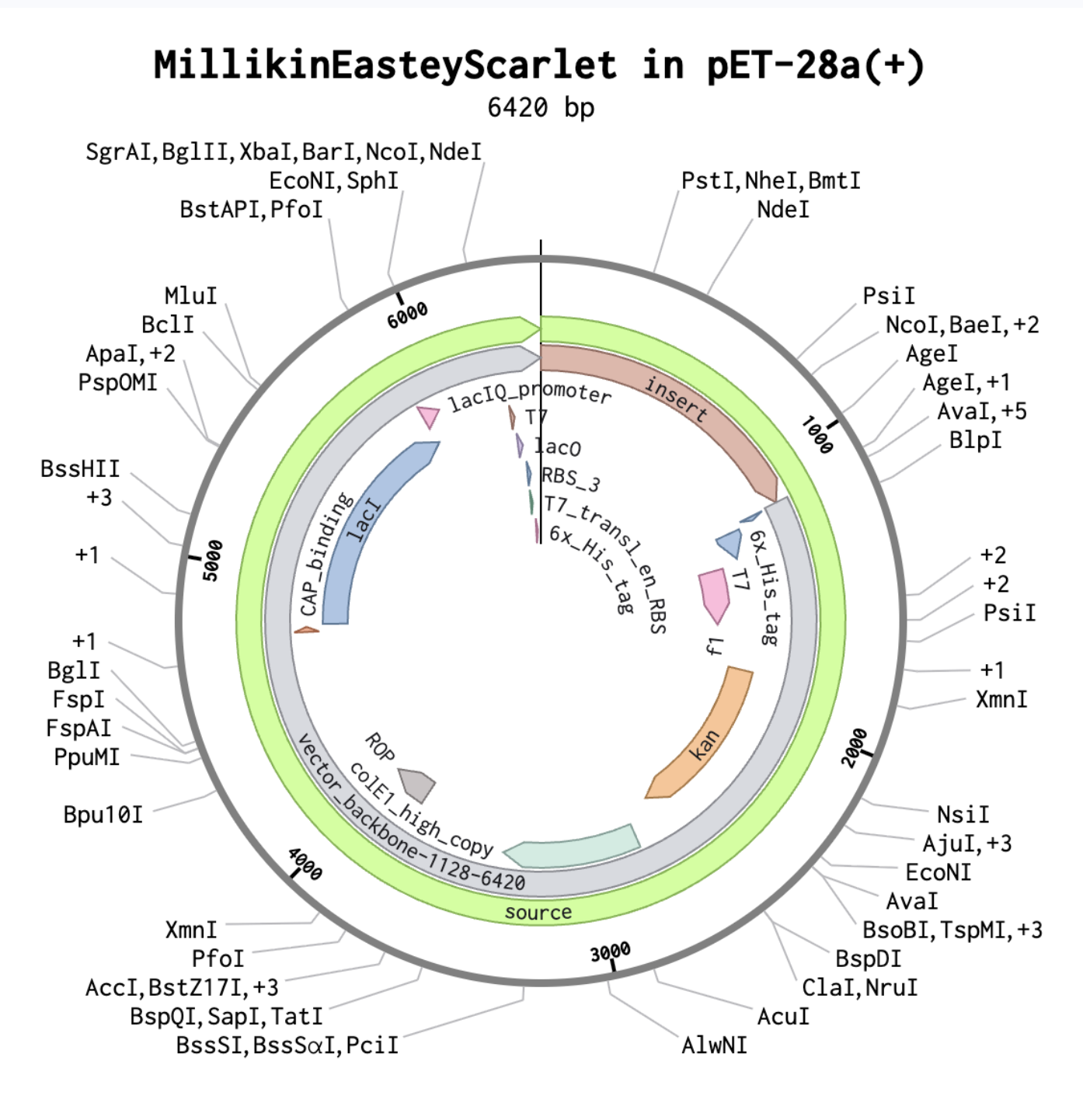
I checked this out with our BUGSS lab crew and asked if everything seemed good. Our awesome TA Amanda said maybe Kanamycin resistance was what we used before. So, I was thinking about switching Ampicillin Resistance Gene for Kanamycin Resistance Gene, and then I looked up the pET28a-backbone and according to the IGEM Registry of Standard Biological Parts at https://parts.igem.org/Part:BBa_K3521004 “pET28a-Backbone … contains anti-kanamycin genes.” So, I am likely getting rid of any resistance genes in my construct since they seem redundant with what the pET28a-backbone already has.
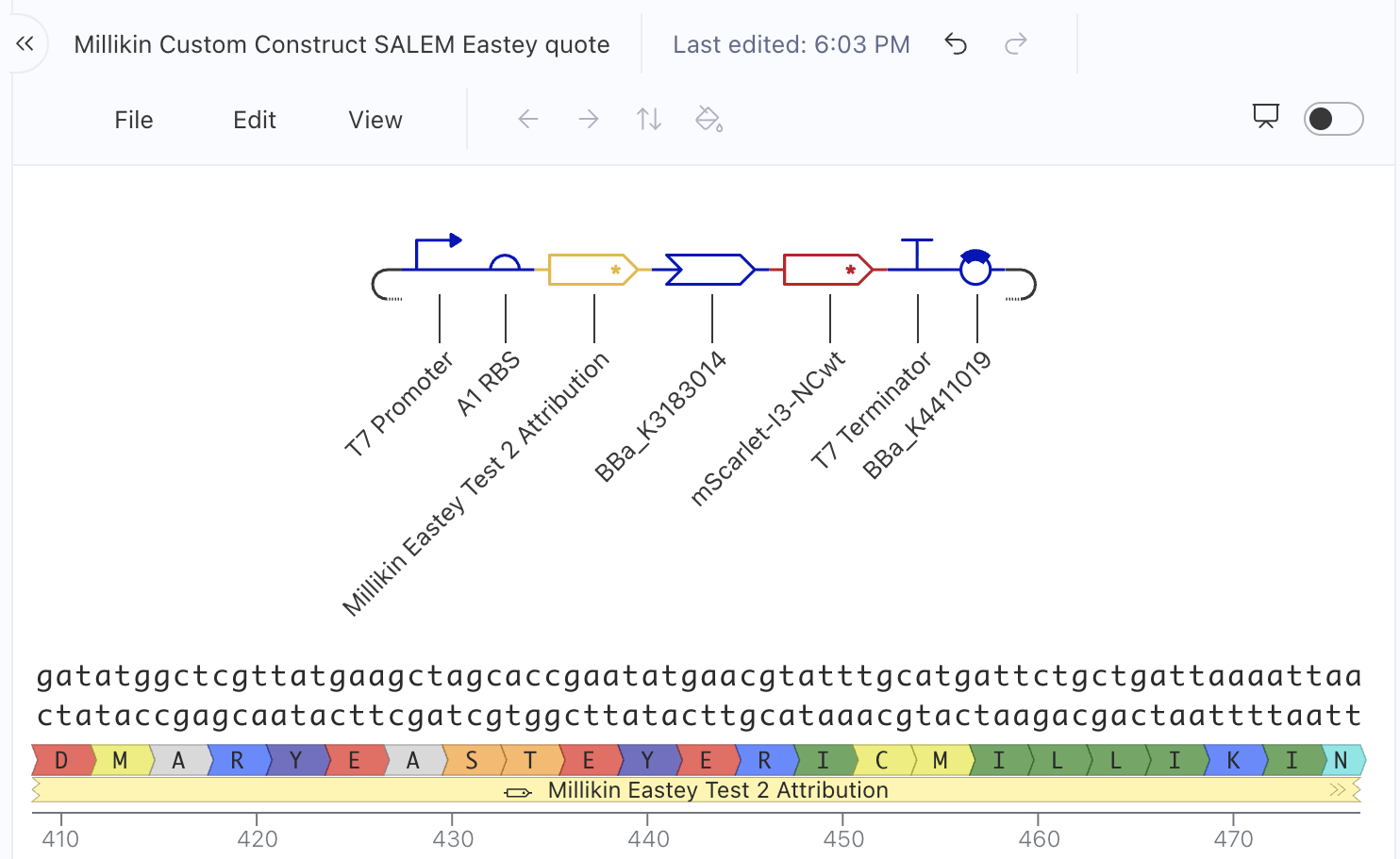
Updated construct plan April 17, 2026:
This is now out of date! I am keeping it here for documentation and explanation purposes
From left to right, this version had:
T7 promoter
A1 RBS
Millikin Eastey Test 2 Attribution (my code insert, now with MARYEASTEYERICMILLIKIN attribution after the quote, which you can see at the bottom of the screenshot above)
BBa_K3183014 (Gly-Ser-Gly Protein Domain Linker for E. coli)
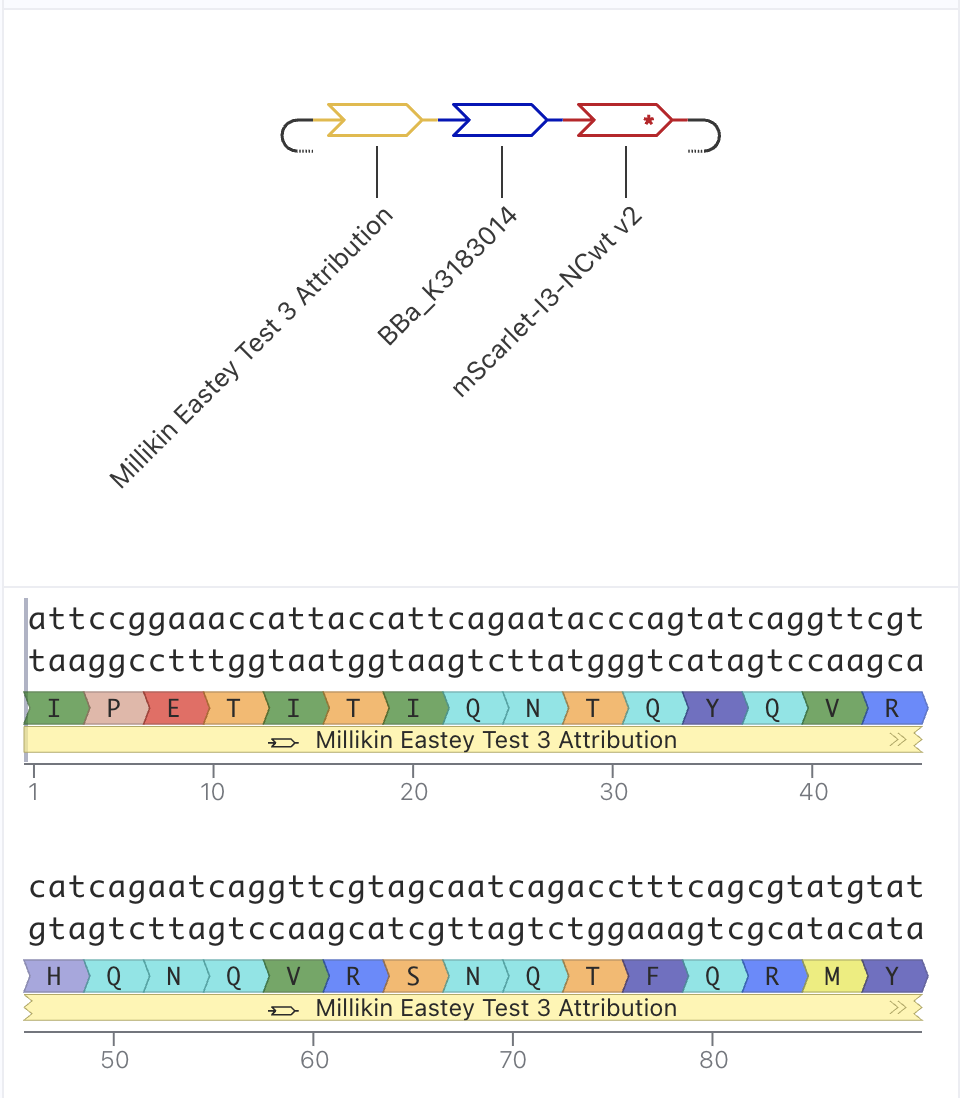
This is my latest/final version! I met with our awesome Lisa Scheifele, BUGSS Lab Executive Director, who helped me improve this! Basically, I still had some redundancies between my Asimov Kernel construct and the pET28a-backbone/
This is my updated Mary Eastey code, CDS (-start -stop) for Asimov Kernel:
And this is my construct design from Asimov Kernel, much simpler than before, as before I was including several parts that were redundant with the PET-28a(+) backbone.
Place Twist Bioscience order.
I put it into the “2026 HTGAA Ordering: DNA, Reagents, Consumables” spreadsheet on April 23 and my awesome node lead Amanda submitted the forms on April 25. I then got the Twist Bioscience quote on May 7. Things are progressing smoothly.
Below is an image from Benchling showing a plasmid map of the Asimov Kernel construct inserted into the pET-28a(+) as ordered from Twist Bioscience:
Test the color(s) by hand by streaking on agar with inoculating loop
I will initially test the color(s) by hand by streaking on an agar plate with an inoculating loop in the BUGSS Lab. I can also measure the fluorescent proteins with fluorometry and ImageJ, as we did previously at BUGSS Lab. These are shown below.
Finalize a design for 7 petri dishes
I have been making regular updates to my agar art design, to improve its look, to strengthen its concept, and to display my latest Opentrons coding techniques. My most recent design for the center petri dish has a star made of arrows; this is both based on the SBOL visual glyph for promoter – https://sbolstandard.org/docs/SBOL-Visual-2.2.pdf – as well as the chaos star of Michael Moorcock fantasy novels and the later used in chaos magick by Peter J. Carroll (1953 – 22 April 2026) who recently passed away. A large animated GIF of a recent version of that central star is in section 10 below.
It has been updated for Ginkgo cloud lab use, so I am developing my own system to convert images into color coordinates. I have been writing some Unix shell scripts to run in terminal that use ImageMagick to crop and scale images, convert them into colors that we have at BUGSS Lab for Opentrons art, and then extract the color information and coordinates for use in Python scripting for Opentrons.
Below is a screenshot of one of my scripts running in Mac Terminal, showing extracted color coordinates for green fluorescent protein.
Use Opentrons liquid handling robot to create the artwork
For this I will largely use the previous system we used at BUGSS Lab for Opentrons agar art, with the primary Opentrons system modification being that I will be using my new scripts for creating color coordinates for use within Python code for Opentrons.
Below is a previous Opentrons self-portrait, in a photo by Joel Tyson, showing some of the work I will be building on:
Rotate in digital video editing to test the animation effect
I have had success with my early-aim system for digital rotation of agar art photography to create animated videos. Below is a recent example from late May showing some of my new Opentrons code, featuring variable size dots, here going from 0.1 to 0.25 microliters:
Some of the synthetic biology techniques most relevant to my project
Bioethical Considerations
DNA Gel Art: DNA Editing
DNA Gel Art: DNA Construct Design
DNA Gel Art: Databases
Lab Automation: Creating Code for Laboratory Automation
Lab Automation: Using Liquid Handling Robots (e.g., Opentrons)
Lab Automation: Designing a Twist Order
Protein Design: Use of Asimov Kernel
Protein Design: Use of Benchling
Protein Design: Databases
Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project. (min. 4 sentences)
I am using protein databases and Asimov Kernel for DNA Construct Design for purchasing through a Twist Order. I particularly enjoyed working with Asimov Kernel for construct design because I found its graphical interface, where I was able to build a construct out of glyphs, seemed sort of like an occult process where one might use a series of magical symbols to construct a golem or other synthetic being.
I am also Creating Code for Laboratory Automation for Using Liquid Handling Robots (Opentrons). I have particularly enjoyed writing my own Unix shell scripts and ImageMagick programs for converting my digital image files into color coordinates for Opentrons code. These new scripts I have written have allowed me to more quickly prototype new designs. I expect to keep building on these in the future. I have used ImageMagick in the past; I was pleased that I was able to use it for this project to carry on my magical themes.
Identify any How To Grow (Almost) Anything Industry Council companies which are associated with your final project (optional)
Associated with Aim 1 of my final project would probably be:
Epibone https://www.epibone.com/ “facial bone product … customized for patients via a CT scan … with stem cells extracted from patients’ abdominal fat”
Mycoworks https://www.mycoworks.com/ “create a platform for the highest quality materials using Fine Mycelium”
Upside Foods https://upsidefoods.com/ “Cultivated meat is meat grown directly from animal cells”
SECTION 5: Results & Quantitative Expectations
What aspect of your final project did you choose to validate?
I have designed a DNA construct to express the fluorescent protein mScarlet-I3-NCwt, ordered it via Twist Bioscience, and will ultimately test its fluorescence by using it in agar art created with an Opentrons liquid handling robot. Prior to testing it with the Opentrons robot, I will perform other tests such as streaking it on an agar plate.
Write down a detailed protocol of how you validated this aspect of your final project.
I have validated the design of my construct within Asimov Kernel by running simulations such as checking for protein concentrations over time as well as running a biosecurity sequence scanner.
I have validated my custom Unix shell scripts and my resulting Opentrons Python code by running Opentrons simulations within Google Colab notebooks.
I will validate the fluorescence of my construct by streaking on an agar plate before using it in the Opentrons robot.
I can also compare its fluorescence to other fluorescent proteins by using fluorometry and ImageJ, as we previously did in our lab. This will be most useful in later aims of this ongoing project, as I create more fluorescent proteins.
What synthetic biology techniques did you utilize in validating this aspect of your final project?
From my list of techniques above:
I used Asimov Kernel for my construct design, running simulations, and biosecurity scanning.
I created code for laboratory automation and for Opentrons Liquid Handling Robots and used Google Colab notebooks to run simulations to validate my code.
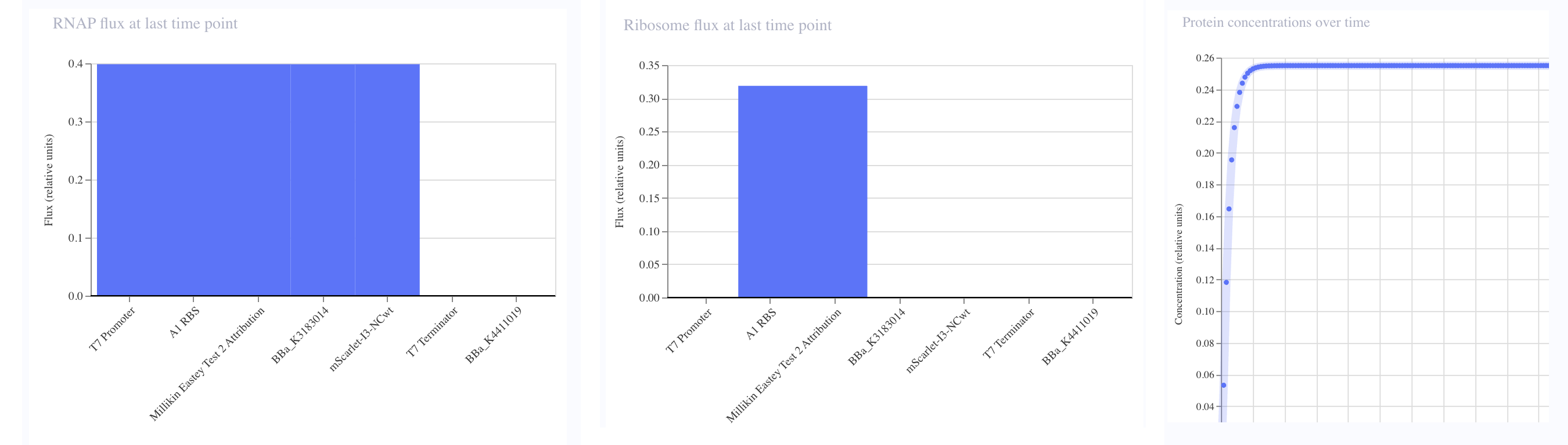
You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data.
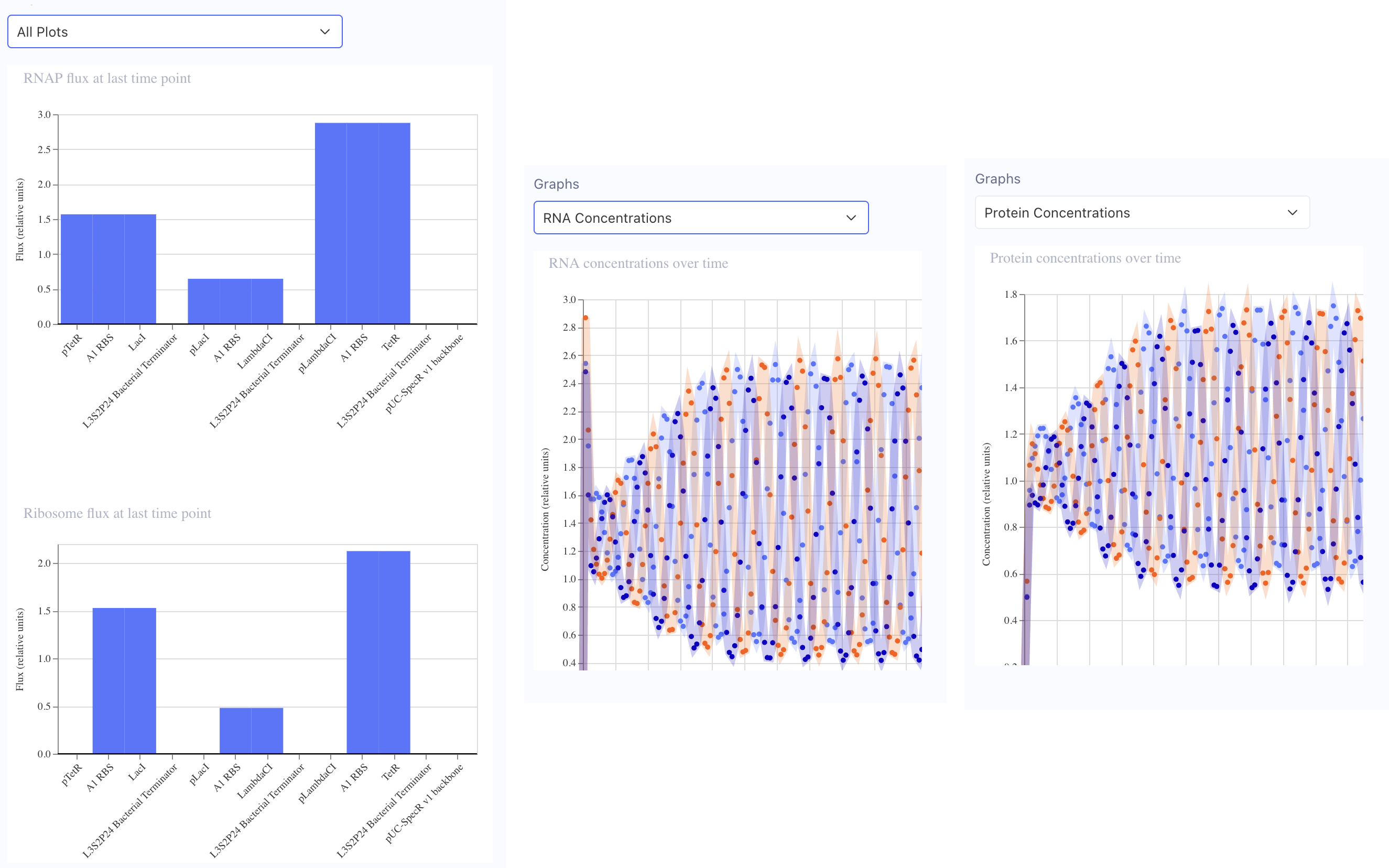
I ran several construct simulations in Asimov Kernel. For example, in my simulation of one of my constructs with the coded Eastey quote and mScarlet-I3-NCwt fluorescent protein, the screenshots below from Asimov Kernel show RNAP Flux and Ribosome Flux seemingly pretty well-balanced, and the protein concentration over time seems fast enough that experiment results are pretty quick, but not so fast that there would be worries that something toxic might grow too quickly.
I also ran Asimov Kernel’s Biosecurity sequence scanner using SecureDNA. Below is a screenshot showing “No flagged sequences detected.”
I also performed countless validations of my Opentrons code using simulations within Google Colab notebooks, as I added new features to my series of custom scripts for converting my digital artwork into Opentrons robot coordinates.
Did you encounter any unexpected challenge(s) when performing your validation?
The major challenge for me with Asimov Kernel was probably just my learning curve with it as a new tool for me. I also had learning curve challenges with the Opentrons simulations when they were new to me. At this point toward the end of this project, the challenges with my Opentrons simulations have been the sorts of challenges that are typical of problem solving with an iterative project as I have added new features to my scripts, such as variable volume dot sizes.
SECTION 6: ADDITIONAL INFORMATION
List all references cited in this assignment (bullet-point list)
Hamidi, F., Dusman, L., Booy, L. “Infinite Transformations in a Suitcase: Encountering Human-DNA Interaction through Poetry-infused Wine.” (2024). In Proceedings of the ACM Conference on Creativity and Cognition. https://doi.org/10.1145/3689050.3707674
Ryan, J. C. (2017). Biological processes as writerly?: An ecological critique of DNA-based poetry. Environmental Humanities, 9(1), 129–148. https://doi.org/10.1215/22011919-3829163
Su, Y., Walker, J. R., Park, Y., et al. (2022). Engineered amber-emitting NanoLuc luciferase and its use for immunobioluminescence imaging in vivo. Journal of the American Chemical Society, 144(17), 7875–7882. https://doi.org/10.1021/jacs.2c02320
Create a supply list and budget for your project (bullet-point list)
Main budget areas are:
BUGSS Lab membership (to cover lab space, time, and supplies): $100 a month or $1,000 a year
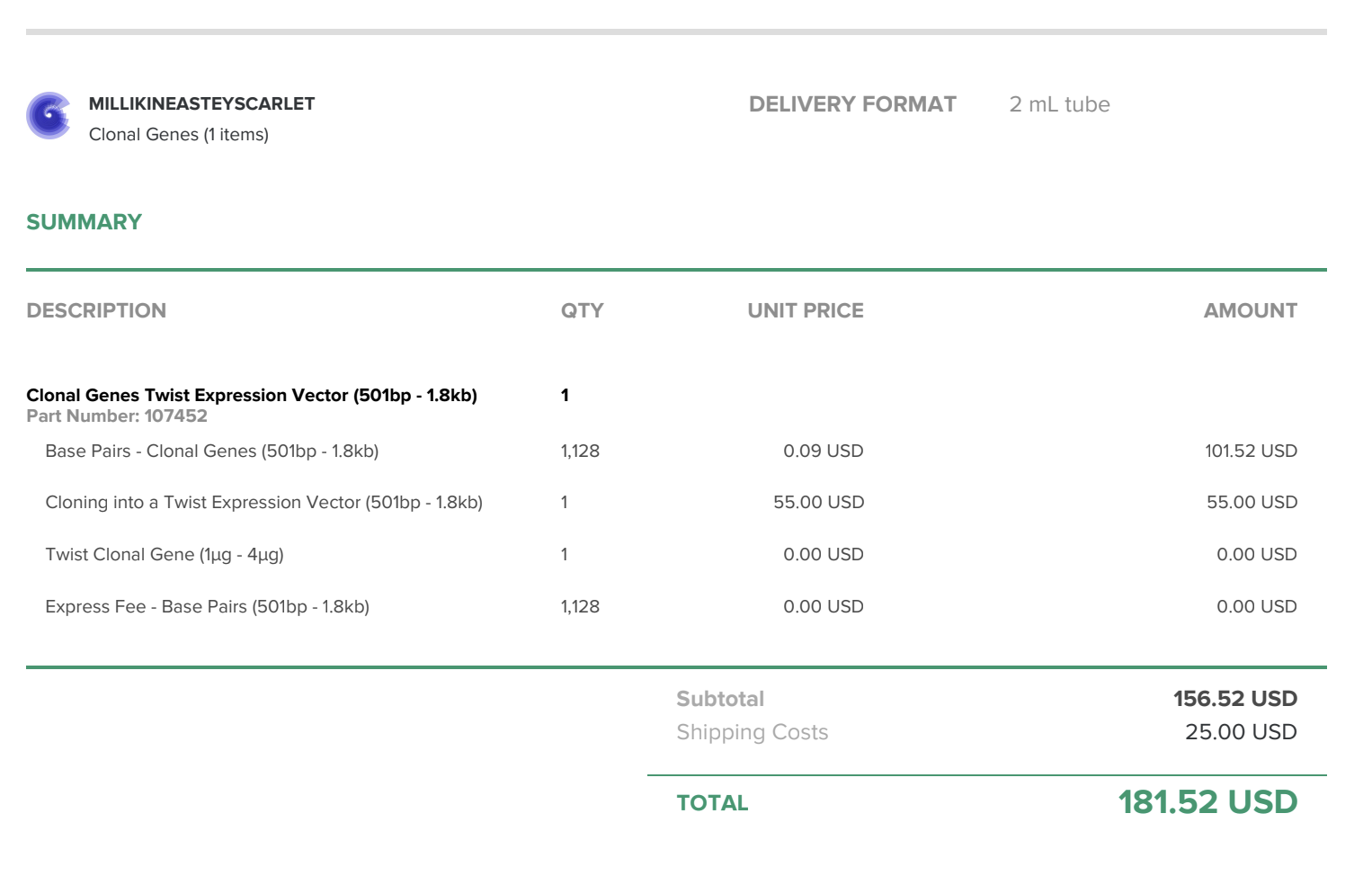
Twist order (for my custom DNA constructs): $181.52
Below is a screenshot of my first DNA order from Twist Bioscience:
Thank you! Big thanks on this project go to:
Director Dr. David Sun Kong, Head TA Ronan Donovan, Prof. George Church, Prof. Joseph Jacobson, and everyone else at HTGAA (How to Grow (Almost) Anything)
Amanda Mainello-Land, Joel Tyson, and Lisa Scheifele at BUGSS (Baltimore Underground Science Space)
Juhi Dhanesha, Mantis Harper-Blanco, Marian Valdivia, and Violeta Vilcapoma, lab partners and collaborators working with BUGSS as part of HTGAA
Ingrid Stump, Prospect Research Analyst at Virginia Museum of Fine Arts
Everybody who joined the 12-hour global HTGAA presentations!