Week 2 HW: DNA read, write & edit

Homework 02
Part 1: Benchling & In-silico Gel Art
- I successfully made a Benchling account and imported the Lambda DNA.
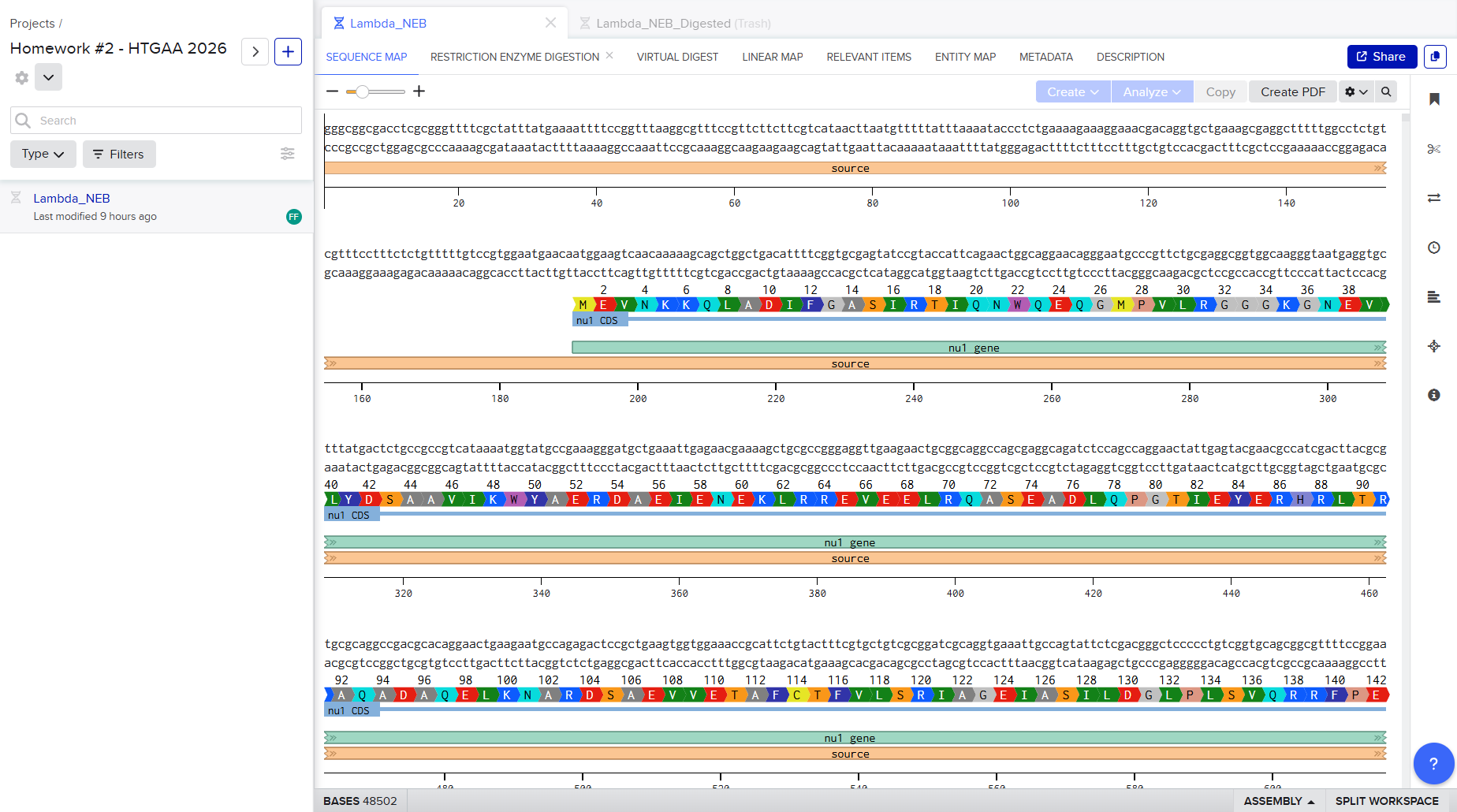
- Simulate Restriction Enzyme Digestion with the following Enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
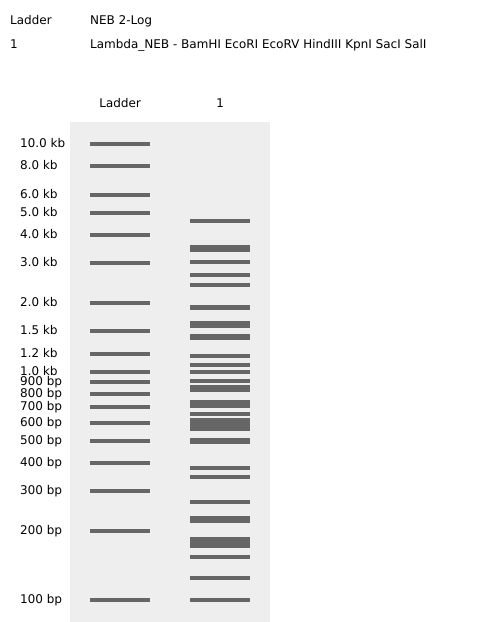
Restriction Enzyme Digestion Simulation using enzymes EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI using Benchling.
- Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
For my in-silico Gel Art I wanted to initially make a star! Sadly, after using Ronan’s website to visualize my idea, I realized that it would be a bit complicated using the listed Restriction Enzymes.
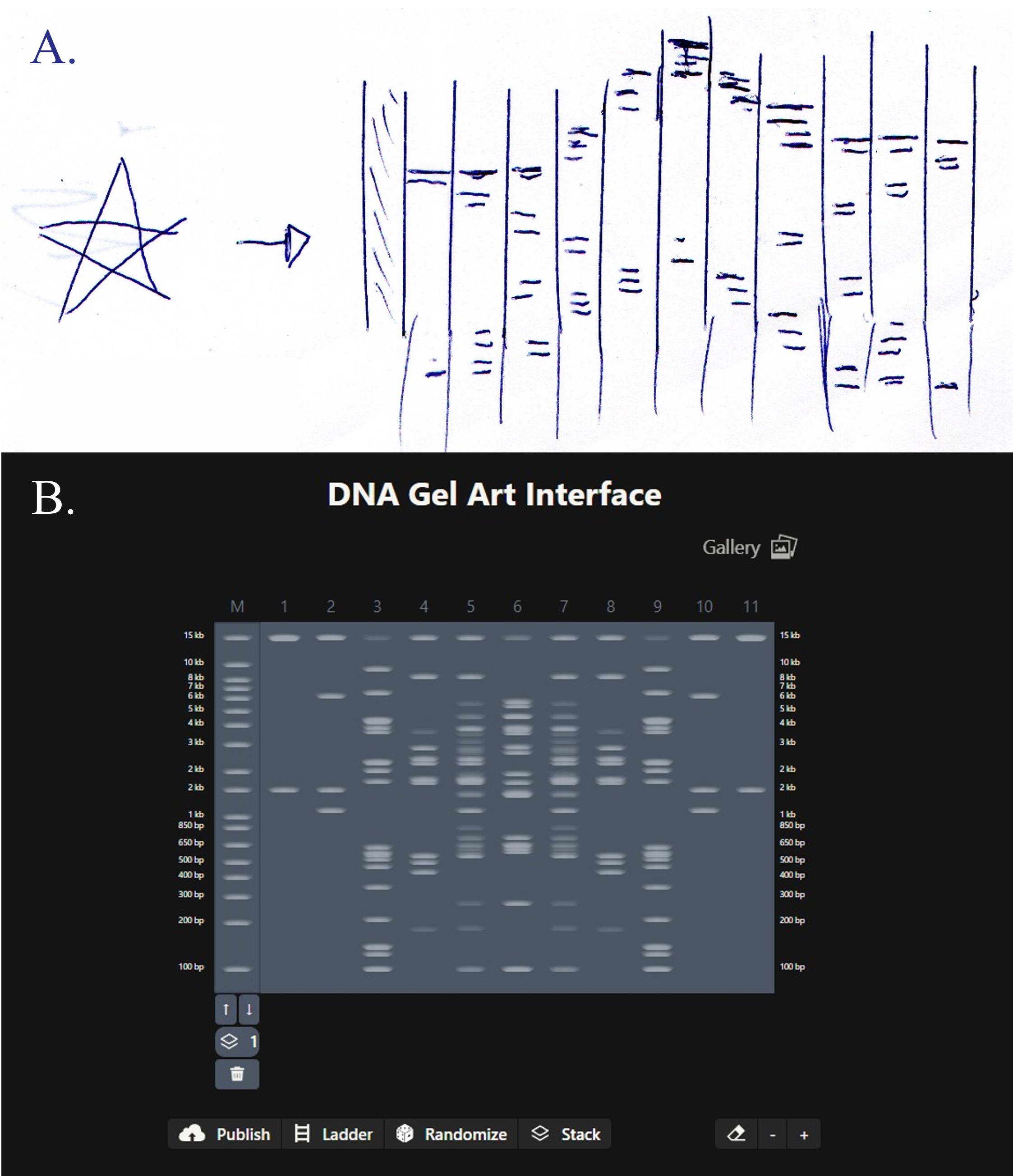
Here is a rough initial sketch for the star and my attempt to do it on Ronan’s website tool
So, I ended up making some tulips instead! You can check out my design on Ronan’s website too!
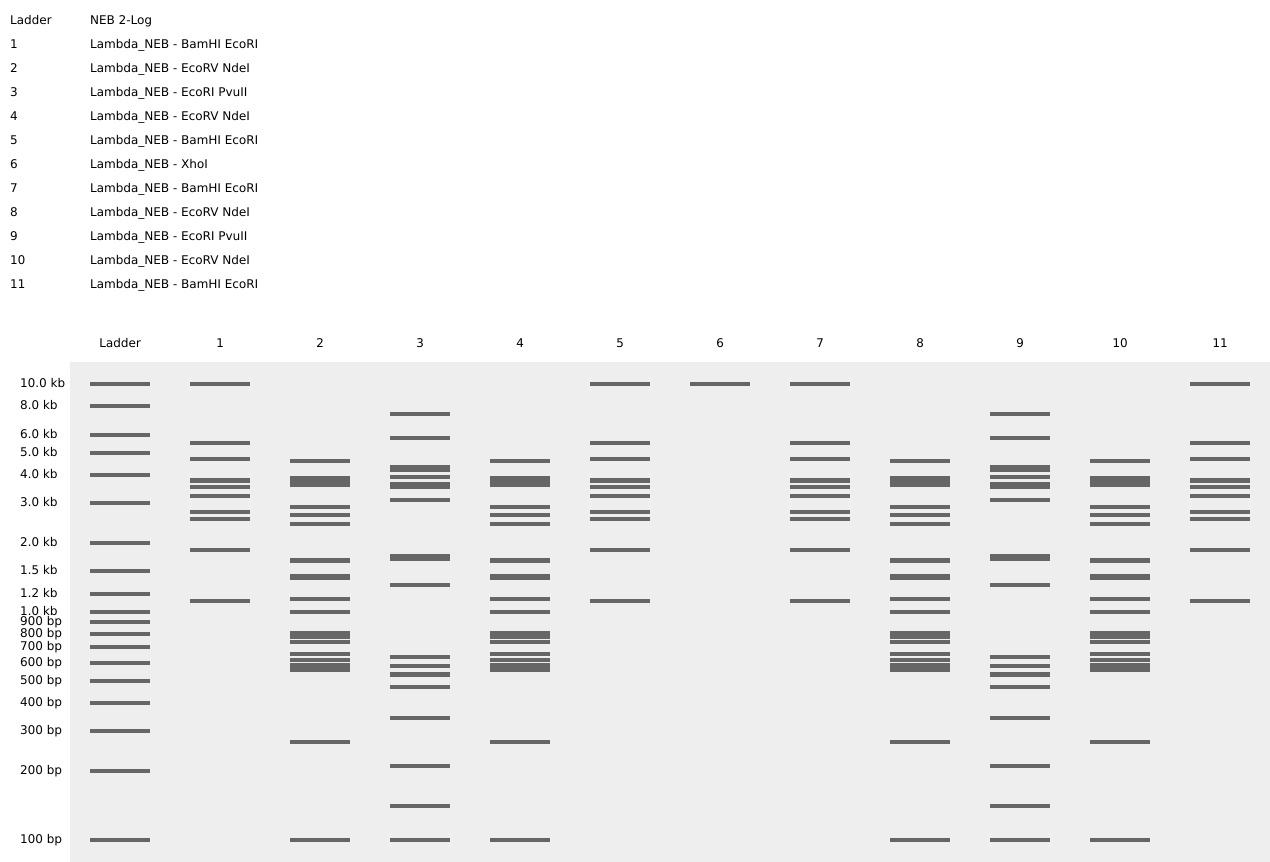
Here is a picture of the tulips design using Benchling!
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I skipped this one since I do not have Lab access.
Part 3: DNA Design Challenge
3.1. Choose your protein.
I have chosen the Chitinase enzyme from the bacterium Bacillus thuringiensis (NCBI Accession: WCH14858.1).
I found this protein interesting because of its potential in environmental conservation and biotechnology. This enzyme is capable of degrading chitin, which is a primary component of fungal cell walls and insect exoskeletons. Based on the literature, the chitinase protein is particularly efficient due to its modular structure, which typically includes a catalytic domain and chitin-binding domains that enhance its hydrolytic activity (1). Because of that, this protein becomes a very powerful tool for biological control: it can act synergistically with Cry proteins to perforate the peritrophic matrix of insect pests, increasing the efficiency of biopesticides. Additionally, I selected this specific protein because Bacillus thuringiensis is a safe organism to handle in a Level 1 biosafety laboratory (BSL-1), making it a practical and efficient candidate for recombinant protein production in E. coli.
>WCH14858.1 chitinase [Bacillus thuringiensis] MLNKFKFFCCILVMFLLLPLSPFQAQAANNLGSKLLVGYWHNFDNGTGIIKLKDVSPKWDVINVSFGETGGDRSTVEFSPVYGTDAEFKSDISYLKSKGKKIVLSIGGQNGVVLLPDNAAKDRFINSIQSLIDKYGFDGIDIDLESGIYLNGNDTNFKNPTTPQIVNLISAIRTISDHYGPDFLLSMAPETAYVQGGYSAYGSIWGAYLPIIYGVKDKLTYIHVQHYNAGSGIGMDGNNYNQGTADYEVAMADMLLHGFPVGGNANNIFPALRSDQVMIGLPAAPAAAPSGGYISPTEMKKALNYIIKGVPFGGKYKLSNQSGYPAFRGLMSWSINWDAKNNFEFSSNYRTYFDGLSLQK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
To determine the nucleotide sequence that corresponds to the chitinase protein I went to the original Bacillus thuringiensis genome.
[Original sequence of Bacillus thuringiensis from its genome Bacillus thuringiensis - Nucleotide - NCBI]
Chitinase protein DNA sequence
atgttaaacaagttcaaatttttttgttgtattttagtaatgttcttacttctaccgttatcccctttccaagcacaagcagcaaacaatttaggttcaaaattactcgttggatactggcataattttgataacggtactggcattattaaattaaaagacgtttcaccaaaatgggatgtaatcaatgtatcttttggtgaaactggtggtgatcgttccactgttgaattttctcctgtgtatggtacagatgcagaattcaaatcagatatttcttatttaaaaagtaaaggaaagaaaatagttctttcaataggtggacaaaatggggtcgttttacttcctgacaatgccgctaaggatcgttttattaattccatacaatctctgatcgataaatacggttttgacggaatagatattgaccttgaatcaggtatttacttaaacggaaatgacactaacttcaaaaacccaactacccctcaaatcgtaaatcttatttcagctattcgaacaatctcagatcattatggtccagattttctattaagcatggcccctgaaacagcttatgttcaaggcggatatagcgcatatggaagcatatggggtgcatatttaccaattatttatggagtgaaagataaactaacatacattcacgttcaacactacaacgctggtagcgggattggaatggacggtaataactacaatcaaggtactgcagactacgaggtcgctatggcagatatgctcttacatggttttcctgtaggtggtaatgcaaataacattttcccagctcttcgttcagatcaagtcatgattgggcttccagcagcaccagcggcagctccaagtggtggatacatttcgccaactgaaatgaaaaaagctttaaattatatcattaaaggagttccattcggaggaaagtataaactttctaaccagagtggctatcctgcattccgcggcctaatgtcttggtctattaattgggatgcaaaaaacaactttgaattctctagtaactatagaacatattttgatggtctttccttgcaaaaataa
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
I need to optimize codon usage because, although the genetic code is redundant, different organisms have distinct ‘codon biases.’ Since I am using a sequence from Bacillus thuringiensis, I have optimized it for Escherichia coli K-12 using Benchling’s Codon Optimization tool to ensure that the host cell can translate it efficiently. I chose the K-12 strain specifically because it is the gold standard in synthetic biology laboratories since it is a safe, non-pathogenic, and well-characterized model that guarantees reliable folding for my chitinase enzyme.
Chitinase protein DNA sequence Codon-Optimization
ATGCTGAACAAATTTAAATTTTTTTGCTGCATTCTGGTGATGTTTCTGCTGCTGCCGCTGAGCCCGTTTCAGGCGCAGGCGGCGAACAACCTGGGCAGCAAACTGCTGGTGGGCTATTGGCATAACTTTGATAACGGCACCGGCATTATTAAACTGAAAGATGTGAGCCCGAAATGGGATGTGATTAACGTGAGCTTTGGCGAAACCGGCGGCGATCGCAGCACCGTGGAATTTAGCCCGGTGTATGGCACCGATGCGGAATTTAAAAGCGATATTAGCTATCTGAAAAGCAAAGGCAAAAAAATTGTGCTGAGCATTGGCGGCCAGAACGGCGTGGTGCTGCTGCCGGATAACGCGGCGAAAGATCGCTTTATTAACAGCATTCAGAGCCTGATTGATAAATATGGCTTTGATGGCATTGATATTGATCTGGAAAGCGGCATTTATCTGAACGGCAACGATACCAACTTTAAAAACCCGACCACCCCGCAGATTGTGAACCTGATTAGCGCGATTCGCACCATTAGCGATCATTATGGCCCGGATTTTCTGCTGAGCATGGCGCCGGAAACCGCGTATGTGCAGGGCGGCTATAGCGCGTATGGCAGCATTTGGGGCGCGTATCTGCCGATTATTTATGGCGTGAAAGATAAACTGACCTATATTCATGTGCAGCATTATAACGCGGGCAGCGGCATTGGCATGGATGGCAACAACTATAACCAGGGCACCGCGGATTATGAAGTGGCGATGGCGGATATGCTGCTGCATGGCTTTCCGGTGGGCGGCAACGCGAACAACATTTTTCCGGCGCTGCGCAGCGATCAGGTGATGATTGGCCTGCCGGCGGCGCCGGCGGCGGCGCCGAGCGGCGGCTATATTAGCCCGACCGAAATGAAAAAAGCGCTGAACTATATTATTAAAGGCGTGCCGTTTGGCGGCAAATATAAACTGAGCAACCAGAGCGGCTATCCGGCGTTTCGCGGCCTGATGAGCTGGAGCATTAACTGGGATGCGAAAAACAACTTTGAATTTAGCAGCAACTATCGCACCTATTTTGATGGCCTGAGCCTGCAGAAATAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To produce chitinase from my designed sequence, I can use either cell-dependent or cell-free methods. In a cell-dependent approach, I would insert the DNA into a host like E. coli K-12, where the cell’s own machinery handles the work: RNA polymerase transcribes the DNA into mRNA, and then ribosomes translate that message into the final enzyme. On the other hand, cell-free protein synthesis allows me to skip the living cell entirely by using just the necessary biological “parts” (like enzymes and ribosomes) in a tube. This last approach is a much faster way to prototype the protein without keeping bacteria alive, although I really have a space in my heart for bacterial cultures.
3.5. [Optional] How does it work in nature/biological systems?
- Describe how a single gene codes for multiple proteins at the transcriptional level.
From what I’ve understood, a single gene can produce different proteins through mechanisms like alternative splicing, where the cell mixes and matches different sections of the message (exons) to create several versions of a protein from the same DNA template. In bacteria like Bacillus thuringiensis, they also use polycistronic operons, which group several related genes under a single promoter. This allows the bacteria to produce a whole set of coordinated enzymes all at once.
- Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!
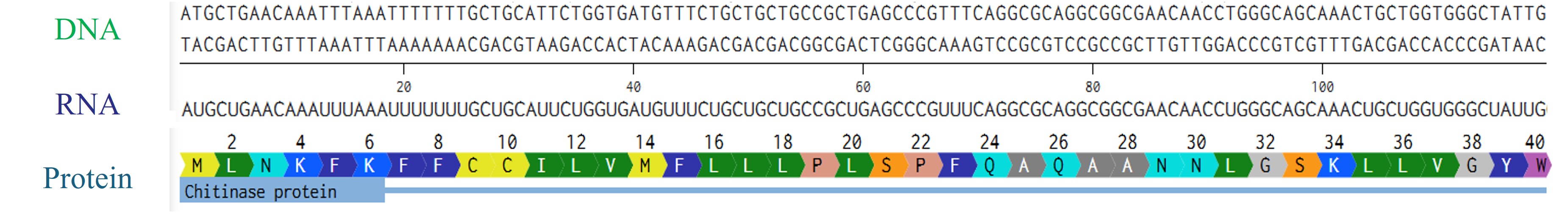
Rearranged snapshot of Chitinase protein information flow from DNA to RNA to protein. Captured from Fabri’s Benchling and arranged in PowerPoint
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
I’ll make a sequence that will make E. coli glow fluorescent blue under UV light by always expressing sfBFP (a blue fluorescent protein):
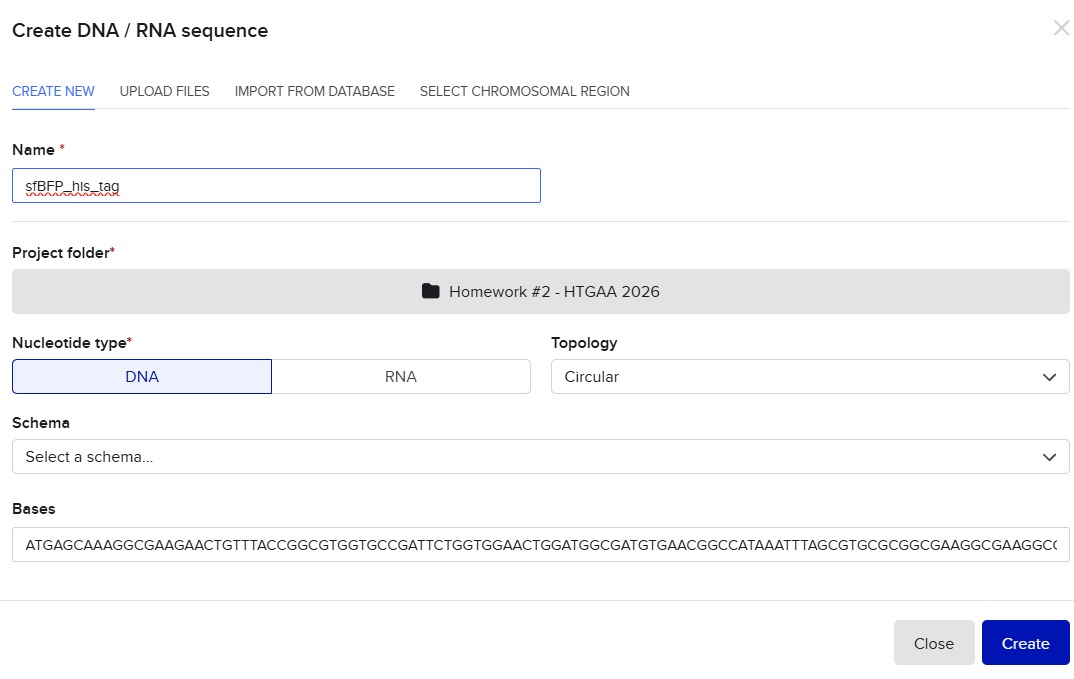
Screenshot of the creation of the sfBFP sequence in Benchling
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
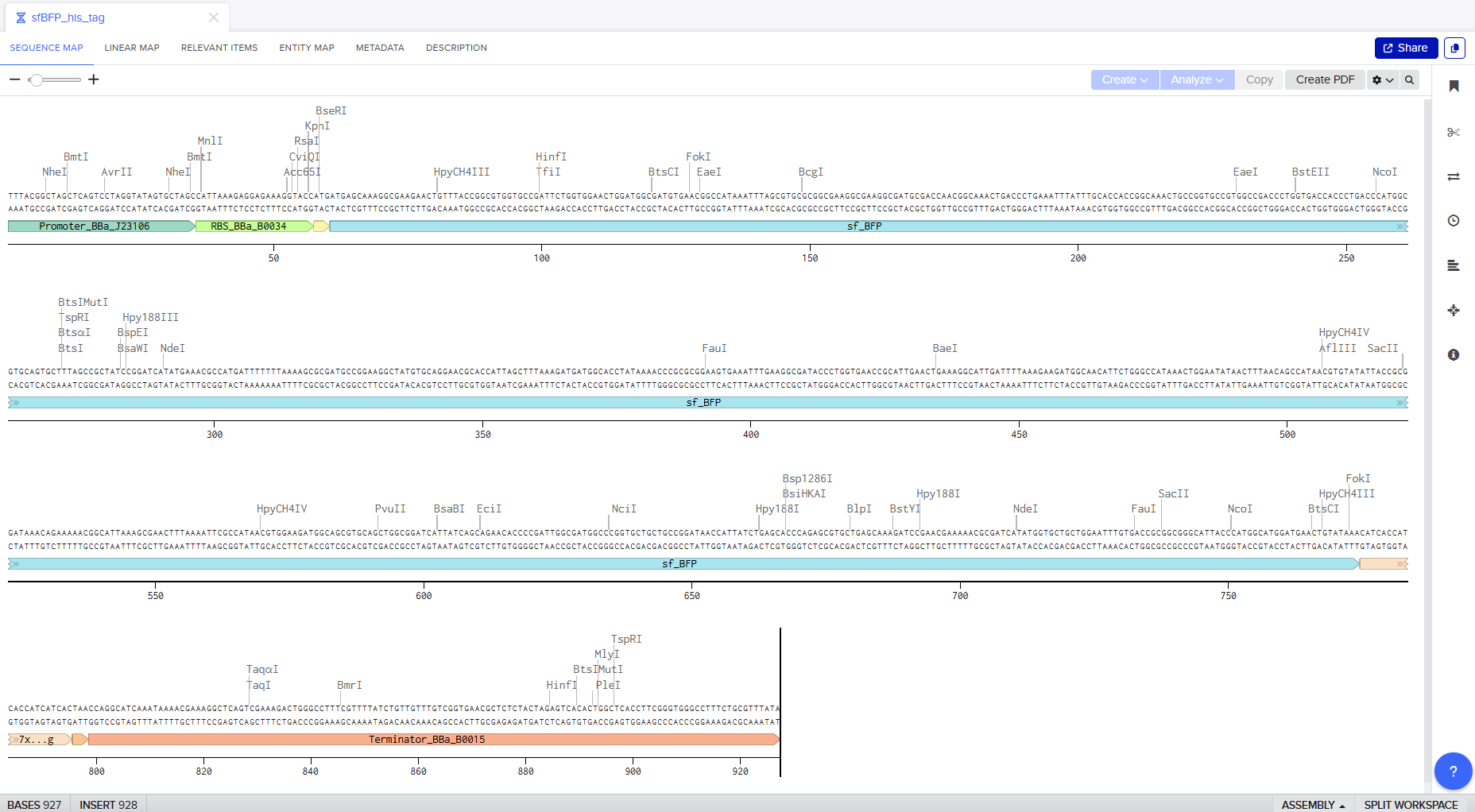
Screenshot of the whole sequence with its annotations!
- Promoter (BBa_J23106):
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC - RBS (BBa_B0034 with spacers for optimal expression):
CATTAAAGAGGAGAAAGGTACC - Start Codon:
ATG - Coding Sequence (Codon optimized DNA sfBFP):
ATGAGCAAAGGCGAAGAACTGTTTACCGGCGTGGTGCCGATTCTGGTGGAACTGGATGGCGATGTGAACGGCCATAAATTTAGCGTGCGCGGCGAAGGCGAAGGCGATGCGACCAACGGCAAACTGACCCTGAAATTTATTTGCACCACCGGCAAACTGCCGGTGCCGTGGCCGACCCTGGTGACCACCCTGACCCATGGCGTGCAGTGCTTTAGCCGCTATCCGGATCATATGAAACGCCATGATTTTTTTAAAAGCGCGATGCCGGAAGGCTATGTGCAGGAACGCACCATTAGCTTTAAAGATGATGGCACCTATAAAACCCGCGCGGAAGTGAAATTTGAAGGCGATACCCTGGTGAACCGCATTGAACTGAAAGGCATTGATTTTAAAGAAGATGGCAACATTCTGGGCCATAAACTGGAATATAACTTTAACAGCCATAACGTGTATATTACCGCGGATAAACAGAAAAACGGCATTAAAGCGAACTTTAAAATTCGCCATAACGTGGAAGATGGCAGCGTGCAGCTGGCGGATCATTATCAGCAGAACACCCCGATTGGCGATGGCCCGGTGCTGCTGCCGGATAACCATTATCTGAGCACCCAGAGCGTGCTGAGCAAAGATCCGAACGAAAAACGCGATCATATGGTGCTGCTGGAATTTGTGACCGCGGCGGGCATTACCCATGGCATGGATGAACTGTATAAA - 7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli):
CATCACCATCACCATCATCAC - Stop Codon:
TAA - Terminator (BBa_B0015):
CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
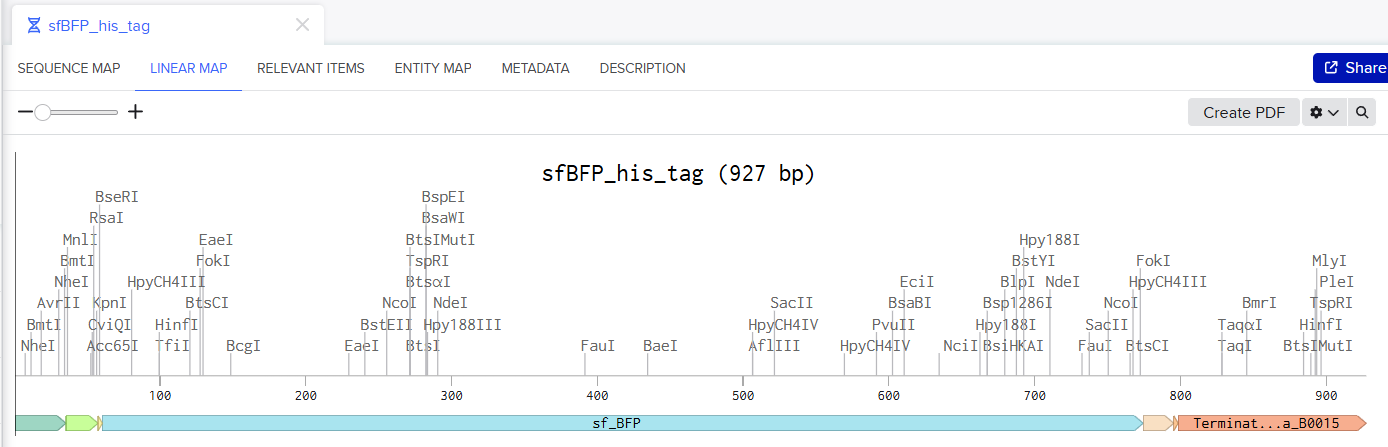
Screenshot of the Linear map of Constitutive sfBFP DNA and here is the Benchling Link
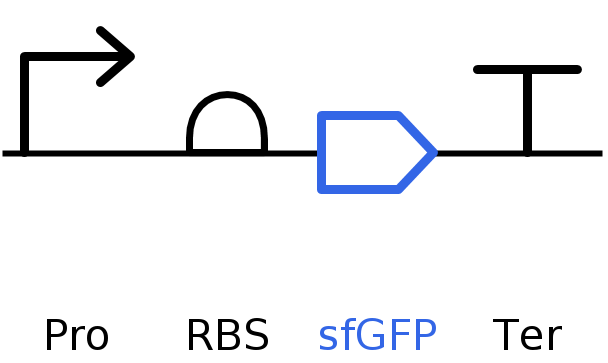
SBOL of the Linear map of Constitutive sfBFP DNA.
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.5. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
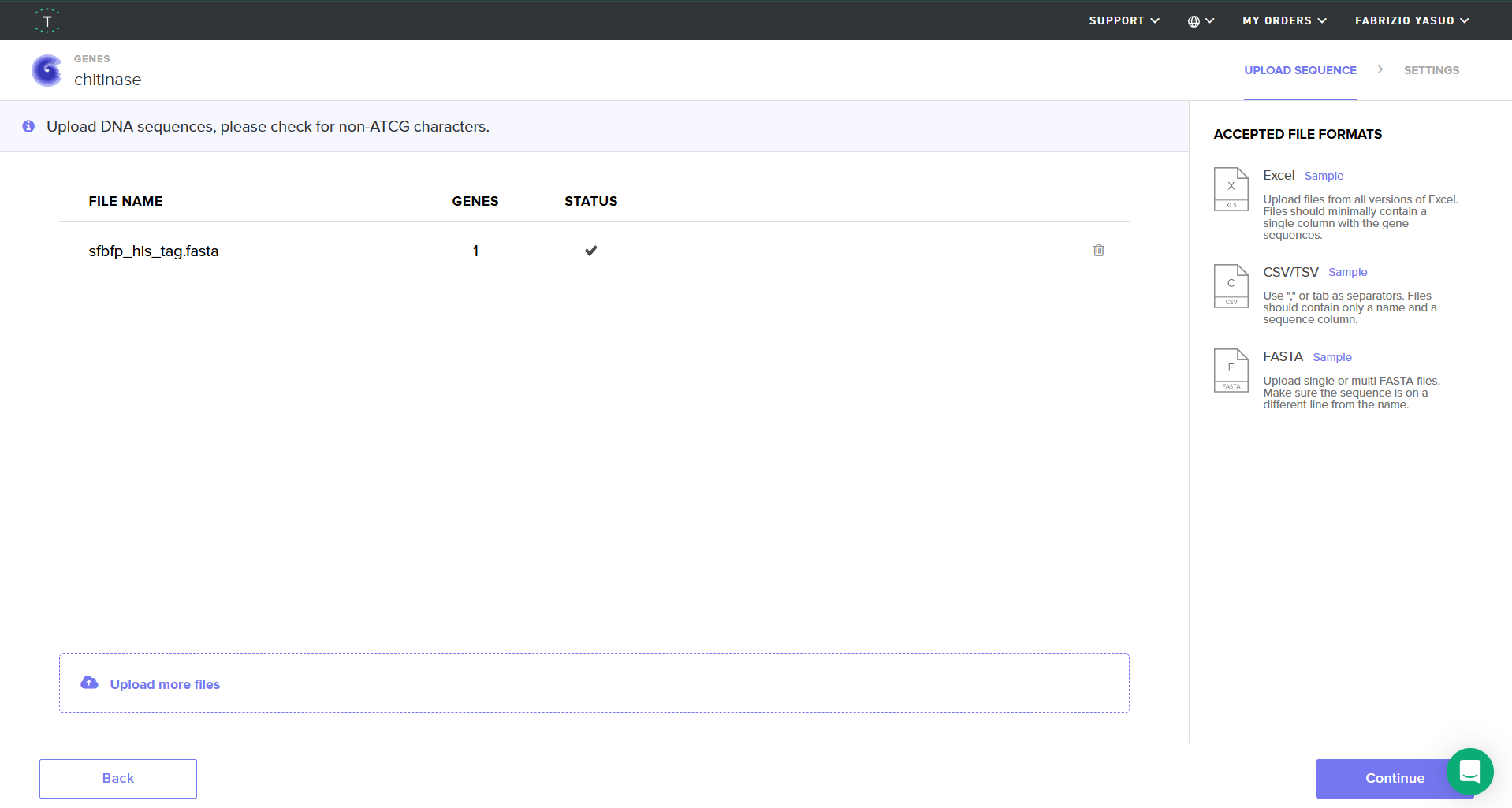
Screenshot of my uploaded sfBFP FASTA file in Twist
4.6. Choose Your Vector
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
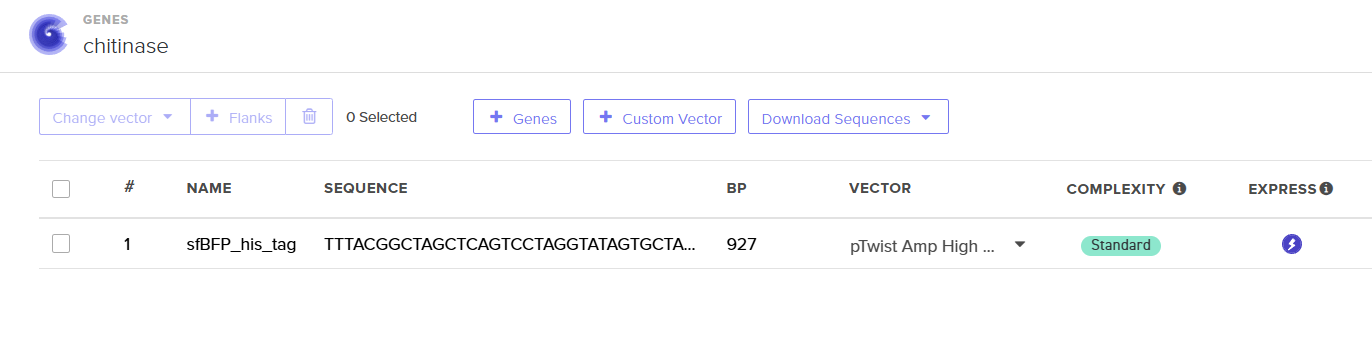
Screenshot of sfBFP with pTwist Amp High Copy vector
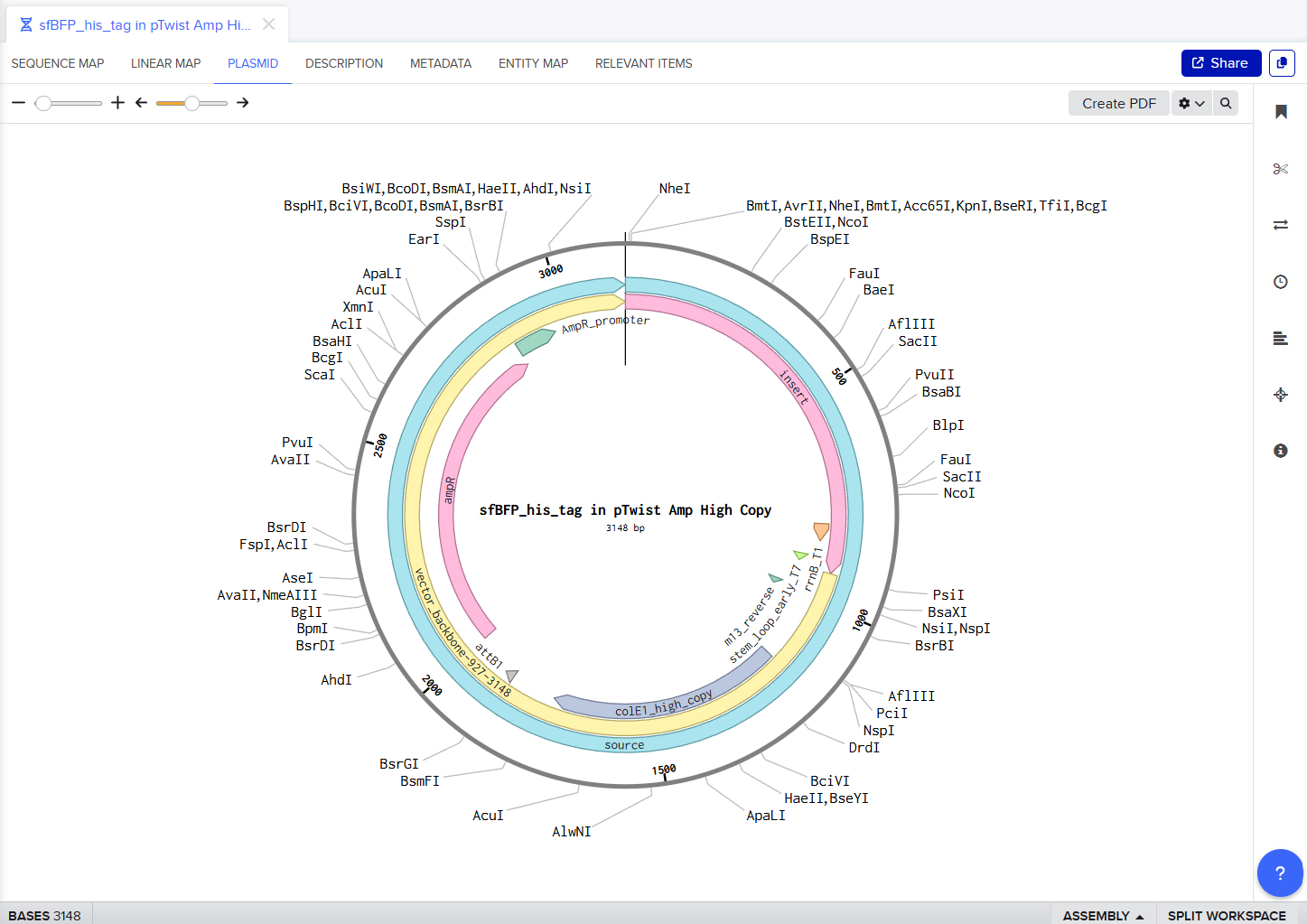
My Twist Ready Plasmid!!
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I want to sequence eDNA from river water samples collected at different points in different regions, especially near my hometown. Rivers collect DNA from fish, amphibians, and even terrestrial animals that drink from or live near the water. By sequencing the DNA, I can perform a biodiversity assessment to detect invasive species (like the trout in some Andean rivers) and/or monitor the presence of endangered amphibians without the need for traditional trapping methods.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?\
I would use Illumina (Next-Generation Sequencing) because its massive parallelization would allow me to read millions of sequences from hundreds of species in a single run, which is perfect for complex environmental samples (e.g., in rivers).
Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
Illumina’s NGS is second-generation. That’s because it uses synthesis-based sequencing on a solid surface rather than reading single long molecules.
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
My input would be filtered river water DNA. Preparation involves metabarcoding (amplifying specific markers like 16S or COI) and adapter ligation to attach fragments to the flow cell.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Illumina’s NGS has many steps but these are the essential ones that make the work itself. First, DNA fragments are attached to a flow cell where they form dense clusters through bridge amplification to ensure the detection signal is strong enough. Next, fluorescently labeled nucleotides are added one by one, and a high-resolution camera records the specific color flash emitted as each base is incorporated into the strand. Finally, the software interprets these light patterns and decodes them into a digital DNA sequence through base calling. (2)
- What is the output of your chosen sequencing technology?
The output of Illumina’s NGS is a FASTQ file containing millions of digital reads that identify the species present in the river samples. Once I get the file I can analyze it. with bioinformatics and get results.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would like to synthesize a genetic biosensor designed to detect heavy metal contamination, such as mercury, in river water. By placing this circuit into a safe host like E. coli K-12, the bacteria could “glow” or change color when it senses toxins, acting as a real-time environmental monitor to help protect the river’s biodiversity.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?\
I would love to use Twist Bioscience’s Silicon-based Synthesis to perform the DNA synthesis because of its incredible scalability and its promise of making DNA synthesis better and faster. (3)
Also answer the following questions:
- What are the essential steps of your chosen sequencing methods?
The steps that Twist follows use silicon chips to print thousands of genes simultaneously, which significantly reduces costs and improves precision. First, the digital sequence is uploaded and ‘printed’ onto a silicon chip; using phosphoramidite chemistry, the machine builds thousands of short DNA strands, known as oligonucleotides, base by base. Second, these short oligos are harvested from the chip and gathered together. Finally, the fragments are enzymatically assembled to form the complete, full-length biosensor circuit, ensuring high precision and scalability.
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The main limits are that very complex designs can significantly increase the turnaround time and the cost of production. Additionally, sequences with difficult content, such as high GC-rich regions, can lower the synthesis success rate.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would like to edit the chitinase genes in native river bacteria to make them more efficient at degrading organic waste. This would help prevent fungal outbreaks and the accumulation of debris, keeping the river ecosystem balanced and clean in a natural way.
(ii) What technology or technologies would you use to perform these DNA edits and why?\
I would use CRISPR-Cas9 because it is the most precise, well-known, and easy-to-design tool for genome engineering in bacteria. The system works by using a guide RNA (gRNA) that leads the Cas9 nuclease to a specific target in the chitinase gene to create a cut. By providing a DNA repair template, I can then insert a more efficient version of the enzyme into the genome.
Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
This technology edits DNA by acting like a pair of molecular scissors. It follows three main steps: first, the guide RNA identifies and binds to a specific target sequence in the genome. Second, the Cas9 nuclease creates a double-strand break at that exact location. Finally, the cell’s natural repair machinery goes and fixes the break; by providing a DNA repair template, the cell can be tricked into incorporating a new, more efficient chitinase sequence during this repair process.
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
I would need to digitally design a specific gRNA that is perfectly complementary to the chitinase gene to avoid off-target cuts. Additionally, the required inputs for the experiment include the Cas9 protein (or a plasmid encoding it), the custom synthetic gRNA, a DNA donor template containing the desired edit, and the target bacterial cells that will be transformed with these components!
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The biggest limitation of this method is the risk of off-target cuts, where the Cas9 might cut a similar DNA sequence elsewhere in the genome by mistake. Additionally, the efficiency of the edit depends a lot on the cell’s repair mechanism; in some bacteria, the rate of successful “homology-directed repair” can be low, meaning many cells might fail to incorporate the new gene correctly.
References
- Martínez-Zavala, S. A., Barboza-Pérez, U. E., Hernández-Guzmán, G., Bideshi, D. K., & Barboza-Corona, J. E. (2020). Chitinases of Bacillus thuringiensis: Phylogeny, Modular Structure, and Applied Potentials. Frontiers in Microbiology, 10, 3032. https://doi.org/10.3389/fmicb.2019.03032
- Next-Generation Sequencing (NGS) | Explore the technology.
- Twist Bioscience. High quality gene synthesis - Twist Bioscience. Gene synthesis