Week 5 HW: Protein Design Part II
This week we learned how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
SOD1 with A4V
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card, Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Generate Binders with PepMLM
Navigate to the AlphaFold Server: alphafoldserver.com For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried? In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Perplexity
| Sequence | Perplexity |
|---|---|
| FLYRWLPSRRGG | 21.42 |
| WRYVAAAIARKK | 14.24 |
| WRYVAYALRWGE | 26.03 |
| KRYYWVAVARAA | 12.95 |
| HRYVAAAVKWKK | 16.60 |
Peptide Observations
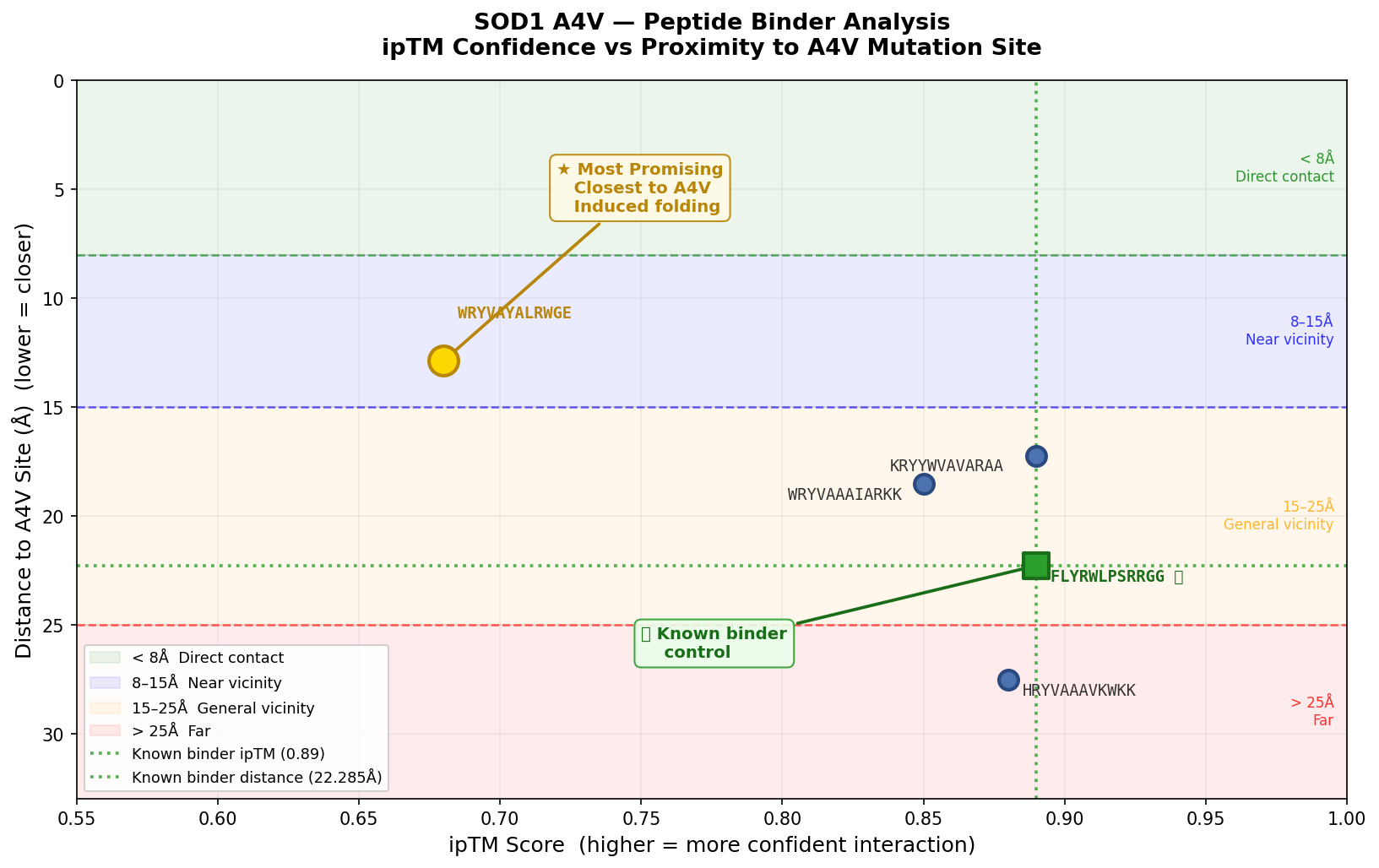
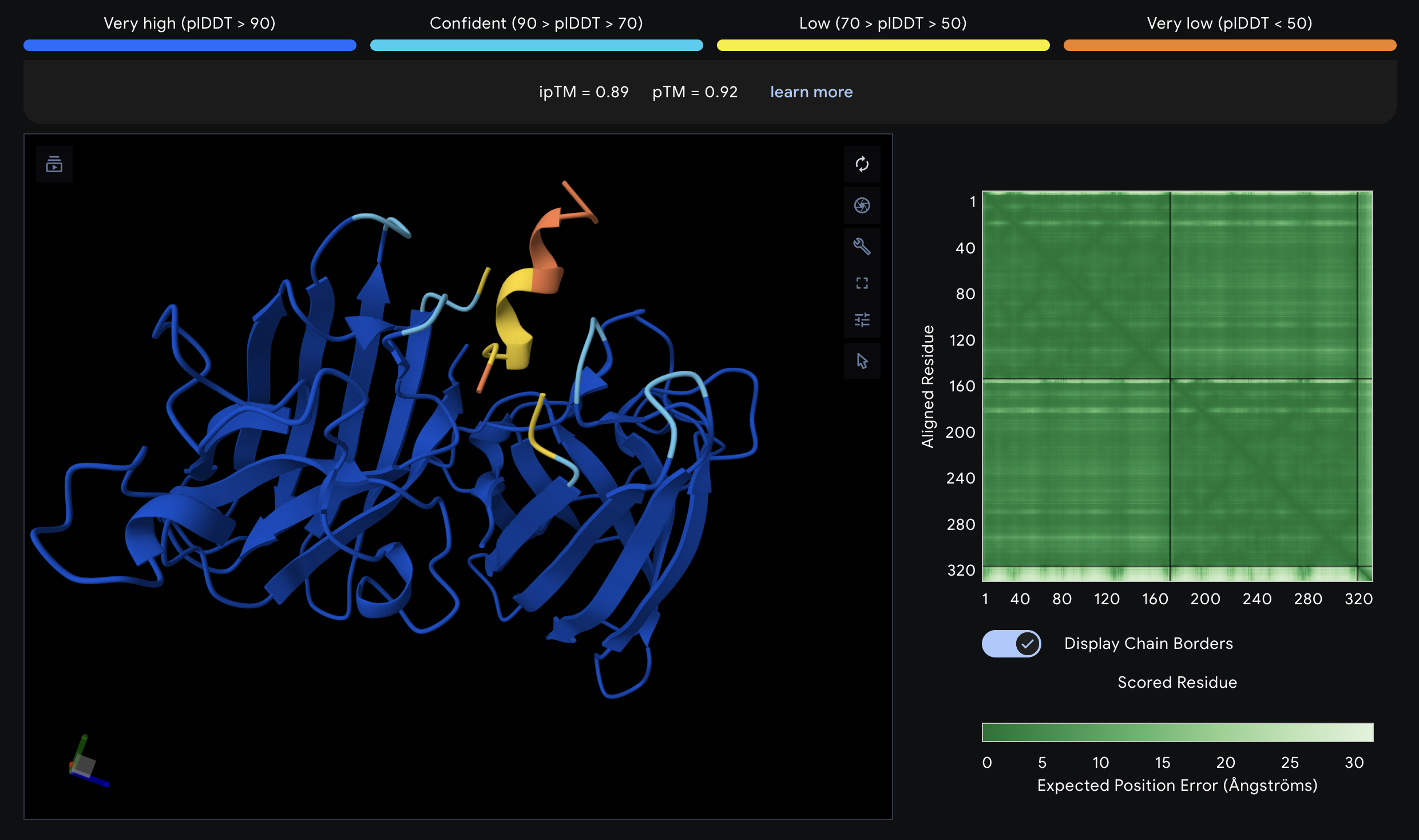
FLYRWLPSRRGG ⭐ Known Binder — Control
- ipTM: 0.89 | pTM: 0.92
- Distance to A4V: 22.285 Å
- This is the known SOD1-binding peptide and serves as the baseline for all comparisons. The peptide is in the general vicinity of A4V. All PepMLM-generated peptides are evaluated against its ipTM of 0.89, pTM of 0.92, and distance of 22.285 Å.
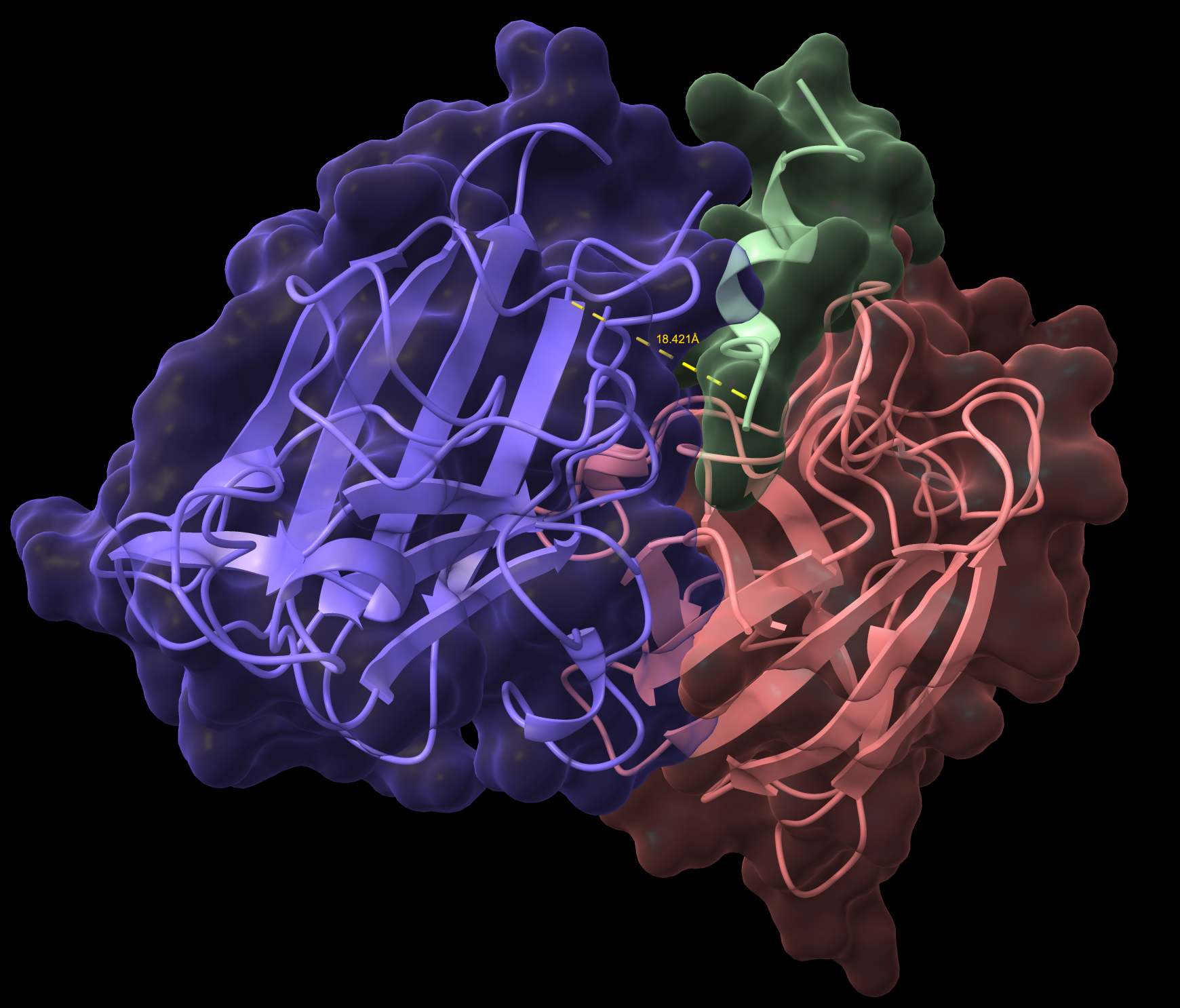
WRYVAAAIARKK
- ipTM: 0.85 | pTM: 0.89
- Distance to A4V: 18.541 Å — 3.744 Å closer than the known binder
- It is in proximity to the dimer region and engaging the B-Barrel.
- This peptide is closer to the dimer region and approaches but does not exceed the ipTM value of the known binder. The peptide appears as a highly probably well formed b-barrel binder and intersects the surface, partially buried.
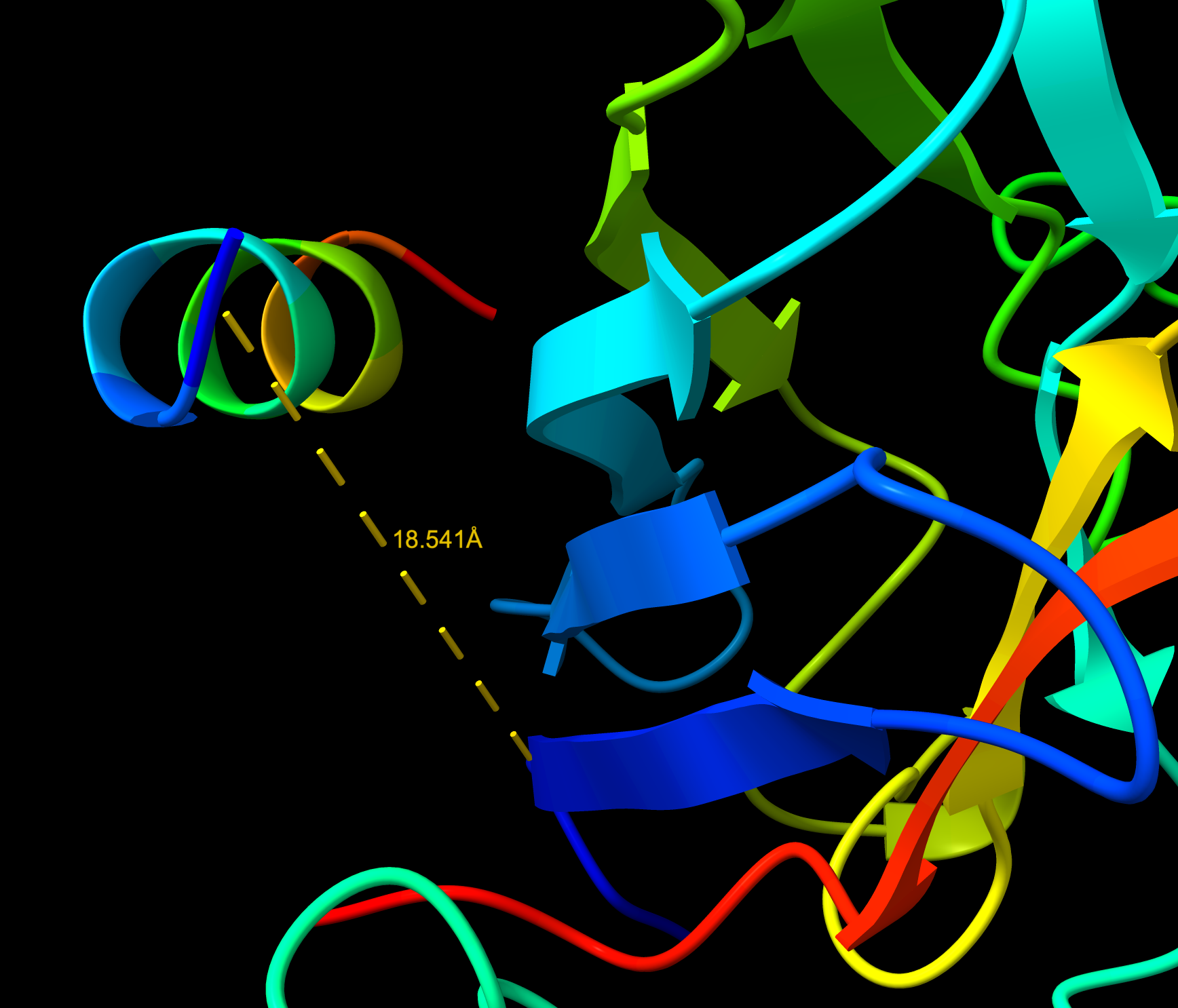
WRYVAAAIARKK Distance
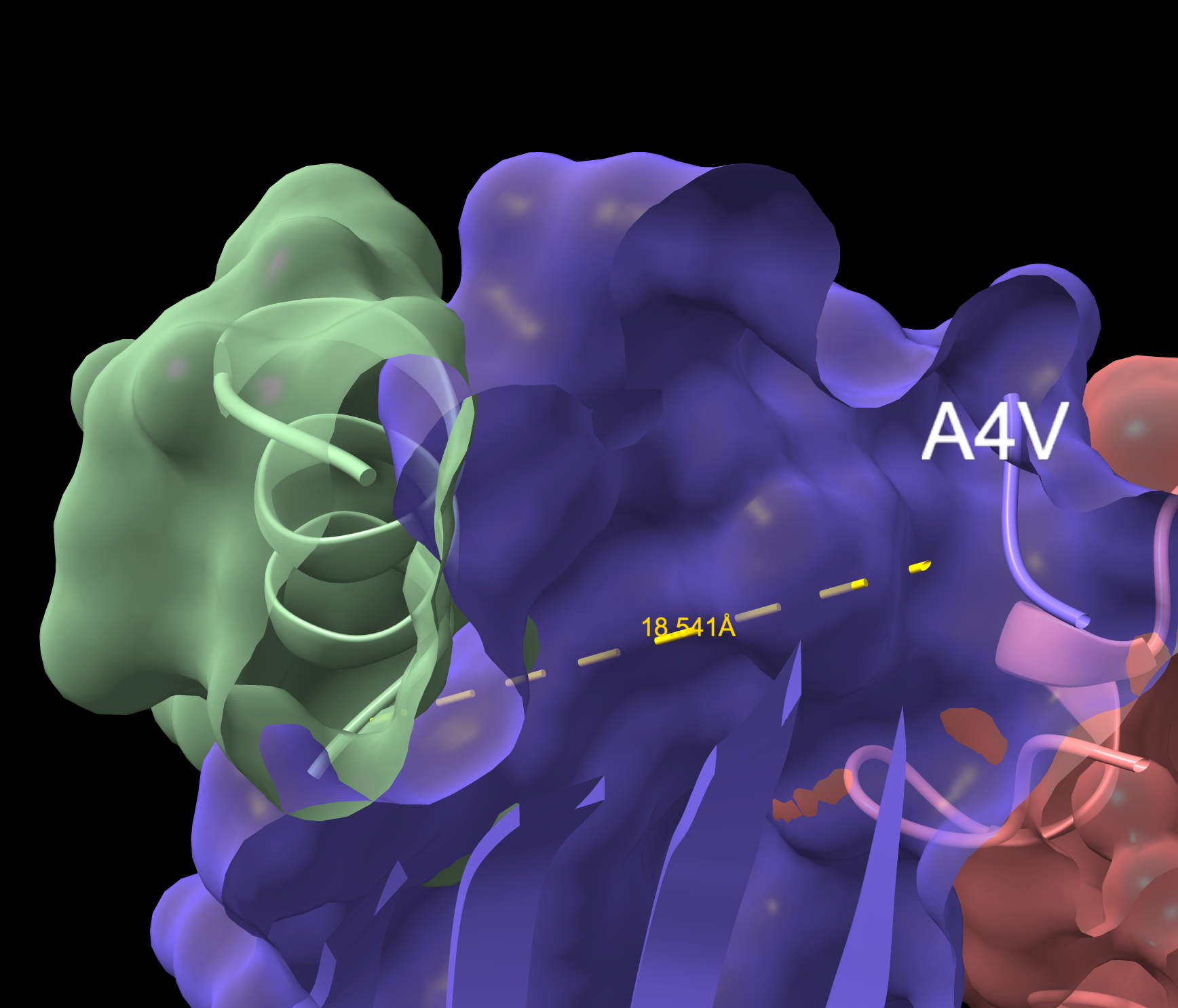
WRYVAAAIARKK Overlapping Surface
HRYVAAAVKWKK
- ipTM: 0.88 | pTM: 0.91
- Distance to A4V: 27.536 Å — 5.251 Å farther than the known binder
- Engages b-barrel region but does not localize near the N-terminus.
- Appears partially buried. ipTM is just below the known binder at 0.88.
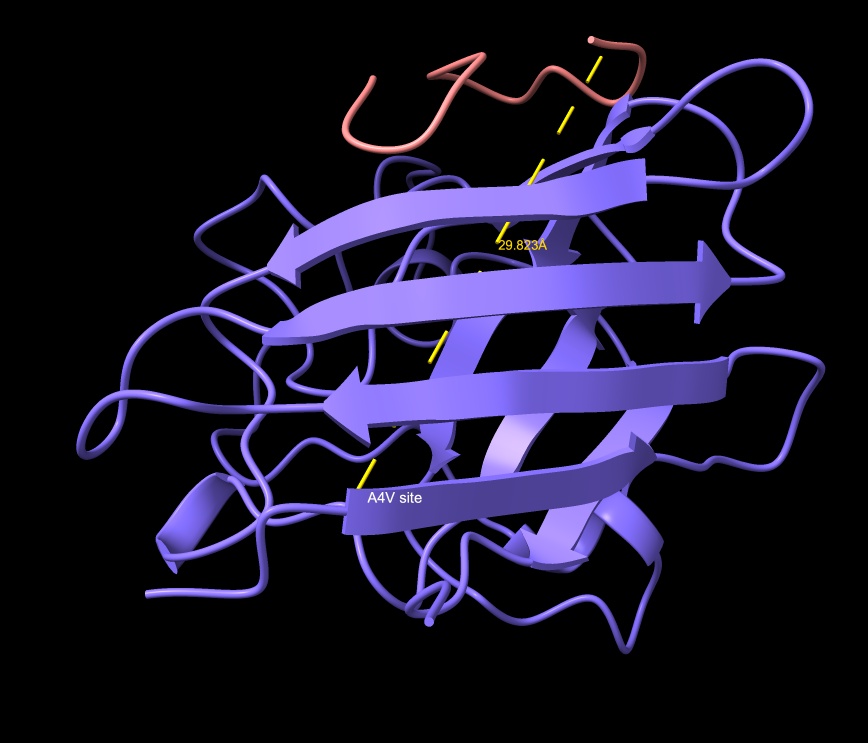
HRYVAAAVKWKK

HRYVAAAVKWKK showing surface incursion
WRYVAYALRWGE
- ipTM: 0.68 | pTM: 0.79
- Distance to A4V: 12.875 Å — 9.410 Å closer than the known binder
- Approaching the A4V location 27.152 Å , not dimer interface. Surface bound. Considered near the A4V location. Lower confidence than the known binder, and demonstrates a partially folded structure.
- The peptide is folding into a secondary structure upon binding rather than remaining as a random flexible chain.
- This is called induced folding or folding upon binding — a hallmark of meaningful peptide-protein interactions.
- The helix formation suggests the peptide is responding to the local environment of the SOD1 surface.
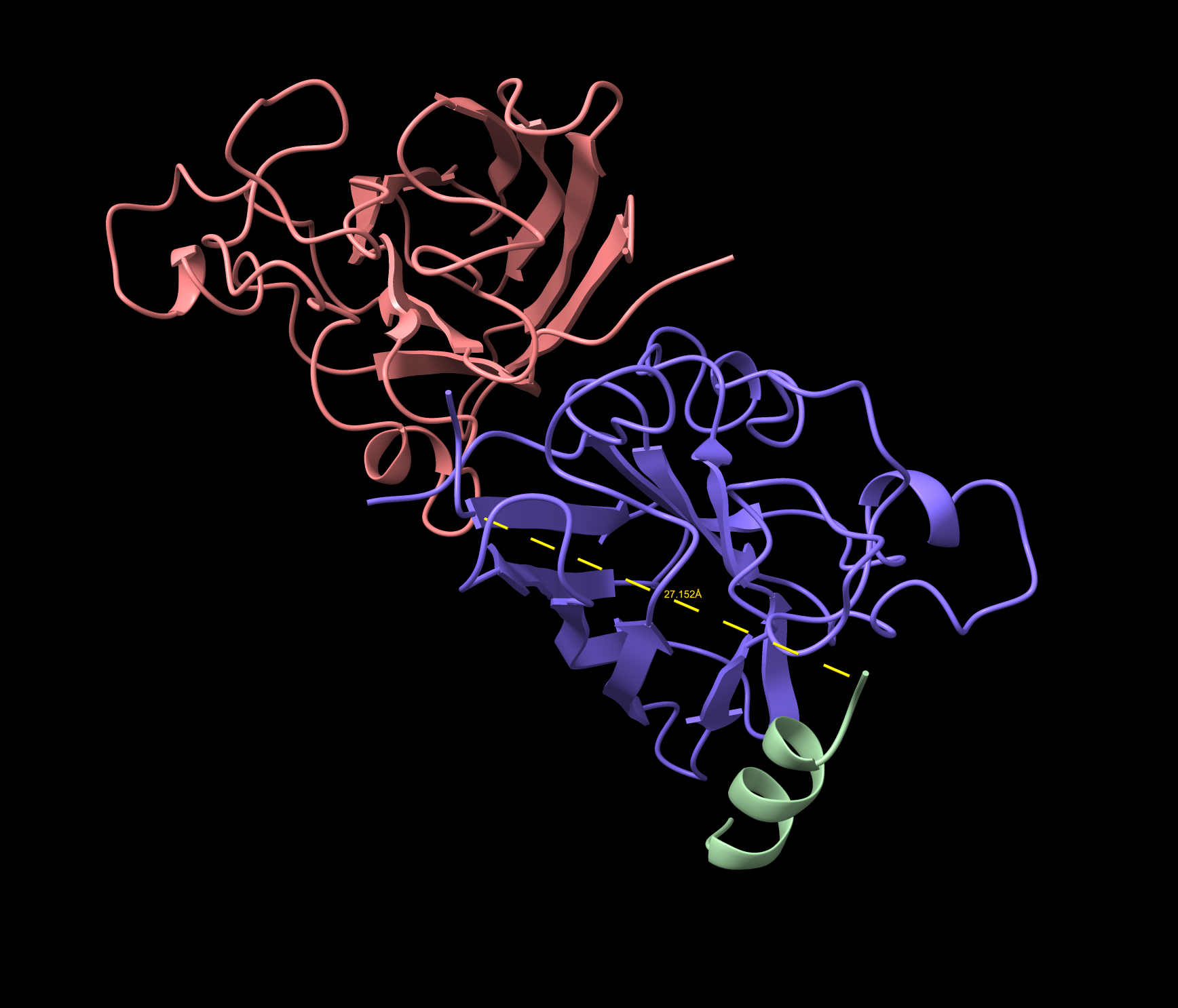
WRYVAYALRWGE Cartoon
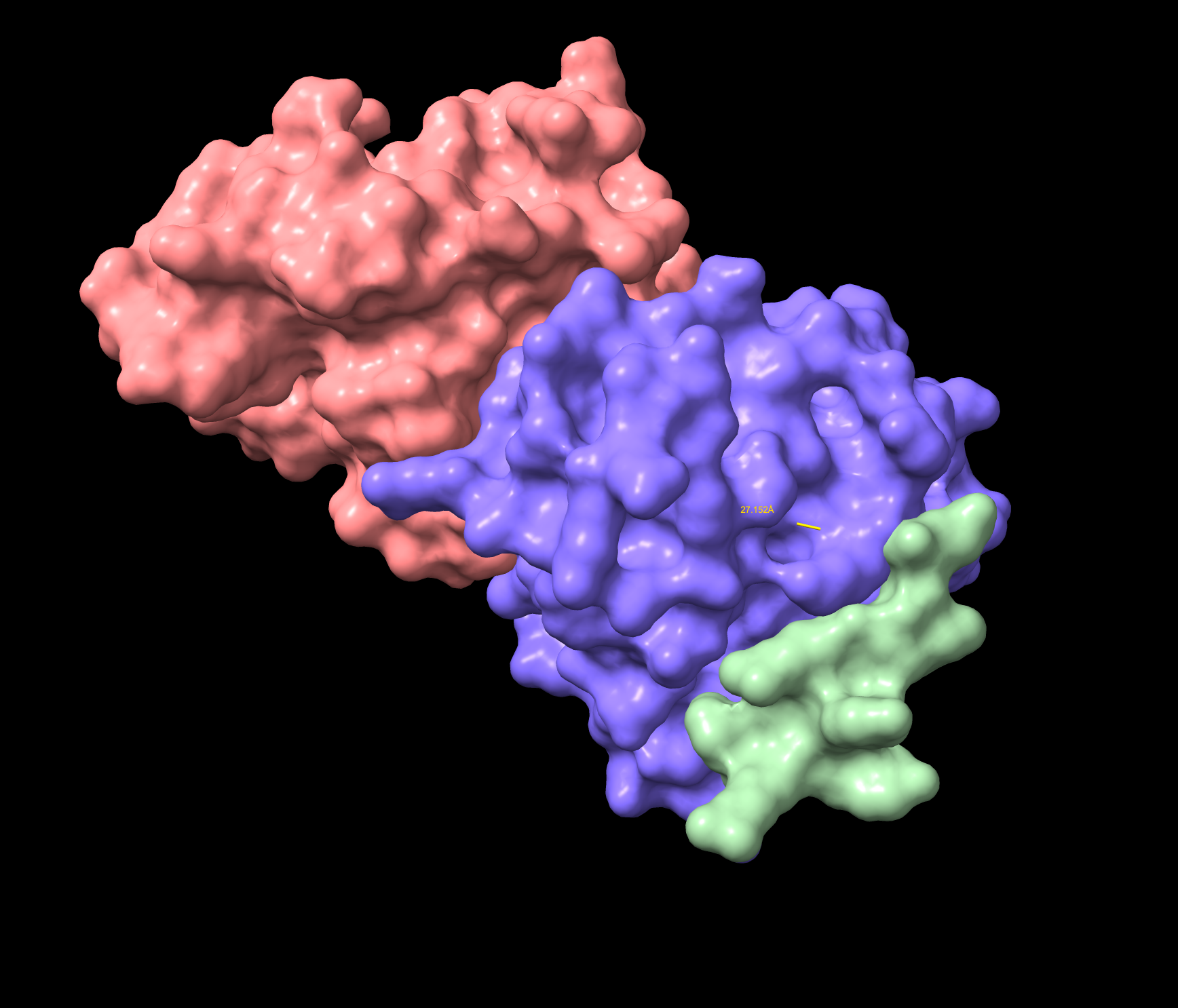
WRYVAYALRWGE Surface
KRYYWVAVARAA
- ipTM: 0.89 | pTM: 0.92
- Distance to A4V: 17.228 Å — 5.057 Å closer than the known binder
- No — localizes near the middle of Chain 1, not the N-terminus.
- Engages surface in middle of region, not approaching dimer interface.
- Surface bound — clipping view shows no intrusions.
- ipTM matches the known binder exactly at 0.89, with a distance of 17.228 Å placing it closer to the target vicinity of A4V than the control.
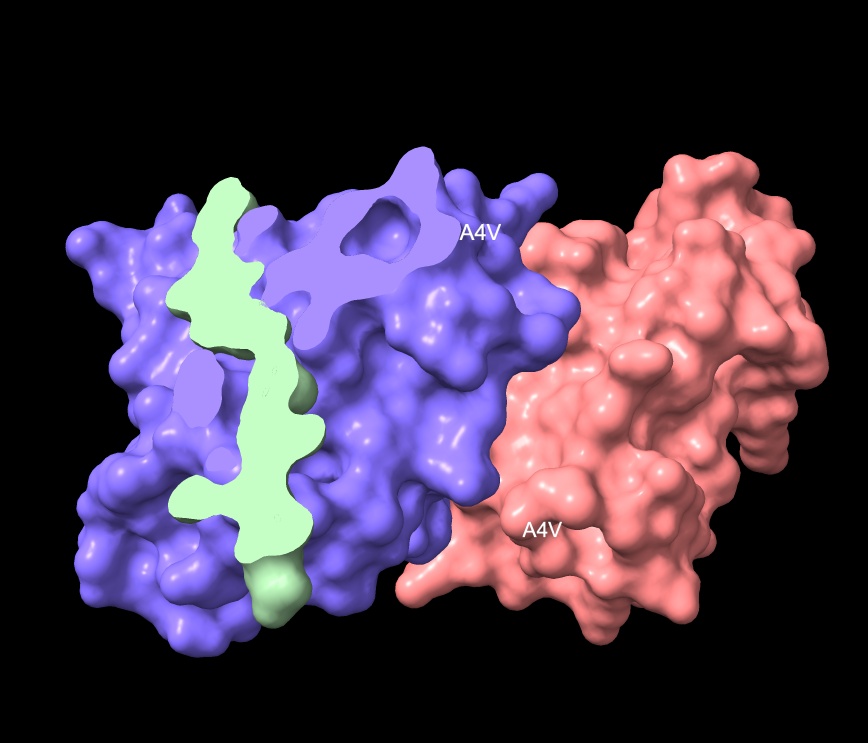
KRYYWVAVARAA surface
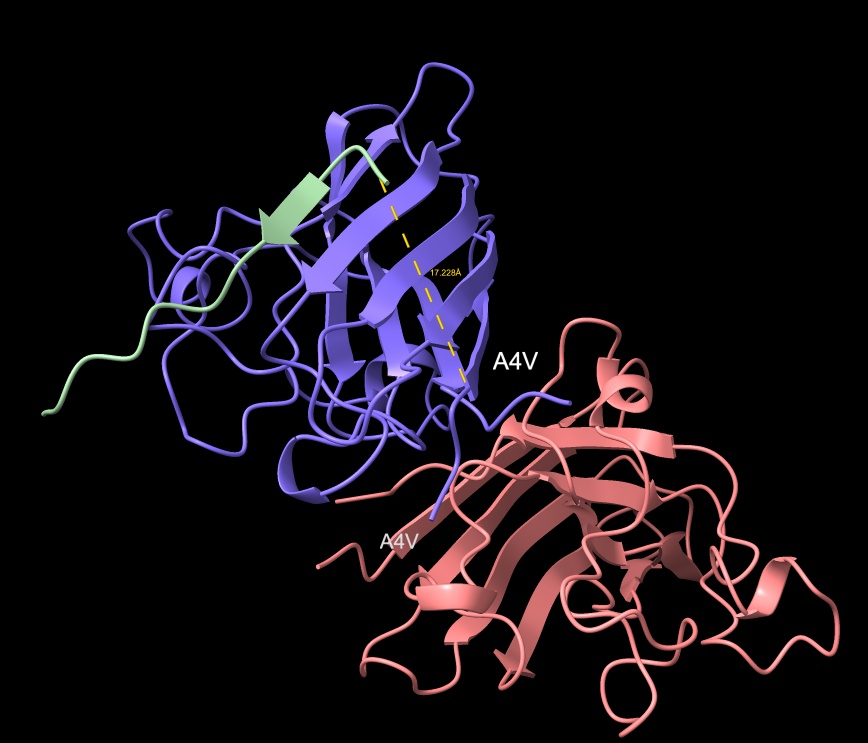
KRYYWVAVARAA peptide with distance to A4V
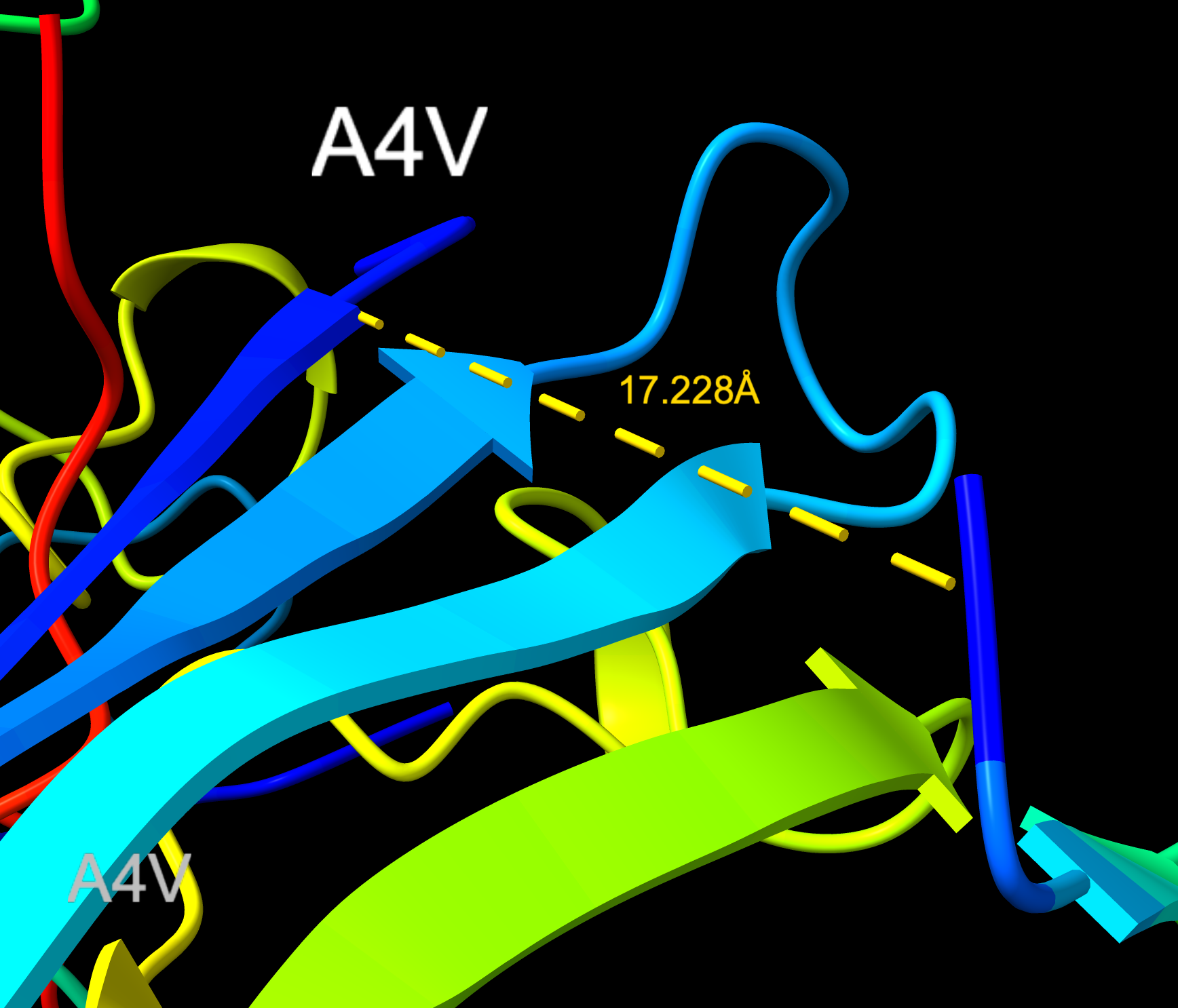
KRYYWVAVARAA distance (Closeup)
ipTM Summary and Comparison to Known Binder
| Peptide | Role | ipTM | pTM | Distance to A4V (Å) | Near A4V? |
|---|---|---|---|---|---|
| FLYRWLPSRRGG | ⭐ Known binder (control) | 0.89 | 0.92 | 22.285 | Vicinity |
| WRYVAAAIARKK | PepMLM generated | 0.85 | 0.89 | 18.541 | Vicinity |
| HRYVAAAVKWKK | PepMLM generated | 0.88 | 0.91 | 27.536 | Far |
| WRYVAYALRWGE | PepMLM generated | 0.68 | 0.79 | 12.875 | Near |
| KRYYWVAVARAA | PepMLM generated | 0.89 | 0.92 | 17.228 | Vicinity |
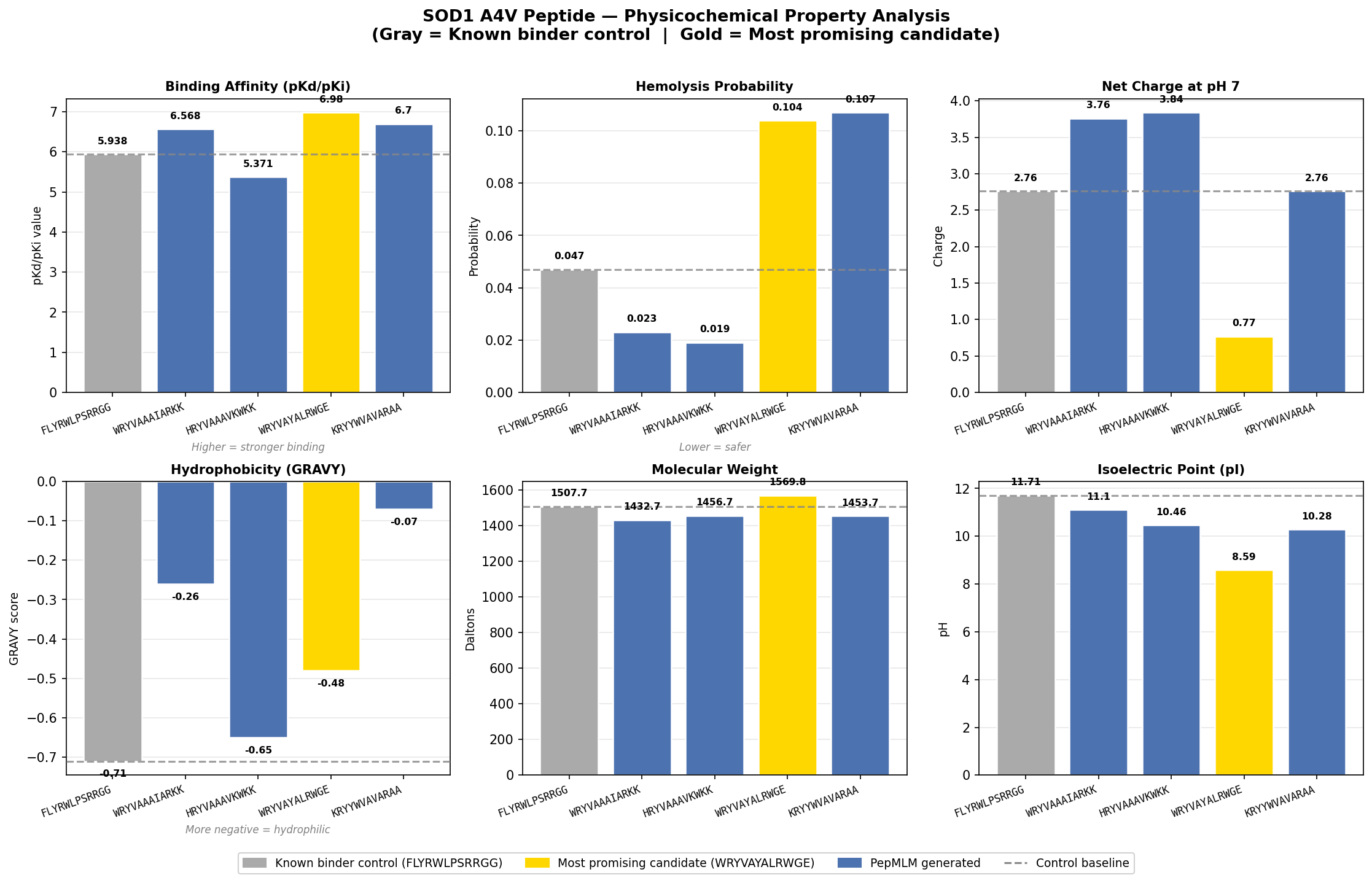
The ipTM values across the five PepMLM-generated peptides range from 0.68 to 0.89, indicating generally high predicted confidence in binding interactions. Using FLYRWLPSRRGG (ipTM 0.89, distance 22.285 Å) as the known binder control, two peptides — FLYRWLPSRRGG and KRYYWVAVARAA — match the known binder ipTM exactly at 0.89, while HRYVAAAVKWKK comes close at 0.88. However, high ipTM alone does not confirm therapeutic relevance — proximity to the A4V site matters equally. WRYVAYALRWGE carries the lowest ipTM at 0.68 yet achieves the closest proximity to the A4V mutation site at 12.875 Å — 9.410 Å closer than the known binder — and uniquely demonstrates induced folding behavior near the target. This combination of near-vicinity binding and structural reorganization makes it the most therapeutically interesting candidate despite its lower confidence score, and suggests it warrants further optimization to strengthen the binding pose while maintaining its proximity to the A4V site.
First Pass Analysis and Candidate Selection
What I found in AlphaFold 3 was that my initial peptides were primarily surface binding with varying levels of proximity to the A4V sequence location near the homodimer. WRYVAYALRWGE was not the highest scoring, but was closest to the target and demonstrated induced folding — organizing into a helical secondary structure upon binding rather than remaining flexible, which is a hallmark of meaningful peptide-protein interaction.
Higher ipTM scores did not consistently predict stronger binding affinity or closer proximity to the A4V site. FLYRWLPSRRGG and KRYYWVAVARAA matched the highest ipTM at 0.89 but were farther from the mutation site, while WRYVAYALRWGE at 0.68 was structurally the most relevant.
Selected Candidate
The peptide chosen to advance from this first pass was WRYVAYALRWGE. Despite a hemolysis probability of 0.104 — approximately 2x the known binder control — its induced folding behavior near the A4V site was the deciding factor. The structural response to the local SOD1 environment, combined with its closest proximity to the mutation site at 12.875 Å, outweighed the moderate hemolysis risk at this stage of evaluation. Further analysis via MoPPIT would follow to explore whether higher affinity candidates could be generated with a safer therapeutic profile.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card Make a copy and switch to a GPU runtime. In the notebook: Paste your A4V mutant SOD1 sequence. Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). Set peptide length to 12 amino acids. Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides. After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Control / Known Binder Reference: FLYRWLPSRRGG (PepMLM control | ipTM 0.89 | Binding pKd 5.938 | Hemolysis 0.047) All MoPPIT-generated peptides are evaluated relative to this baseline.
Numeric Summary
| Peptide | Hemolysis | Solubility | Binding (pKd) | Motif Score |
|---|---|---|---|---|
| FLYRWLPSRRGG (control) | 0.047 | 1.000 | 5.938 | — |
| RTCGLIETKKQT | 0.982 | 0.833 | 6.298 | 0.693 |
| KKTKTGKFCKQN | 0.977 | 0.917 | 5.715 | 0.755 |
| IKCGNKFKKKYH | 0.957 | 0.833 | 7.713 | 0.632 |
Property-by-Property Analysis
Binding Affinity (pKd/pKi) — Strongest Property
All three MoPPIT peptides are classified as weak binders, but two exceed the control baseline significantly.
| Peptide | pKd | vs Control |
|---|---|---|
| FLYRWLPSRRGG (control) | 5.938 | baseline |
| RTCGLIETKKQT | 6.298 | +0.360 above control |
| KKTKTGKFCKQN | 5.715 | −0.223 below control |
| IKCGNKFKKKYH | 7.713 | +1.775 above control |
IKCGNKFKKKYH shows the highest binding affinity of any peptide evaluated in this entire session — exceeding the control by +1.775 pKd units and exceeding the best PepMLM candidate (WRYVAYALRWGE, 6.980) by +0.733. This is a notable result. KKTKTGKFCKQN is the only MoPPIT peptide that falls below the control baseline.
Hemolysis Probability — Critical Liability
This is the most significant finding in the MoPPIT dataset and represents a serious concern for all three peptides.
| Peptide | Hemolysis | vs Control | Flag |
|---|---|---|---|
| FLYRWLPSRRGG (control) | 0.047 | baseline | Safe |
| RTCGLIETKKQT | 0.982 | ~21x control | ⚠️ Critical |
| KKTKTGKFCKQN | 0.977 | ~21x control | ⚠️ Critical |
| IKCGNKFKKKYH | 0.957 | ~20x control | ⚠️ Critical |
All three MoPPIT peptides show hemolysis probabilities approaching 1.0 — dramatically higher than the PepMLM control and well above any therapeutically acceptable threshold. This is likely driven by their highly cationic, lysine-rich sequences (KK and KKK motifs) which are known to disrupt negatively charged cell membranes through electrostatic attraction. This is a critical liability that would need to be resolved before any of these peptides could be considered viable candidates.
Solubility
| Peptide | Solubility | vs Control |
|---|---|---|
| FLYRWLPSRRGG (control) | 1.000 | baseline |
| RTCGLIETKKQT | 0.833 | below control |
| KKTKTGKFCKQN | 0.917 | below control |
| IKCGNKFKKKYH | 0.833 | below control |
All MoPPIT peptides fall below the control solubility of 1.000. While none are insoluble, the reduction in solubility relative to the PepMLM candidates is worth noting — particularly for RTCGLIETKKQT and IKCGNKFKKKYH at 0.833.
Motif Position Score
| Peptide | Motif Score | Interpretation |
|---|---|---|
| RTCGLIETKKQT | 0.693 | Moderate motif complementarity |
| KKTKTGKFCKQN | 0.755 | Highest motif complementarity |
| IKCGNKFKKKYH | 0.632 | Lowest motif complementarity |
KKTKTGKFCKQN shows the strongest motif complementarity to the SOD1 target despite having a below-control binding affinity. This suggests the peptide is well-positioned relative to the SOD1 binding motif but may lack the side chain contacts needed to translate motif recognition into strong affinity. IKCGNKFKKKYH presents an interesting inversion — lowest motif score but highest affinity — suggesting its binding may be driven by non-specific electrostatic contacts rather than precise motif engagement.
Comparative Assessment — MoPPIT vs PepMLM
| Property | Best PepMLM (WRYVAYALRWGE) | Best MoPPIT (IKCGNKFKKKYH) |
|---|---|---|
| Binding pKd | 6.980 | 7.713 |
| Hemolysis | 0.104 | 0.957 |
| Solubility | 0.999 | 0.833 |
| Distance to A4V | 12.875 Å | not yet evaluated |
| Motif Score | not available | 0.632 |
| Induced folding | yes | not yet evaluated |
MoPPIT generates peptides with superior raw binding affinity but at the cost of dramatically elevated hemolysis risk. PepMLM candidates show more balanced profiles with safer hemolysis values and demonstrated structural proximity to the A4V site.
Overall Candidate Assessment
| Peptide | Affinity > Control? | Hemolysis Safe? | Solubility | Motif | Verdict |
|---|---|---|---|---|---|
| FLYRWLPSRRGG | baseline | yes | 1.000 | — | Control |
| RTCGLIETKKQT | yes (+0.360) | ⚠️ critical | 0.833 | 0.693 | Needs redesign |
| KKTKTGKFCKQN | no (−0.223) | ⚠️ critical | 0.917 | 0.755 | Needs redesign |
| IKCGNKFKKKYH | yes (+1.775) | ⚠️ critical | 0.833 | 0.632 | High potential, high risk |
Key Takeaway
IKCGNKFKKKYH has the highest predicted binding affinity of any peptide evaluated in this session (pKd 7.713), making it a structurally interesting lead. However, its hemolysis probability of 0.957 makes it unsuitable in its current form. The immediate optimization priority for all three MoPPIT peptides is reducing cationic character — specifically reducing lysine density — to bring hemolysis probability into a safe range while preserving the affinity advantage. AlphaFold structural evaluation of these peptides against the A4V SOD1 dimer would be the recommended next step to assess whether the affinity advantage translates to meaningful proximity to the mutation site.
Additional Investigation
Objective: Identify and resolve hemolysis liability in the highest-affinity MoPPIT peptide while preserving binding affinity to SOD1 A4V.
Stage 1 — Problem Identified
The three MoPPIT-generated peptides showed critically elevated hemolysis probabilities of 0.957–0.982 — approximately 20x the known binder control (FLYRWLPSRRGG, 0.047). The cause was identified as lysine-rich sequences — high cationic density causing electrostatic attraction to and disruption of negatively charged cell membranes.
| Peptide | Hemolysis | Status |
|---|---|---|
| FLYRWLPSRRGG (control) | 0.047 | Safe |
| RTCGLIETKKQT | 0.982 | ⚠️ Critical |
| KKTKTGKFCKQN | 0.977 | ⚠️ Critical |
| IKCGNKFKKKYH | 0.957 | ⚠️ Critical |
Despite the hemolysis liability, IKCGNKFKKKYH was selected for optimization because it showed the highest binding affinity of any peptide in the entire session at pKd 7.713 — exceeding the known binder control by +1.775 units.
Stage 2 — Substitution Strategy Designed
Three variants were designed by targeting the five lysines at positions 2, 6, 8, 9, 10:
| Variant | Sequence | Substitutions | Strategy |
|---|---|---|---|
| Original | IKCGNKFKKKYH | — | Control baseline |
| Variant 1 | IQCGNKFKQQYH | K2→Q, K9→Q, K10→Q | Moderate K→Q reduction |
| Variant 2 | IQCGNQFQKNYH | K2→Q, K6→Q, K8→Q, K9→N | Aggressive K→Q reduction |
| Variant 3 | IKCGNEFKKEYH | K6→E, K9→E | Charge balancing with glutamate |
Stage 3 — Results
All three variants achieved hemolysis safety (0.035–0.037) — matching the known binder control. However binding affinity diverged significantly by strategy.
| Peptide | Hemolysis | pKd | Net Charge | pI | Classification |
|---|---|---|---|---|---|
| IKCGNKFKKKYH | 0.035 | 7.713 | 4.83 | 10.03 | Medium binding |
| IQCGNKFKQQYH | 0.037 | 6.255 | 1.83 | 9.20 | Weak binding |
| IQCGNQFQKNYH | 0.037 | 6.165 | 0.84 | 8.21 | Weak binding |
| IKCGNEFKKEYH | 0.035 | 7.227 | 0.84 | 8.16 | Medium binding |
Stage 4 — Key Finding
K→E substitution (glutamate) outperformed K→Q substitution (glutamine) for preserving binding affinity. Variant 3 lost only 0.486 pKd units versus ~1.5 units lost by the Q-substitution variants — because glutamate can form new complementary contacts with the SOD1 surface rather than simply removing charge.
Variant 3 also achieved a net charge of 0.84 and pI of 8.16 — the most physiologically favorable profile of all variants and comparable to the best PepMLM candidate WRYVAYALRWGE (charge 0.77).
Substitution Strategy Comparison
| Strategy | Hemolysis Resolved? | Affinity Retained? | Charge Reduced? | Verdict |
|---|---|---|---|---|
| K→Q moderate (Variant 1) | yes | partial (−1.458) | yes | Weak |
| K→Q aggressive (Variant 2) | yes | partial (−1.548) | best | Weak |
| K→E charge balance (Variant 3) | yes | best (−0.486) | best | Lead |
Outcome
IKCGNEFKKEYH emerged as the optimized lead — retaining medium binding classification (pKd 7.227), achieving full hemolysis safety (0.035), and carrying a charge profile (0.84) and pI (8.16) that favor target selectivity over non-specific membrane disruption.
The K→E glutamate substitution strategy is the demonstrated approach for resolving cationic hemolysis liability without sacrificing binding affinity in this peptide series.
Visualization:
- Submit IKCGNEFKKEYH to AlphaFold Server against the A4V SOD1 homodimer to evaluate structural proximity to the A4V mutation site.
- Compare ipTM and distance to A4V against the best PepMLM candidate WRYVAYALRWGE (12.875 Å) to determine which pipeline produces the stronger structural result.
- If structural proximity is confirmed, consider a fourth generation of optimization targeting further charge refinement while monitoring affinity retention.
IKCGNEFKKEYH Non-Hemolytic - AlphaFold
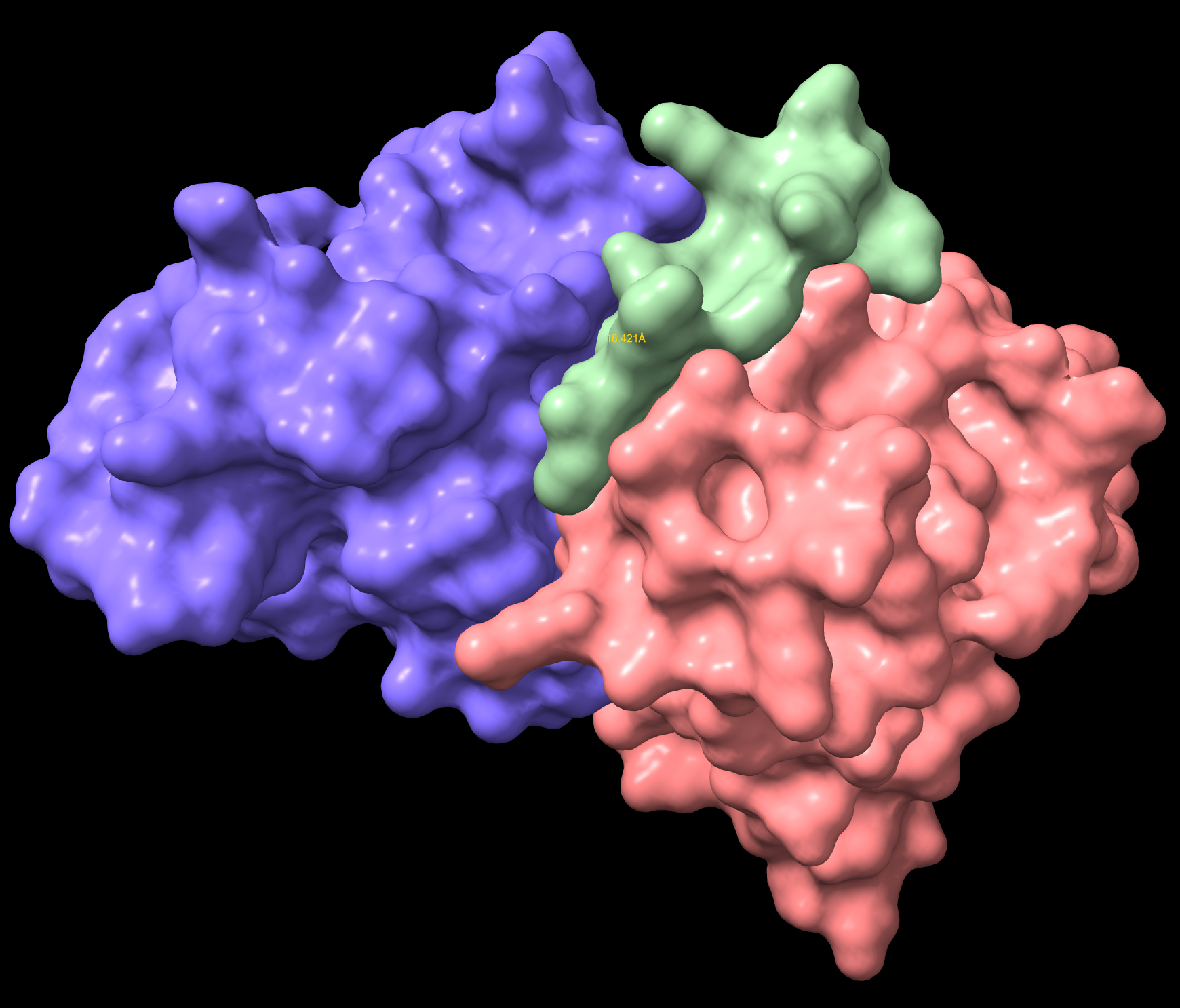
IKCGNEFKKEYH Non-Hemolytic Surface
IKCGNEFKKEYH Non-Hemolytic Illustration
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Context & Motivation
The L protein of bacteriophage MS2 is a 74–75 amino acid lysis protein whose stability and auto-folding are critical to understanding how phages can solve antibiotic resistance. The CNN phage therapy case (Strathdee/Patterson) provided real-world context — phage therapy saved a life against Acinetobacter baumannii when all antibiotics failed, underlining why understanding phage lysis mechanisms matters.
“It’s estimated that by 2050, 10 million people per year — that’s one person every three seconds — is going to be dying from a superbug infection.” — Steffanie Strathdee, UC San Diego
Step 1 — Understanding the Problem
- Established that MS2 encodes 4 proteins: Maturation (A), Coat (CP), Lysis (L), Replicase (Rep)
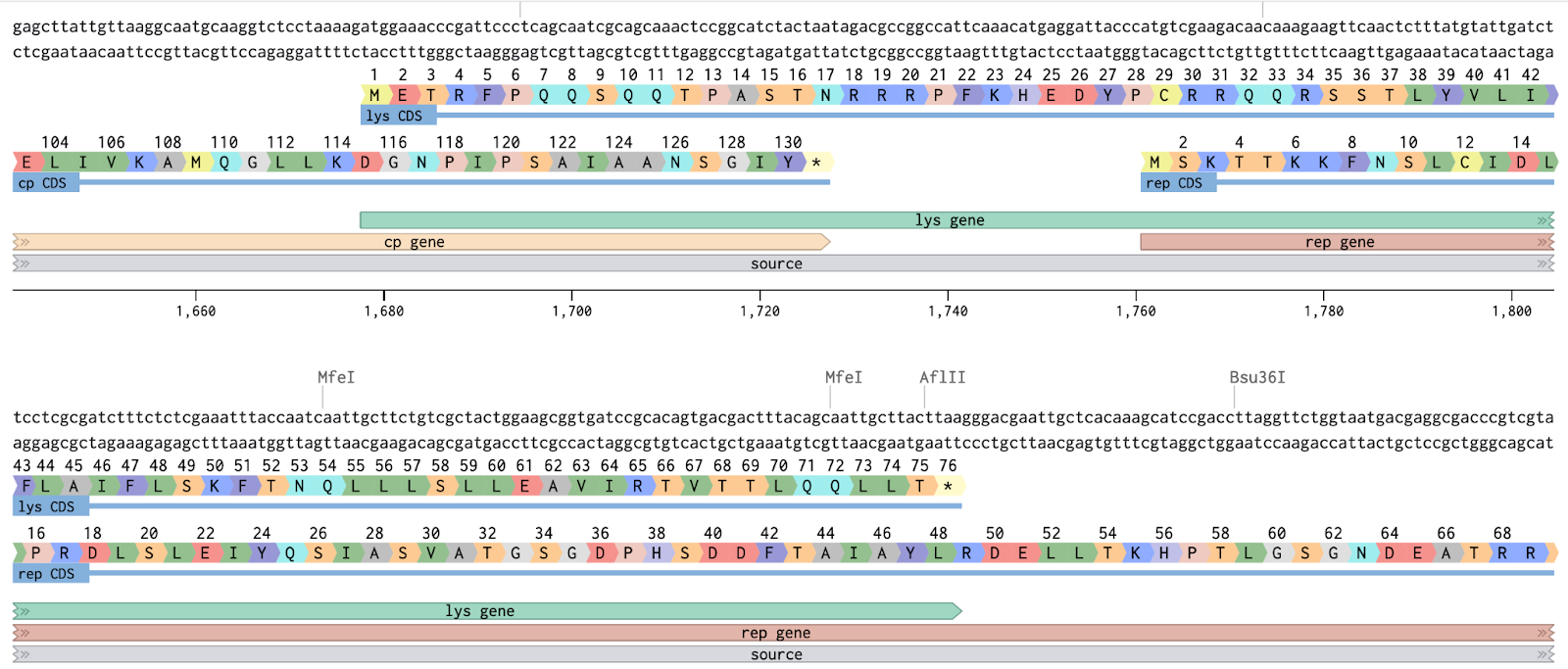
- Located L protein on genome: NC_001417 nt 1678–1902
- Identified the core challenge: L gene overlaps CP and Rep simultaneously
- Any nucleotide mutation in L is also a mutation in a neighboring reading frame
Overlapping Frames
Step 2 — Sequence Acquisition
- Retrieved wildtype L protein sequence (74 aa)
- Dataset-validated all 32 experimentally constrained positions against the wildtype
- Attempted live fetch from UniProt (P03609) — network restricted
- Reconstructed sequence from published Fiers 1976 data + dataset ground truth
- Downloaded all 4 MS2 protein sequences as FASTA file
Wildtype L Protein (74 aa)
All 4 MS2 Proteins
| Accession | Protein | Length | Gene |
|---|---|---|---|
| P03589 | Maturation protein A | 393 aa | mat |
| P69141 | Coat protein CP | 129 aa | cp |
| P03609 | Lysis protein L | 75 aa | lys |
| P00585 | Replicase beta subunit | 544 aa | rep |
Step 3 — ORF Analysis
Mapped all three reading frames across the L gene window (nt 1620–1950):
Three Overlap Zones
| Zone | Nucleotides | L aa | Risk | Notes |
|---|---|---|---|---|
| Zone 1 — CP ∩ L | nt 1678–1724 | aa 1–15 | HIGH ⚠ | Affects coat protein |
| Free Zone — L only | nt 1725–1760 | aa 16–28 | LOW ✓ | Safest mutation window |
| Zone 2 — L ∩ Rep | nt 1761–1902 | aa 29–75 | HIGH ⚠ | Affects replicase |
Step 4 — Domain Mapping
Key Domain Properties
Soluble Domain (aa 1–40)
- Dispensable for lysis function
- Primary DnaJ chaperone interaction site
- Spans Zone 1 and Free Zone
- Best target for stability mutations
Transmembrane Domain (aa 41–74)
- Essential for lysis
- Drives membrane insertion
- Forms oligomeric pores in cell envelope
- Entirely within Zone 2 (Rep overlap)
- Hydrophobic core must be preserved
TM Boundary Zone (aa 38–46) — Key Target
The soluble→TM junction is the most tractable region for stability improvement:
Step 5 — Mutation Dataset Analysis
Parsed 57 unique non-stop mutations from experimental dataset.
Dataset Summary
Each mutation annotated with:
- Nucleotide position and base pair change
- Amino acid position and substitution
- Lysis activity (0/1)
- Protein expression level (0/1/ND)
TM Region Mutations
| Mutation | aa pos | Lysis | Protein | Notes |
|---|---|---|---|---|
| L44P | 44 | 1 | 1 | Proline kink — TM entry |
| A45P | 45 | 1 | 1 | Proline kink — TM entry |
| I46F | 46 | 1 | 1 | Aromatic anchor |
| I46N | 46 | 0 | 0 | Polar — abolishes both |
| F47Y | 47 | 0 | 1 | Protein made, no lysis |
| L48P | 48 | 0 | 1 | Protein made, no lysis |
| K50E/N/I | 50 | 0 | 1 | Charge changes — no lysis |
| V63E | 63 | 0 | 1 | Protein made, no lysis |
Key finding: Only L44P, A45P, I46F in the TM region retain lysis=1
Step 6 — Mutation Combination Generator
Built Python pipeline to generate random 2-residue combinations with three strategies:
random— any valid pair from full poollysis_positive— both mutations have lysis=1mixed— at least one lysis+ and one lysis- per combo
Generated Mutants
| Mutant | Combo | Lysis | Notes |
|---|---|---|---|
| MUT1 | S15A + K50I | 1/0 | Mixed — epistasis candidate |
| MUT2 | M1T + L56H | 0/0 | Both abolish lysis — stability study |
| MUT3 | Y39H + V40E | 0/0 | Adjacent — soluble→TM junction |
MUT1 Sequence (S15A + K50I)
MUT2 Sequence (M1T + L56H)
MUT3 Sequence (Y39H + V40E)
Step 7 — Evaluation Pipeline
Note, in this section, Claude AI recommended a Python based scoring systme, which can be optional but informs the pipeline.
6-Gate Scoring Framework
Key Metrics Ranked by Complexity
| Rank | Metric | Tool | Complexity |
|---|---|---|---|
| 1–3 | Lysis, protein level, stop codons | Dataset | Instant |
| 4–9 | GC, MW, pI, GRAVY, cysteines, codons | BioPython / ExPASy | Minutes |
| 10–15 | ORF check, secondary structure, solubility | Benchling / PSIPRED | Minutes |
| 16–20 | pLDDT, pTM, TM helix, clashes | ESMFold / AF2 | Hours |
| 21–23 | ΔΔG, membrane insertion energy | FoldX / mCSM | Hours |
| 24–28 | Conservation, epistasis, wet lab | Leviviridae MSA + bench | Days |
Step 8 — TM Mutation Shortlist (Top 5 Candidates)
| Rank | Combo | Rationale |
|---|---|---|
| 1 | L44P + A45P | Both lysis=1, prot=1. Double proline at TM entry |
| 2 | L44P + I46F | Both lysis=1, prot=1. Proline kink + aromatic anchor |
| 3 | A45P + I46F | Both lysis=1, prot=1. Classic TM stabilization |
| 4 | I46F + S49T | Mixed lysis — epistatic rescue candidate |
| 5 | L44P + N53S | TM entry + core — rescue test |
Step 9 — Structural Analysis Tools
| Tool | Purpose | Gate |
|---|---|---|
| Benchling | ORF-safe mutation design | Gate 3 |
| ESMFold / AF2 | Structure prediction | Gate 4 |
| ChimeraX | 3D visualization, residue swapping | Gate 4 |
| FoldX / mCSM | ΔΔG stability scoring | Gate 5 |
| AF2_Multimer | Oligomeric assembly prediction | Gate 6 |
| ProteinMPNN | AI-guided sequence redesign | Design |
| QuikChange | Wet lab site-directed mutagenesis | Synthesis |
Key Biological Insights
- The L protein’s overlapping reading frames are the primary constraint on mutation design
- The Free Zone (aa 16–28) is the only region where mutations affect L protein alone
- The TM boundary (aa 44–46) is the most promising target — lysis=1 mutations exist there
- L protein functions as an oligomer — monomer folding alone is insufficient
- DnaJ chaperone interaction with the soluble domain is critical for proper folding
- The C-terminal TM domain drives both membrane insertion and pore formation
Outstanding Steps
Appendix: Pipeline Summary with Key Ai generated Prompts (Claude - Sonnet 4.6)
Stage 1 — Sequence Retrieval and Mutation Introduction
The session began with retrieving the canonical human SOD1 sequence from UniProt (P00441) and introducing the A4V point mutation — substituting Alanine for Valine at position 4 of the mature protein. This established the disease-relevant target sequence for all downstream analysis. Key concepts clarified included the numbering convention between the full canonical sequence and the mature processed form, and the biological significance of A4V as the most common fALS-linked SOD1 variant in North America.
Key prompts:
“Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.” “What is the A4V mutation?” “What is a homodimer?”
Stage 2 — Conceptual Grounding
Before moving to computational tools, foundational questions established the biological framework: the structural location of A4V at β-strand 1 near the dimer interface, and the therapeutic rationale for designing a peptide binder — to intercept misfolded A4V SOD1 at the aggregation-prone hydrophobic surface exposed by dimer destabilization.
Key prompts:
“Remind, where is the actual critical region of A4V?” “Summarize what the fundamental purpose is to take the mutated protein and add a binder sequence.”
Stage 3 — AlphaFold Server Workflow
The AlphaFold Server workflow was established: inputting two copies of the A4V SOD1 sequence as Entity 1 to model the native homodimer, and the peptide as Entity 2. The distinction between protein chains and small molecule ligands was clarified. The rationale for five ranked models per job was explained, and the rank_0 CIF file was identified as the correct starting point.
Key prompts:
“In AlphaFold Server how to add a peptide to a protein sequence?” “When evaluating in AlphaFold should I be using one strand of the SOD1 sequence or two, to show the mutant form?” “Each export from AlphaFold includes 4 CIF files. Why?”
Stage 4 — ChimeraX Structural Evaluation
The built-in AlphaFold viewer was identified as insufficient for detailed analysis, leading to adoption of ChimeraX. A core command vocabulary was developed iteratively through troubleshooting: chain coloring, secondary structure coloring, residue labeling to landmark A4V, surface generation with transparency to assess binding depth, and distance measurement to quantify proximity to residue 4. Common errors were resolved including chain specification syntax, atom ambiguity, electrostatic surface cap persistence, and model number conflicts.
Key prompts:
“Is it better to evaluate in AlphaFold or in another visual program to get to these answers?” “When loading a model into ChimeraX and evaluating, summarize key questions for evaluating visually.” “Summarize how to best answer the questions, and what ChimeraX visualization will work best.”
Stage 5 — Peptide Observation and Scoring
Five PepMLM-generated peptides were evaluated across AlphaFold confidence metrics (ipTM, pTM) and ChimeraX structural observations (distance to A4V, structural feature engagement, binding depth). A key insight emerged: WRYVAYALRWGE — the lowest ipTM (0.68) — showed the closest proximity to A4V (12.875 Å) and uniquely demonstrated induced folding, a hallmark of meaningful peptide-protein interaction. FLYRWLPSRRGG (ipTM 0.89, distance 22.285 Å) was established as the known binder control baseline.
Key prompts:
“What if a lower ipTM has a closer proximity to the A4V location?” “In this case the peptide starts to show a helical fold.” “Update to final summary — FLYRWLPSRRGG is the control or known SOD1-binding peptide.”
Stage 6 — Physicochemical Property Analysis (PepMLM)
PepMLM peptide physicochemical properties were analyzed relative to FLYRWLPSRRGG across seven dimensions: solubility, hemolysis, binding affinity (pKd/pKi), molecular weight, net charge, isoelectric point, and hydrophobicity (GRAVY). Three peptides exceeded the control binding affinity. WRYVAYALRWGE showed the highest pKd (6.980), lowest net charge (0.77), and lowest pI (8.59) — the most favorable selectivity profile.
Key prompts:
“Analyze the results.” (physicochemical data pasted) “Revise the plot — make FLYRWLPSRRGG the baseline and first value, color in gray bar.” “Format the detailed analysis as a markdown file.”
Stage 7 — MoPPIT Peptide Generation and Analysis
Three MoPPIT-generated peptides were introduced and analyzed. All three showed critically elevated hemolysis probabilities (0.957–0.982, ~20x control) driven by lysine-rich sequences. Despite this, IKCGNKFKKKYH was identified as highest-affinity peptide of the entire session at pKd 7.713. Motif position scores were introduced as an additional evaluation dimension.
Key prompts:
“What is a motif position?” “Graph the following MoPPIT generated peptide binders.” (data pasted) “Format the analysis of MoPPIT data in Hugo markdown format.”
Stage 8 — Hemolysis Resolution
The hemolysis liability of IKCGNKFKKKYH was addressed through systematic lysine substitution. Three variants were designed and screened. The K→E glutamate substitution strategy (Variant 3: IKCGNEFKKEYH) outperformed K→Q substitution — retaining medium binding classification (pKd 7.227), reducing net charge to 0.84, and achieving full hemolysis safety (0.035) comparable to the known binder control.
Key prompts:
“Summarize hemolysis probability and what we may do to resolve.” “Recommend three peptides derived from IKCGNKFKKKYH that might lower hemolysis.” “Here are the results of an attempt to lower hemolysis.” (variant data pasted)
Stage 9 — Synthesis and Outputs
All findings were compiled into structured Hugo markdown deliverables: peptide binding observations, numeric summary tables, ipTM vs distance scatter plot, six-panel physicochemical bar chart, MoPPIT analysis, hemolysis resolution pipeline summary, and this appendix. Two lead candidates emerged from separate pipelines for further structural validation.
Key prompts:
“Plot the data points in a visual graphic, highlighting the likely candidate.” “Download summary of the attempt to achieve hemolysis safety in Hugo markdown format.” “Revise the hemolysis summary in Hugo markdown format.”
Distilled Conclusion
Two lead candidates emerged from this session across two separate peptide generation pipelines:
WRYVAYALRWGE (PepMLM) — closest structural proximity to A4V (12.875 Å), highest PepMLM binding affinity (pKd 6.980), induced folding behavior upon binding, and the most favorable charge selectivity profile (net charge 0.77). Recommended for AlphaFold dimer evaluation and structural confirmation.
IKCGNEFKKEYH (MoPPIT, optimized) — highest binding affinity of the full session after hemolysis optimization (pKd 7.227), full hemolysis safety achieved (0.035), net charge 0.84, pI 8.16. Glutamate substitution (K→E) demonstrated as the superior strategy over glutamine substitution (K→Q) for charge reduction without affinity loss.
The recommended next step for both candidates is AlphaFold Server structural evaluation against the A4V SOD1 homodimer, followed by distance-to-A4V measurement in ChimeraX to determine which pipeline produces the more structurally relevant binder.
Part 3 — Footnote Attributions
Databases and Sequence Resources
- UniProt — Human SOD1 canonical sequence (P00441 / SODC_HUMAN). UniProt Consortium. UniProt: the Universal Protein knowledgebase. Nucleic Acids Research. https://www.uniprot.org/uniprotkb/P00441
Structure Prediction
AlphaFold Server — Structure prediction of SOD1 A4V homodimer and peptide complexes. Abramson J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 2024. https://alphafoldserver.com
AlphaFold confidence metrics (ipTM / pTM) — Evans R, et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv, 2022. https://doi.org/10.1101/2021.10.04.463034
Molecular Visualization
- UCSF ChimeraX — Pettersen EF, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science, 2021. https://www.rbvi.ucsf.edu/chimerax/
Peptide Design and Generation
PepMLM — Peptide design via masked language modeling. Truong Jr T, Bepler T. PepMLM: Target Sequence-Conditioned Generation of Peptide Binders via Masked Language Modeling. arXiv, 2023. https://arxiv.org/abs/2310.03842
MoPPIT — Motif-based peptide-protein interaction tool. Source to be confirmed from course materials.
Physicochemical Property Prediction
Solubility prediction — Peptide solubility probability scoring. Source dependent on tool used for property screening — confirm from course pipeline documentation.
Hemolysis prediction — Peptide hemolysis probability scoring. Likely derived from HemoPI or equivalent hemolysis prediction server. Gautam A, et al. HemoPI: a server to predict and design hemolytic peptides. Journal of Translational Medicine, 2014. https://webs.iiitd.edu.in/raghava/hemopi/
Binding affinity (pKd/pKi) — Peptide binding affinity prediction. Source dependent on tool used — confirm from course pipeline documentation.
GRAVY score (hydrophobicity) — Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology, 1982. 157(1):105–132.
Isoelectric point (pI) prediction — Bjellqvist B, et al. The focusing positions of polypeptides in immobilized pH gradients can be predicted from their amino acid sequences. Electrophoresis, 1993.
Disease and Biology Context
SOD1 and fALS — Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature, 1993. 362:59–62.
A4V mutation and ALS — Cudkowicz ME, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Annals of Neurology, 1997. 41(2):210–221.
SOD1 misfolding and aggregation — Banci L, et al. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nature Chemical Biology, 2013.
Induced folding / folding upon binding — Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nature Reviews Molecular Cell Biology, 2005. 6:197–208.
Structural Biology Concepts
Greek key β-barrel topology — Richardson JS. The anatomy and taxonomy of protein structure. Advances in Protein Chemistry, 1981. 34:167–339.
Protein distance thresholds and contact definition — Keskin O, et al. Principles of protein-protein interactions. Chemical Reviews, 2008. 108(4):1225–1244.
Lysine-mediated membrane disruption and hemolysis — Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nature Reviews Microbiology, 2005. 3:238–250.
Additional References
- Fiers W. et al. (1976) Complete nucleotide sequence of bacteriophage MS2 RNA. Nature 260:500–507
- Kastelein R.A. et al. (1982) Lysis gene expression of RNA phage MS2. Nature 295:35–41
- Beremand & Blumenthal (1979) Overlapping genes in RNA phage. Cell 18:257–266
- PMC10688784 — In vitro characterization of the phage lysis protein MS2-L
- PMC554659 — The amino terminal half of the MS2-coded lysis protein is dispensable
- CNN Phage Therapy Article — Strathdee/Patterson case study