Week 4 HW: Protein Design Part I
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Part A - Conceptual Questions
For my homework, I initated a conversation with Claude Ai using Sonnet v4.6. My prompts use a method I use to start with a question, allow me to provide my answer, and receive an evaluation of my response with reinforcing key learning concepts. (Expand to see detailed responses to my answers.). I find this approach to be more interactive and leads to better knowledge retention.
Question 1: How many amino acid molecules are in a 500g piece of meat?
Using the given parameters — 500g of meat, average amino acid weight of 100 Daltons.
Question 2:Why do humans eat beef but not become a cow? Eat fish but not become a fish?
We digest and metabolize proteins rather than absorb and mutate. Our proteins are protected and encoded.
Question 3: Why are there only 20 natural amino acids?
Because that represents a large number of variations — when combined with every possible sequence, the combinations are exponentially high in the billions.
Question 4: Can you make other non-natural amino acids? Design some new ones.
Your answer: Yes — evolution converged on 20, but others may exist outside evolutionary pressure, possibly arising from light spectrum properties or geological timescales.
Question 5: Where did amino acids come from before enzymes that make them, and before life started?
Your answer: Geological, light energy, electrical, and even gravitational forces were all at play resulting in amino acid formations before our understanding of life emerged.
Question 6: If you make an α-helix using D-amino acids, what handedness would you expect?
An alpha-helix creates a right-handed coil.
Question 7: Can you discover additional helices in proteins?
Yes, since a protein may have many evolutionary and disrupted or folded variations.
Question 8: Why are most molecular helices right-handed?
Due to molecular electrical charge initiating primary bonds resulting in a right-handed twist, with left-handed helices possible under favorable conditions.
Question 9: Why do β-sheets tend to aggregate? What is the driving force?
β-sheets aggregate because they are flat and linear in design with bonding properties, repeating in a pattern or weave.
Question 10: Why do amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid diseases form β-sheets due to misfolding pathology. Since they are β-folds they are sticky and thermodynamically strong, difficult to clear — the same properties that would make an excellent material such as a synthetic cement.
Question 11: Design a β-sheet motif that forms a well-ordered structure.
A motif that acts as a 3-dimensional weave on the x, y, and z axis — resulting in a textile stronger than a simple x,y weave, useful in environments requiring strong resistant materials like Kevlar or heat resistant tiles.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I selected GFP https://www.uniprot.org/uniprotkb/P42212/entry
It is a widely studied protein with highly visual properties and application to biosensors, relevant to my final project scope.
Identify the amino acid sequence of your protein.
The amino acid sequence is MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is: 238 amino acids. The most common amino acid is: G, which appears 22 times.
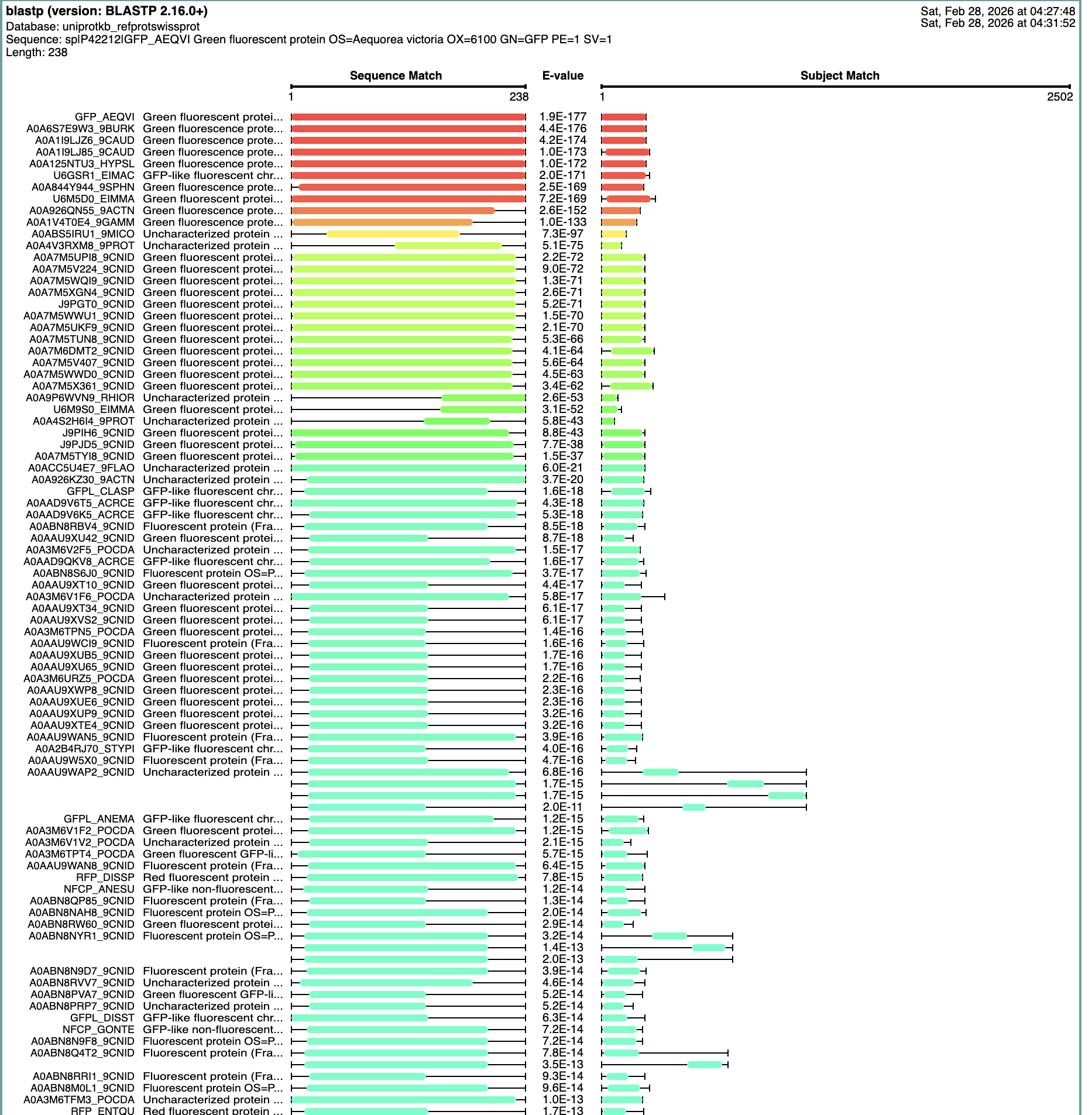
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
The Blast Protein Existence menu showed 152 results with homology.
Does your protein belong to any protein family?
Yes, this is a member of the Green Fluorescent Protein (GFP) Family
Identify the structure page of your protein in RCSB
https://www.rcsb.org/groups/sequence/polymer_entity/P42212
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
In 1996 the protein structure was solved. It is a good quality structure with a resolution of 2.4 Å A primary characteristic is the β-barrel fold with the chromophore inside, which helps to protect from damage.
https://pmc.ncbi.nlm.nih.gov/articles/PMC3739439/
Here are some very interesting sites related to this protein, that I will be revisiting.
https://www.proteinspotlight.org/back_issues/011
https://www.ekac.org/transgenicindex.html
Are there any other molecules in the solved structure apart from protein?
Chromophore (CRO) formed and protected inside. Water molecules (HOH)
Does your protein belong to any structure classification family? Green Fluorescent Proteins, with 633 structures.
Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
The structure has more sheets, indicated by amino acids, in yellow. The barrel shape is helical but the structure is formed in sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The amino acids create a hydrophilic barrel shape that positively attract and retain water, creating a protective surface. Inside of the barrel is the hydrophobic chromophore that is protected until it is triggered by light to release fluorescent illumination.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
The surface is primarily hydrophilic but also has permeability via holes (binding pockets) to allow for controlled hydration, to protect the chromophore, which enables light photons to be absorbed and emitted as fluorescence.
Part C1: Protein Language Modeling
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
- M48 has the single highest probability of a recurring sequence.
- Region 20-27 has an overall high model score
- Region 3 contains a strong outlier
Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- My initial run showed a very dense plot.
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
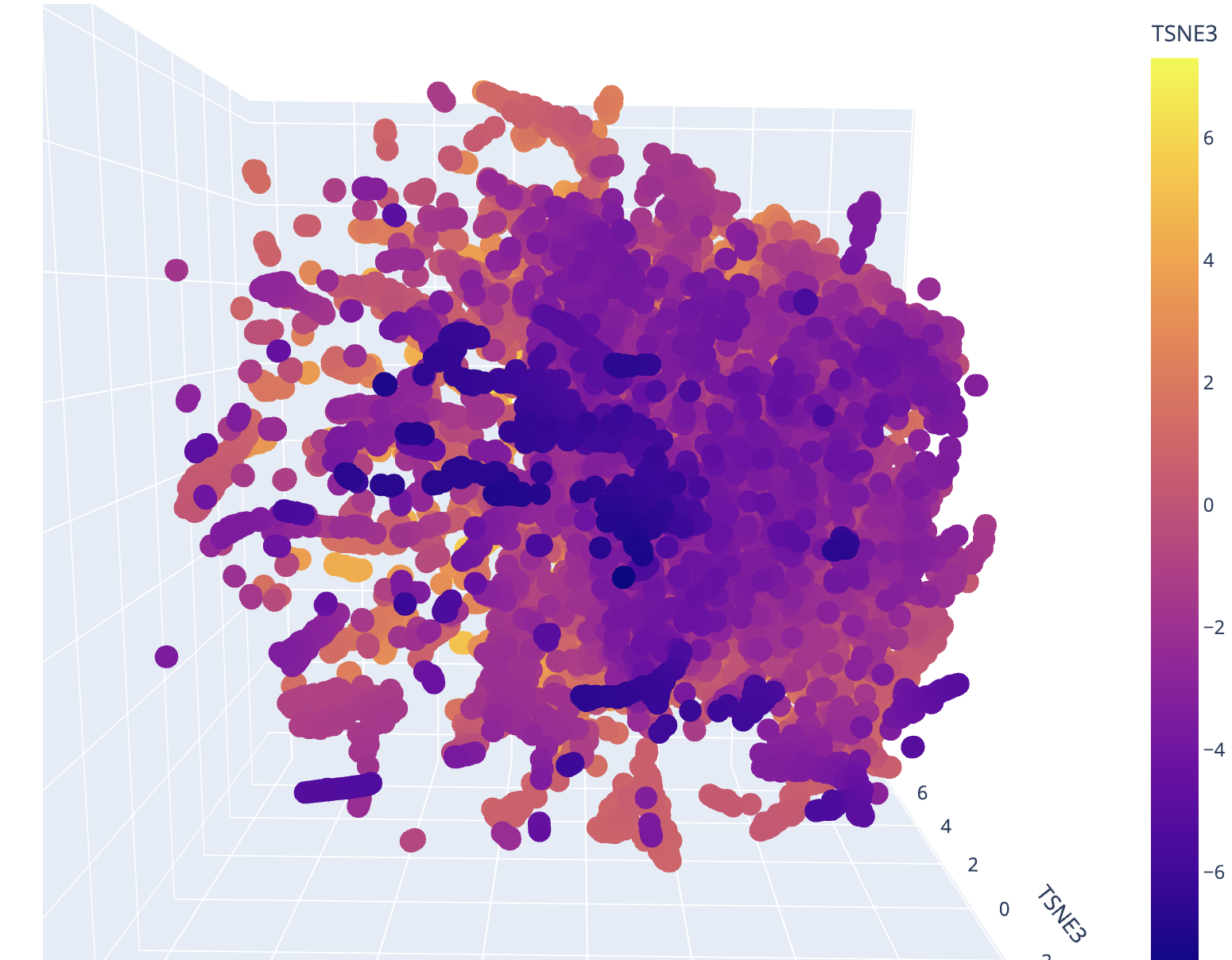
- Analyze the different formed neighborhoods: do they approximate similar proteins?
- I reduced the complexity to generate a plot that includes my selected protein.
- The plot shows similar proteins based on a wide range of dimensions, so they don’t always relate to similar proteins, just similar shared amino acids with higher probability of a match. In some instances, the proteins line up much more predictably, such as a high match in a linear progression.
- Place your protein in the resulting map and explain its position and similarity to its neighbors.
- My selected protein has a near neighbor of Clostridium botulinum which is in the family of Botulinum Neurotoxins. What is intersting is that a protein that creates biofluorescence in jellyfish is in proximity to a protein that creates a neurotoxin. This seems to be a function of evolutionary design of organisms that rely on this close relationship.
Part C2: Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
- Yes, the folded protein closely matches my original structure, but there are some degraded areas of the barrel formation shown with a confidence gradient (green is good, red is bad)
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
- Yes, the structure seems resilient to mutations, even folding better in the α-helix regions.
Part C3: Protein Generation
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
- I initially ran the Inverse Folding function, using default settings.
- It predicted low confidence in the mutation scan:
- It produced a model based on default settings, that was unexpected. (sea slug)
- I realized that I need to enter a new PDB ID for my selected protein.
- I ran it and received an expected result:
I then applied a mutation to my GFP based on a Claude AI inquiry to ’turn the GFP to blue fluorescence'
- Y66H (Tyr→His) — replaces the phenol ring with an imidazole ring, shifting emission from ~509 nm (green) to ~448 nm (blue)
- Y145F (Tyr→Phe) — the “enhanced” BFP (EBFP) stabilizer, improves brightness and folding
- F64L — improves folding at 37°C (same as EGFP)
I ran the new sequence through the mutation scan:
- I had Gemini help to write code that appends this new mutation sequence to the RDP target list.
- Once a prediction was made, I applied the sequence to the ESM to see if it would produce a result.
Input this sequence into ESMFold and compare the predicted structure to your original.
- Here is the mutated, inverse folded, and visualised with ESMFold:
Part D. Group Brainstorm on Bacteriophage Engineering
- Find a group of ~3–4 students
- Read through Phage Reading resources
- Review bacteriophage goals
- Brainstorm Session
- Choose One or two main goals
- One Page Proposal
- Which tools
- Why tools may help to solve sub-problem
- One or two potential pitfalls
- Schematic of Pipeline
- Group’s short plan for engineering a bacteriophage
- Post plan here
Part D - Plan
Hypothesis:
I believe we can focus on the cationic properties, or positive electrical charges that are present in the amino acid sequence. By substituting amino acids that enable more positive charge strengthening electrostatic attraction, we may create more binding activity. Lysis timing can be tuned in either direction by manipulating charge density.
Experimental Pipeline
Phase 1 — Discovery
- UniProt
- Retrieve canonical L-protein sequence
- Confirm Region 1, 2, and 3 boundaries
- BLAST
- Search for homologous sequences across phage strains
- Identify conservation and variability at target residues
- PyMOL
- Render 3D structural model
- Apply polarity-based color coding to each region
Phase 2 — Mutation Analysis
- PyMOL
- Isolate target residues
- Examine local chemical environment and spatial context
- ESM2
- Mask target residues and score substitution probability
- Generate per-residue probability data for C2, C3, C4
- Heatmap
- Synthesize BLAST conservation and ESM2 probability scores
- Overlay onto PyMOL structure to confirm target sites
- ESMFold
- Predict 3D structure of each mutant sequence
- Generate pLDDT confidence scores per residue
- PyMOL
- Import ESMFold outputs
- Render side-by-side comparison of C1 baseline vs C2, C3, C4
Phase 3 — Synthesis
- Codon Optimization
- Optimize mutant sequences for E. coli expression
- Verify no unintended mRNA secondary structures introduced
- Twist Bioscience
- Submit all four constructs for gene synthesis
- Confirm synthesis feasibility and receive gene fragments
Phase 4 — Plasmid Design
- Benchling
- Design annotated circular plasmid constructs for C1–C4
- Include promoter, RBS, insert, terminator, and selection marker
- Review Gate
- Confirm correct reading frame and insert orientation
- Verify no unintended open reading frames
- Confirm host compatibility before proceeding
Phase 5 — Execution
- Opentrons OT-2
- Run liquid handling protocol for all four constructs
- Collect lysis timing, plaque formation, and MurA activity data
- Compare all results against C1 baseline
Potential Pitfalls
- My hypothesis focuses on Region 1 (faces cytoplasm, cationic/hydrophilic) and Region 3 (amphipathic, faces periplasm) to control timing of MurA enzyme inhibition.
Region 1 and Region 3
- Polarity change risk
- Too much polarity change could cause the phage to bind and become entrapped
Region 2
- Avoid mutagenesis
- Very well defined helical fold
- Subject to disruption with minor change to structure