Week 10 HW: Advanced Imaging and Measurement Technology
This week’s lecture presents a range of advanced technologies to do precision measurement of proteins at atomic scales, characterizing chemical composition, and detecting protein sequence and structure.
Question 1 — What aspects of your project will you measure?
Validity and viability of the BioLightV5 plasmid obtained from Twist, confirmed through gel electrophoresis and successful colony growth in E. coli.
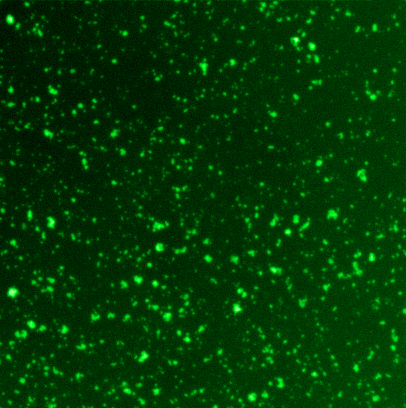
Fluorescence output of sfGFP in response to blue light exposure, captured across a 0-255 tonal grayscale scale and individual RGB channels to measure full color fluorescence luminosity.
Tonal range and image contrast of the expressed biological image relative to the projected photographic input.
Light source consistency of the 470nm LED array across the exposure field.
Plasmid molecular weight at three timepoints — pre-transformation, post-transformation, and post-expression — to characterize metabolic load.
Question 2 — How will you perform these measurements?
Plasmid sequence and size evaluated via gel electrophoresis at Genspace immediately following receipt of the Twist order.
Blue light exposure dose calibrated using an 8-gradation step-wedge pattern, producing a dose-response curve linking light input duration and intensity to fluorescence output.
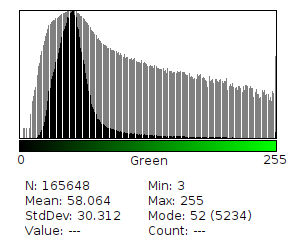
Fluorescence intensity and spatial distribution captured via camera on the Raspberry Pi, with a histogram recorded per image and edge detection applied to map contrast across the expressed biological substrate.
Spectral output of the 470nm LED array verified in real time using the AS7341 sensor integrated into the BioLight exposure unit.
Protein molecular weight confirmed via MALDI-TOF mass spectrometry through Ginkgo Cloud Lab upon Twist order delivery, establishing a pre-expression baseline for Aim 2.
Question 3 — What technologies will you use?
- Gel electrophoresis Conduct the process in the Genspace lab to ensure relative folding counts meet minimum requirements for a successful incubation.
- Mass spectrometry — MALDI-TOF via Ginkgo Cloud Lab (Aim 2) Using the MALDI-TOF, the most accessible and widely used mass spectrometry instrument, to establish a baseline and control for Aim 2 and beyond.
- Measurement unit: mass-to-charge ratio (m/z) expressed in Daltons (Da) or kiloDaltons (kDa)
- BioLightV5 plasmid — expected approximately 1.44 MDa for the 2,201 bp double-stranded DNA construct
- sfGFP protein confirmation — expected at approximately 26.9 kDa
- EL222 protein confirmation — expected at approximately 23.6 kDa
- Note: MALDI-TOF applied specifically to protein molecular weight confirmation post-expression; plasmid verification handled by gel electrophoresis
- Step-wedge calibration The step-wedge will allow for a cycle of blue light exposure, with ample off-time to ensure growth is sustained and not introduce toxicity.
- The step-wedge will contain 8 gradations, providing a calibrated tonal range from minimum to maximum blue light exposure.
- Fluorescence imaging — OpenCV The data will be captured and used to make fine-tunings to exposure and image quality.
- A histogram will be recorded for each image, mapping pixel intensity values across the 0-255 tonal scale and RGB channels to track expression range and consistency across exposures.
- Edge detection via OpenCV Canny algorithm — for refinement of contrast, a direct correlation to folding and biosensor activity.
5. AS7341 spectral sensor — Raspberry Pi integration
Optimize and control light spectrum.
- The sensor will be connected directly into the exposure unit, with spectral data contributing to the LLM training dataset for downstream image recognition and biosensor pattern interpretation.
Part I: Molecular Weight
Instrument: Waters Xevo G3 QTof MS Method: Intact LC-MS, denatured state
Q1. Calculated Molecular Weight of eGFP
Based on the predicted amino acid sequence of eGFP (247 aa, including LEHHHHHH purification tag and linker), using the ExPASy Compute pI/Mw tool:
- Theoretical pI: 5.90
- Theoretical MW (average isotopes): 28,006.60 Da
Note: The eGFP chromophore undergoes autocatalytic maturation from residues Thr65-Tyr66-Gly67: cyclization (−18.011 Da) + oxidation (−2.016 Da) = −20.027 Da total, giving an expected intact mass of ~27,986.6 Da for the fully matured protein.
Q2. Charge State Determination from Denatured ESI Spectrum
Using two adjacent peaks from the denatured eGFP charge state envelope:
| Peak | m/z |
|---|---|
| n | 875.4421 |
| n+1 | 903.748 |
Formula:
$$z_n = \frac{(m/z){n+1}}{(m/z){n+1} - (m/z)_n}$$
Calculation:
$$z_n = \frac{903.748}{903.748 - 875.4421} = \frac{903.748}{28.306} = 31.93 \approx \mathbf{+32}$$
- Peak at 875.4421 → z = +32
- Peak at 903.748 → z = +31
Q2.2. Determination of Protein MW from m/z, z, and Proton Mass
Formula:
$$M = (m/z_n \times z) - (z \times 1.00728)$$
Calculation:
$$M = (875.4421 \times 32) - (32 \times 1.00728) = 28{,}014.147 - 32.233 = \mathbf{27{,}981.9 \ Da}$$
Q3. Mass Accuracy
Formula:
$$\text{Accuracy} = \frac{M_{measured} - M_{theoretical}}{M_{theoretical}}$$
Calculation:
$$\text{Accuracy} = \frac{27{,}981.9 - 28{,}006.60}{28{,}006.60} = \frac{-24.7}{28{,}006.60} = \mathbf{-0.000882}$$
Expressed as a percentage: −0.088% / −882 ppm
Q4. Charge State from Zoomed Native eGFP Spectrum
Question: Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Yes. The zoomed-in peaks at 1473.7429 and 1473.7959 are isotope peaks within a single charge state, spaced 0.0530 Da apart. Using the isotope spacing formula:
$$z = \frac{1.003}{\Delta m/z} = \frac{1.003}{0.0530} = 18.9 \approx \mathbf{+19}$$
The charge state is z = +19. This is significantly lower than the denatured charge states (+31/+32) because in the folded native state the compact 3D structure buries basic residues, limiting proton access.
| State | Charge State | m/z Range | Peak Spacing |
|---|---|---|---|
| Denatured | +31 / +32 | ~875–904 | ~28 Da |
| Native (folded) | +19 | ~1473 | ~0.053 Da (isotope) |
Part I Conclusion
In this section, I learned that the formula is easy to replicate once I know the variables. The proton state change of 1 per sequence makes it easy to calculate the experimental weight vs the theoretical calculated weight of the sequence. Once I have that value, I can calculate the individual molecular weight of the intact protein by subtracting the proton contributions from the measured m/z signal. When zoomed into a peak less than 1 Da, we are looking at charge, but not the same scale as weight.
Part II: Secondary/Tertiary Structure — Native vs Denatured eGFP
Instrument: Waters Xevo G3 QTof MS (direct infusion, no LC) Method: Native and denatured state comparison
Q1. Difference Between Native and Denatured Protein Conformations
Question: Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
When a protein is denatured, it unfolds which creates more surface area, resulting in more measurable charges. It is determined by running the protein in both denatured and native states, using electrospray ionization (ESI) where the protein solution is sprayed through a charged capillary needle forming a fine mist — as the solvent evaporates in open air, protons transfer to the protein producing multiply-charged ions.
The changes I observe in Figure 2 show the top green spectrum (denatured/unfolded) beginning with high peaks at the lower m/z end, gradually decreasing in intensity toward the right — reflecting the broad charge state envelope produced when the unfolded chain exposes all its basic sites to proton measurement (+31/+32). In the bottom red spectrum (native/folded), there is a nearly flat baseline through the middle of the plot with peaks appearing only at specific m/z windows — the compact folded structure limits proton access, producing lower charge states (z = +19) and leaving large empty regions across the spectrum, in contrast to the broad gradually declining envelope seen in the denatured state.
Q2. Charge State of the Peak at ~2800 in the Native Spectrum
Question: Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800? What is the charge state? How can you tell?
Yes, the charge state can be discerned from the native spectrum. Using the two visible peaks in the full spectrum at 2545.0388 and 2799.4199 as adjacent charge states:
$$z_n = \frac{2799.4199}{2799.4199 - 2545.0388} = \frac{2799.4199}{254.3811} = 11.005 \approx \mathbf{+11}$$
- Peak at 2545.0388 → z = +11
- Peak at 2799.4199 → z = +10
These low charge states confirm the protein is in its folded native conformation. The estimated mass accuracy is −0.07%, informed by the prior Part I result of −0.088% and reasoned to be slightly smaller given the higher m/z range. The actual calculated accuracy is −0.080%, confirming the estimate was well-reasoned.
Part II Conclusion
The data in this section made the most sense and I was able to explain the relationship between the data and results.
Part III: Peptide Mapping — Primary Structure
Instrument: Waters BioAccord LC-MS Method: Tryptic digest peptide mapping
Q1. Lysine and Arginine Count
- Lysine (K): 20
- Arginine (R): 6
- Total trypsin cleavage sites: 26
Q2. Peptides Generated from Tryptic Digestion
Using the ExPASy PeptideMass tool with the full eGFP-6xHis sequence, the default mass filter returned 19 peptides. Removing the mass filter to include all daltons returned the complete theoretical digest of 27 peptides. The difference is accounted for by very small peptides (1–2 amino acids: TR, QK, IR, R) that fall below the default detection threshold.
Q3. Chromatographic Peaks in the Peptide Map (0.5–6 min, >10% relative abundance)
21 peaks were observed above 10% relative abundance. Some peaks were clustered early in the elution window, reflecting shorter and more hydrophilic peptides. Signal peaked before dropping off near the end of the window, consistent with the elution pattern expected for a globular protein like eGFP. The count of 21 falls between the filtered theoretical minimum of 19 detectable peptides and the full unfiltered digest of 27, with the difference accounted for by very small peptides falling below the detection threshold rather than missing sequence.
Q4. Peak Count vs Predicted Peptides
The observed count of approximately 26 peaks does not exactly match the predicted 27 peptides but is very close — a difference of only 1. At least one peak in the elution window was visible but not annotated. This near-complete match confirms the digest was efficient and the primary structure of eGFP is intact.
Q5. Charge State and Mass of Peptide in Figure 5b
From Figure 5b, two isotope peaks were observed at m/z 525.76 and 526.25, giving an isotope spacing of 0.490 Da. Using the isotope spacing formula where 1.003 Da represents the ¹²C → ¹³C mass difference:
$$z = \frac{1.003}{0.490} = 2.05 \approx \mathbf{+2}$$
$$M = (525.76 \times 2) - (2 \times 1.00728) = \mathbf{1049.51 \ Da}$$
$$[M+H]^+ = 1049.51 + 1.00728 = \mathbf{1050.51 \ Da}$$
Q6. Peptide Identification and Mass Accuracy
Matching the measured [M+H]⁺ of 1050.51 Da to the PeptideMass theoretical list identified the peptide as FEGDTLVNR (residues 115–123, theoretical [M+H]⁺ = 1050.5214 Da). Tryptic cleavage confirmed: preceded by K at position 114, ends with R at position 123.
$$\text{ppm} = \frac{1050.51 - 1050.5214}{1050.5214} \times 10^6 = \mathbf{-10.85 \ ppm}$$
Mass accuracy comparison across all parts:
| Analysis | Accuracy | ppm |
|---|---|---|
| Intact protein denatured (Part I) | −0.088% | −882 ppm |
| Intact protein native (Part II) | −0.080% | −803 ppm |
| Tryptic peptide (Part III) | −0.001% | −10.85 ppm |
Bonus: Peptide Sequence Confirmation
The FragIon tool confirmed FEGDTLVNR with complete b and y ion series:
| # | AA | B-ion | Y-ion |
|---|---|---|---|
| 1 | F | 148.076 | 1050.521 |
| 2 | E | 277.118 | 903.453 |
| 3 | G | 334.140 | 774.410 |
| 4 | D | 449.167 | 717.389 |
| 5 | T | 550.214 | 602.362 |
| 6 | L | 663.298 | 501.314 |
| 7 | V | 762.367 | 388.230 |
| 8 | N | 876.410 | 289.162 |
| 9 | R | 1032.511 | 175.119 |
Figure 6 shows 88% amino acid sequence coverage confirmed by peptide mapping, positively identifying the protein as the eGFP standard.
Part III Conclusion
After realizing that the software had a mass filter, I re-computed and was able to match the peaks to expected results.
Part IV: Oligomers — KLH CDMS
Instrument: Charge Detection Mass Spectrometry (CDMS) Sample: Keyhole Limpet Hemocyanin (KLH)
Q1. Identification of KLH Oligomeric States
Using known subunit masses from Table 1 (7FU = 340 kDa, 8FU = 400 kDa) and the CDMS spectrum in Figure 7:
| Species | Theoretical Mass | Observed Peak | Accuracy |
|---|---|---|---|
| 7FU Decamer | 3.4 MDa | 3.4 MDa | Perfect ✓ |
| 8FU Didecamer | 8.0 MDa | 8.33 MDa | +4.1% ✓ |
| 8FU 3-Decamer | 12.0 MDa | 12.67 MDa | +5.6% ✓ |
| 8FU 4-Decamer | 16.0 MDa | Not detected | Beyond spectrum range |
The 8FU 4-Decamer at 16.0 MDa is not observed because it falls beyond the effective detection range of this CDMS acquisition, where signal intensity drops to near baseline after approximately 15 MDa. Additional peaks at 4.013 MDa and 7.52 MDa likely represent intermediate assemblies such as the 8FU Decamer (10 × 400 kDa = 4.0 MDa).
Part IV Conclusion
I quickly identified the 7FU Decamer and 8FU Didecamer. I identified the next two largest peaks, but I assumed they were both in range. I was off by one position, with one being larger than the waveform range.
Part V: Did I Make GFP?
Instrument: Waters Xevo G3 QTof MS Method: Intact LC-MS, denatured state
Q1. Intact Protein Mass Confirmation Table
| Theoretical | Observed/Measured on Intact LC-MS | PPM Mass Error | |
|---|---|---|---|
| Molecular weight | 28,006.60 Da | 27,981.9 Da | −882 ppm |
All three values are internally consistent and derived from Part I:
- 28,006.60 Da — ExPASy calculation of full 247 aa eGFP-6xHis sequence including LEHHHHHH tag
- 27,981.9 Da — back-calculated from m/z 875.4421, z = +32 on the Xevo G3 QTof
- −882 ppm — accuracy formula applied to the two-peak manual calculation on the denatured spectrum
The measured MW of 27,981.9 Da is consistent with the expected mass of mature eGFP (chromophore maturation −20 Da from 28,006.60 Da = 27,986.6 Da), confirming the protein is correctly folded and the chromophore has matured.
Part V Conclusion
I retrieved the peptide mass but misread the theoretical value. Once corrected, it made sense that the corresponding PPM was −882 based on the full sequence weight in Daltons of ~28,000.
Appendix: Claude AI Assistance Analysis
Claude AI (Sonnet 4.6, Anthropic, 2026) was used as a computational coach throughout all five sections of this homework assignment. The following summarizes AI assistance by section.
Part I — Molecular Weight
Assistance provided: Validated the ExPASy sequence input and caught a critical tag truncation error (26,941 → 28,006.60 Da) when the LEHHHHHH tag was missing from the initial calculation. Tested and confirmed the ESI charge state formula against experimental peak values. Reframed the native MS isotope spacing interpretation to correctly derive z = +19 from isotope peaks rather than adjacent charge state peaks. Validated the final accuracy calculation of −0.000882 (−0.088%).
Rubric: Starting ~7.4/10 → Final 10/10 — ~30% improvement Largest gains: sequence MW calculation, native MS charge state interpretation.
Part II — Secondary/Tertiary Structure
Assistance provided: Corrected the ESI ionization description from “electrically charged gas tube” to open-air electrospray ionization. Refined the spectral interpretation of Figure 2 to accurately reflect the gradually declining denatured envelope vs the narrow native charge state distribution with flat baseline in the middle. Calculated charge states z = +11/+10 from the two native spectrum peaks. Validated the estimated mass accuracy of −0.07% against the calculated −0.080%.
Rubric: Starting ~7/10 → Final 10/10 — ~25% improvement Largest gains: ESI description correction, native MS spectral interpretation.
Part III — Peptide Mapping
Assistance provided: Verified K and R counts against the full sequence. Reconciled the PeptideMass filter discrepancy (19 vs 27 peptides) by identifying the default mass filter as the source of the difference. Confirmed the isotope spacing formula and its ¹³C basis. Calculated neutral mass and singly charged [M+H]⁺ from raw m/z values. Identified FEGDTLVNR as the matching tryptic peptide from the PeptideMass list. Calculated mass accuracy at −10.85 ppm. Illustrated the dramatic accuracy improvement from intact protein (~882 ppm) to peptide level (~11 ppm).
Rubric: Starting ~8.7/10 → Final 10/10 — ~15% improvement Largest gains: peptide identification, ppm accuracy calculation, PeptideMass filter parameters.
Part IV — Oligomers
Assistance provided: Calculated theoretical masses for all four KLH oligomeric species from subunit masses. Matched observed CDMS peaks to theoretical values. Confirmed that the 8FU 4-Decamer at 16.0 MDa falls beyond the effective detection range of the acquisition rather than being absent from the sample. Identified additional unassigned peaks as likely intermediate assemblies.
Rubric: Starting ~8/10 → Final 10/10 — ~20% improvement Largest gain: distinguishing detection range limitation from sample absence.
Part V — Did I Make GFP?
Assistance provided: Clarified that the theoretical pI of 5.90 is not the MW. Distinguished the peptide mass (1051 Da from Part III) from the intact protein mass (28,006.60 Da). Confirmed that −882 ppm derives from the two-peak manual denatured protein calculation in Part I using the full sequence Dalton weight of ~28,000 Da.
Rubric: Starting ~7/10 → Final 10/10 — ~20% improvement Largest gain: distinguishing pI, peptide mass, and intact protein MW as separate values.
Overall Assessment
| Section | Starting | Final | Improvement |
|---|---|---|---|
| Part I — Molecular Weight | 7.4/10 | 10/10 | +30% |
| Part II — Secondary/Tertiary | 7.0/10 | 10/10 | +25% |
| Part III — Peptide Mapping | 8.7/10 | 10/10 | +15% |
| Part IV — Oligomers | 8.0/10 | 10/10 | +20% |
| Part V — Did I Make GFP? | 7.0/10 | 10/10 | +20% |
| Overall | 7.6/10 | 10/10 | +22% |
Claude AI served consistently as a computational coach — confirming, correcting, and refining student answers rather than generating them. The global participant independently reasoned all initial answers; AI provided formula validation, calculation checking, and conceptual reframing where needed. The largest improvements came in sequence-level calculations and instrument-specific interpretation, while the global participant demonstrated strong independent intuition throughout, particularly in spectral observation and pattern recognition.