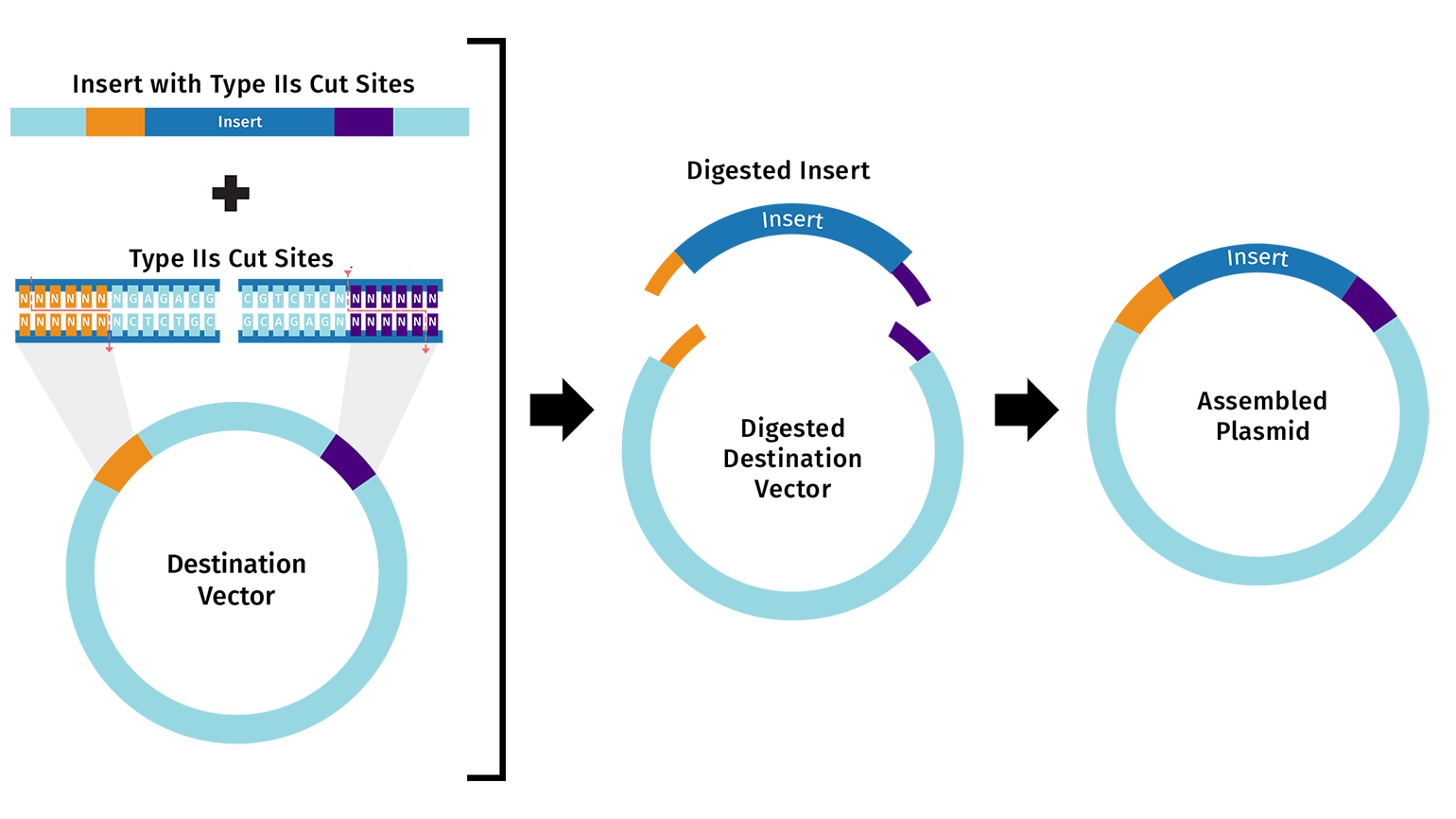A Bioengineering student at UTEC (University of Engineering and Technology) in Peru, passionate about redesigning biology at the molecular level. ASM member actively involved in scientific events and community building. Focused on protein design, synthetic biology, and bioinformatics.
Excited to connect with fellow bio-hackers and innovators! 🧬
Class Assignment 𓅨 First, describe a biological engineering application or tool you want to develop and why. I want to develop a 3D Bio-Art Platform that merges biological growth with interactive synthetic biology. The idea is to use 3D-printed molds and structured agar media to create “living sculptures” that don’t just sit there but actually “feel” and react.
Week 2 Lecture Prep Homework Questions from Professor Jacobson: 1) Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? The error rate of polymerase is 1 in 106 compared to the ~3.2 billion bp of the human genome. This means that the polymerase makes 3200 errors each time it replicates. Biology manages this discrepancy through DNA repair mechanisms, such as real-time proofreading and post-replication mismatch repair (MutS Repair system).
Homework Assignment: Python Script for Opentrons Artwork Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. For my first design I made a colorful butterfly! I first used the Opentrons art page to design it by using the upload image option. Initially the design
Homework: Protein Design I Objective: Learn basic concepts: amino acid structure 3D protein visualization the variety of ML-based design tools Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project). Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
Homework Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Homework Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Some components in the Phusion High-Fideñity PCR Master Mix include the Phusion DNA Polymerase, which is the enzyme that actually builds the new DNA strands with high accuracy. It also contains dNTPs, which are the building blocks (A, T, C, and G) used to synthesize the DNA. There are also buffer salts and magnesium ions (Mg^2+) that provide the right chemical environment for the enzyme to stay stable and work efficiently.
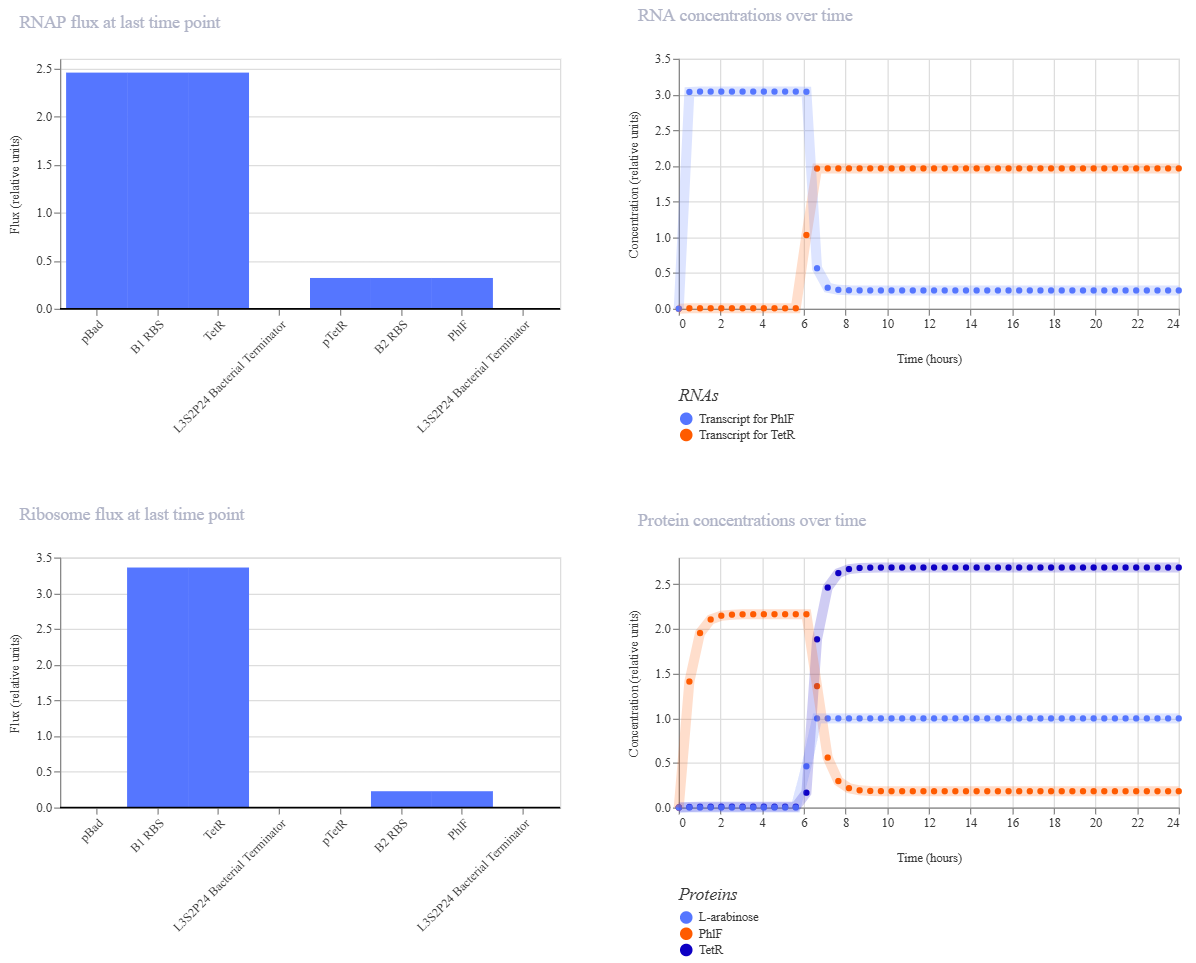
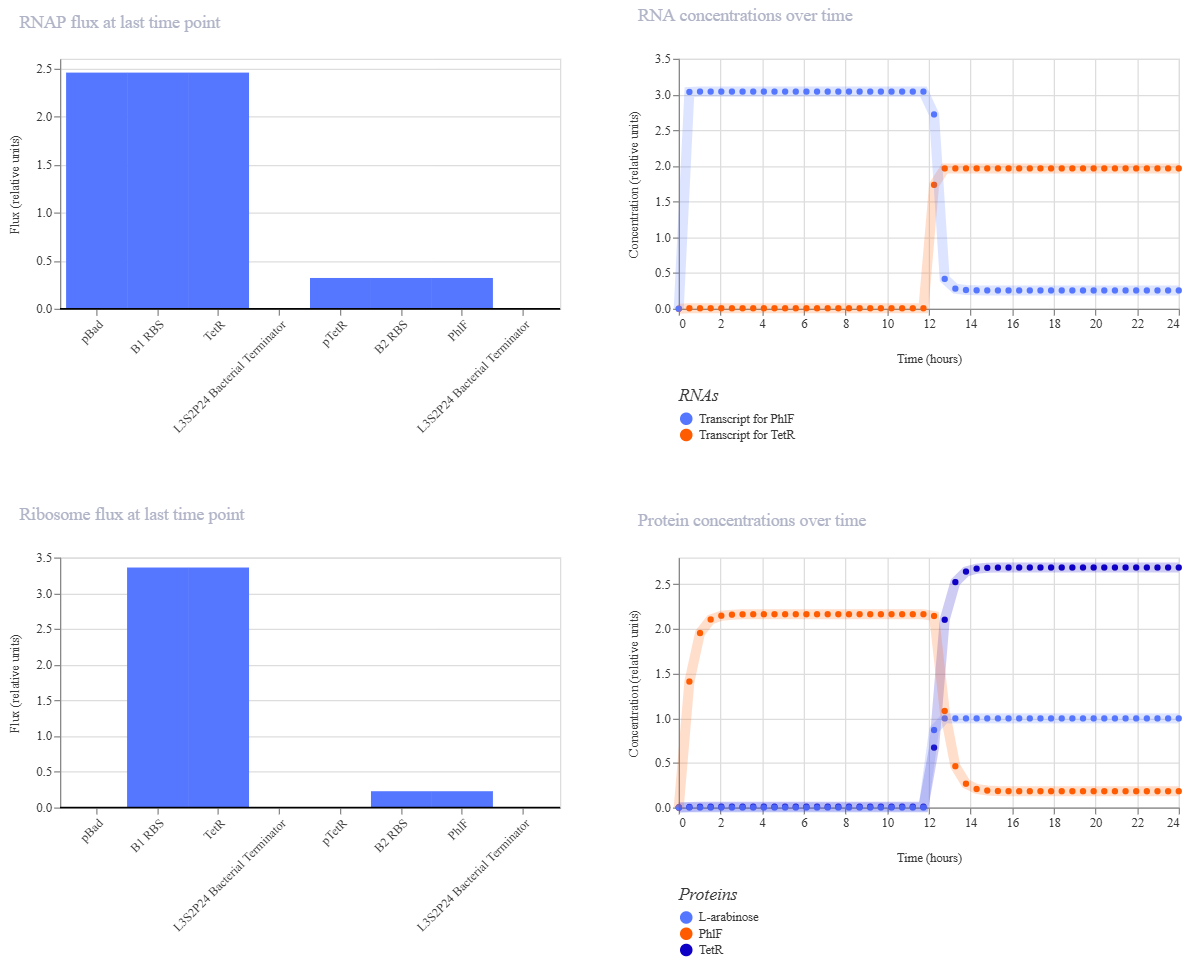
This week covers neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network “perceptron”-like computation and learning.
Homework Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Traditional Boolean circuits are limited because they only understand “on” or “off” (0 or 1), which doesn’t reflect the noisy and analog reality of a cell. IANNs allow for weighted inputs and non-linear integration, meaning the cell can make decisions based on the concentration of signals rather than just their presence. This allows for complex pattern recognition, like identifying a specific metabolic state or a signature of multiple biomarkers, making the decision-making process much more robust and “intelligent” than a simple AND/OR gate.
This week introduces synthesis of proteins using cellular machinery outside of a cell.
Homework Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. The biggest advantage of cell-free systems is that they offer an open environment where you have total control over experimental variables like pH and salt concentrations without a cell membrane getting in the way. This flexibility is especially beneficial when producing antimicrobial peptides or lysis proteins that would normally kill a living host, as well as for high-throughput screening of genetic circuits where you need to test many DNA variants in hours rather than waiting days for cultures to grow.
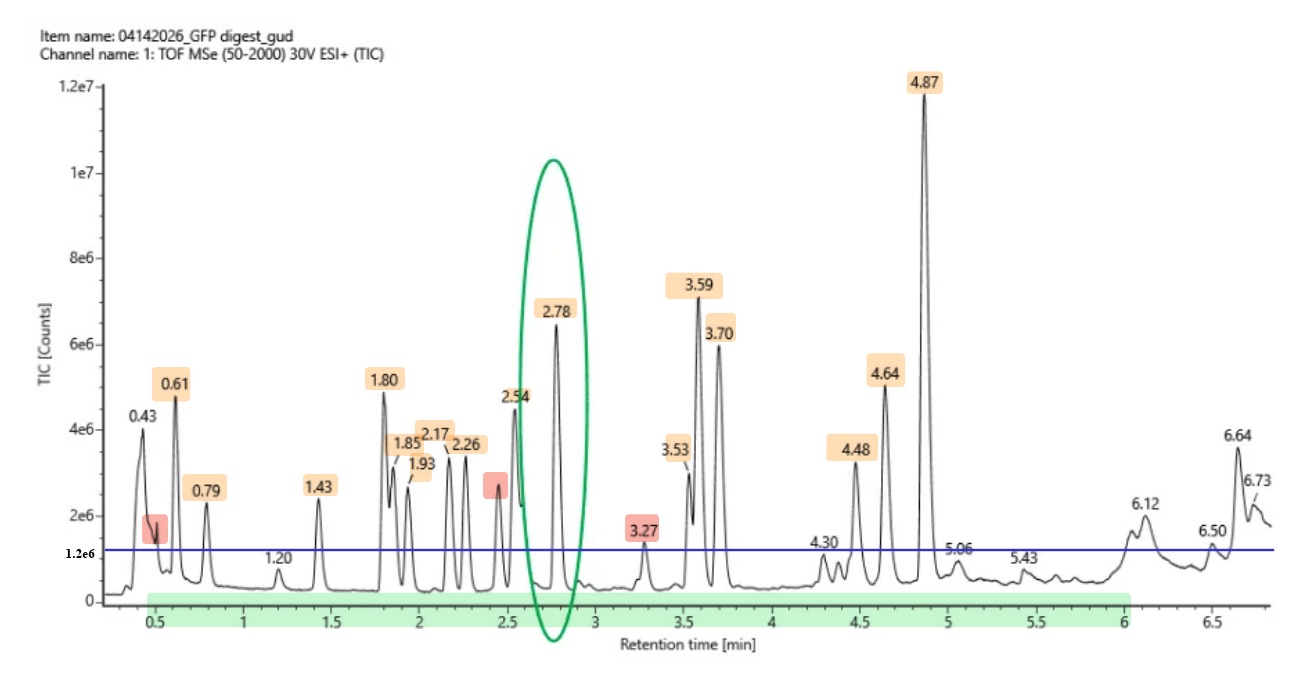
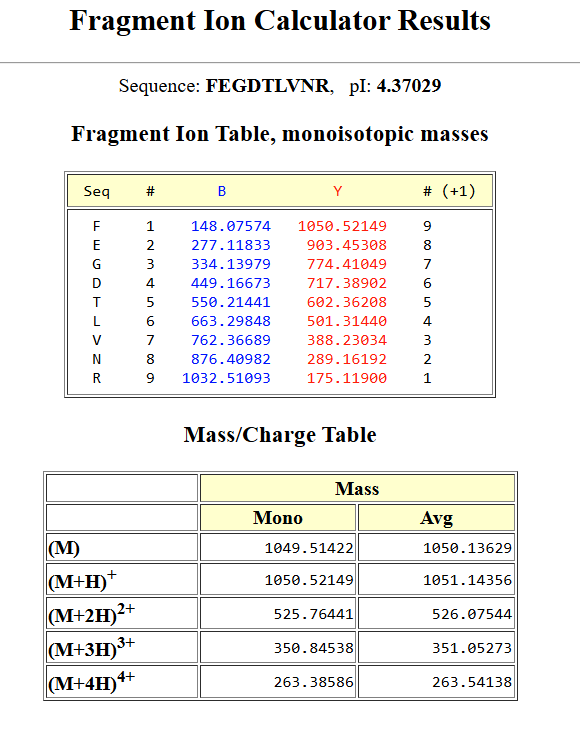
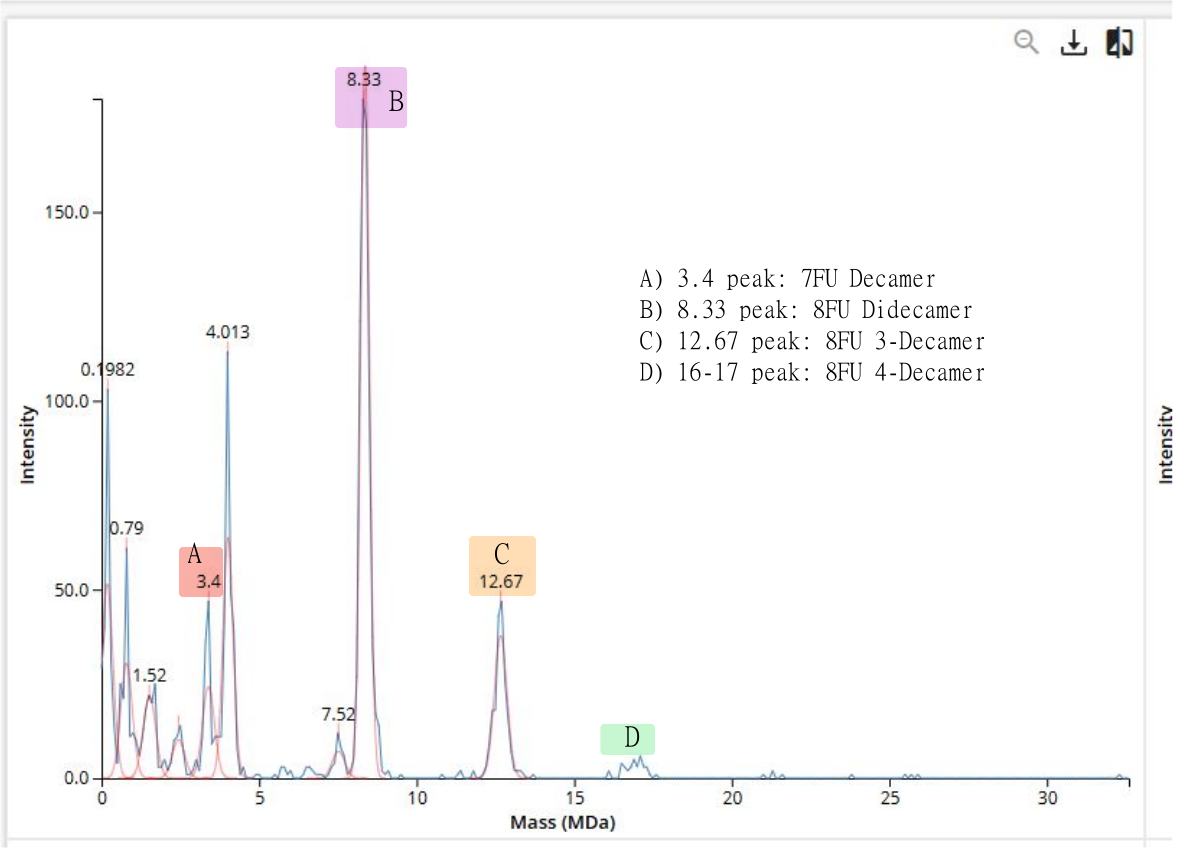
Homework Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Cloud laboratories are making science accessible, affordable, and reproducible. Our aim this semester is to showcase how they can enable human creativity at scale, and how they provide a platform for collaboration and community.
This week focuses on designing, synthesizing, and editing whole genomes, from minimal cells to refactored microbes and synthetic chromosomes.
Homework Important Be sure you’ve seen the updated week 11 homework which is due at the start of the April 28 lecture.
Tip Continue making progress this week on your Individual Final Project and on DNA orders (due Friday midnight ET).
No Lab Assignment this week. Final Project Lab time available If your final project requires lab work, you can schedule a block of lab time this week.
Continued working on the final Individual project.
We wrap up the term looking towards a future of Bio-Design and Bio-Fabrication.
Homework: Finish your Final Project
Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners) Worked on final project and finished the slides on time!
Subsections of Homework
Week 1 HW: Principles and Practices
Class Assignment 𓅨
First, describe a biological engineering application or tool you want to develop and why.
I want to develop a 3D Bio-Art Platform that merges biological growth with interactive synthetic biology. The idea is to use 3D-printed molds and structured agar media to create “living sculptures” that don’t just sit there but actually “feel” and react.
The sculpture uses a quorum sensing circuit to create organic, emergent color gradients as the bacteria colonize the 3D agar structure. However, by engineering the bacteria with inducible promoters sensitive to microcurrents, heat, or other factors, the sculpture reacts to human and environmental touch. When you touch a specific plate, the bacteria trigger a rapid flash of bioluminescence or a sharp color change. It’s a very solarpunk vision where the artwork is a living, sensing entity that bridges the gap between autonomous growth and intentional human interaction.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Some of the main goals include the following:
A. Preventing Malicious Use & Biological Escape (Biosecurity: To ensure that the bacteria used in the sculptures cannot be extracted and repurposed or survive outside the controlled art environment. This could be achieved with the help of:
An Intrinsic Biological Lock: Implementing a strategy where the metabolic reagents and the bacterial chassis are only viable inside a specific chemical or mechanical environment of the 3D bio-art sculpture.
Genetic Safeguards: Using “kill switches” so the organisms are biologically incapable of surviving in the local ecosystem if the sculpture is broken, archived, or discarded.
Access Control & Registry: Establishing a “Bio-Art registry” where any high-expression or highly interactive strain is registered and tracked from the lab to the gallery or art exposition.
B. End User Safety & Interaction Reliability (Biosafety): To guarantee that the interaction between the public and the “living touch” interface is 100% safe, reliable and follows predictable patterns. This could be achieved with the help of:
Interaction Safety Protocols: Establishing clear “bio-etiquette” protocols and adding physical boundaries to prevent accidental ingestion, skin irritation from undesired contact, or environmental transfer during public exhibitions. Also, establishing risk protocols and measures for any accidents or incidents that could happen.
Contamination Control: Implementing a strategy to ensure that the emergent bacteria patterns are not contaminated by other wild-type bacteria from the users’ hands, which could ruin the artistic expression, 3D bio-art sculpture, and the biosafety protocols.
Real-time Stability Monitoring: Integrating “self-reporting” circuits and sensors where bacteria change to a “warning color” (like a bright red or yellow) if the population begins to mutate or if the containment is failing.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Multi-Layered Kill Switches (Technical Strategy that can be applied through international organization like WHO, ASM, etc)
Purpose: Currently, containment is mostly physical. In this strategy, all interactive bio-art must use a “dead-end” genetic design.
Design: Using nutritionally dependent strains that require a synthetic, non-canonical nutrient embedded in the agar. Without this “artificial food,” the bacteria degrades immediately.
Assumptions: We assume that horizontal gene transfer in the environment won’t provide the bacteria a way to bypass this dependency.
Risks: A “success” might make the biology too fragile for long exhibitions but in a controlled manner, while a failure would be the organism finding a natural substitute for the synthetic nutrient, which could lead to unwavering growth.
Action 2: Public Interaction “Bio-Etiquette” Certification (New Requirement that is applied by the responsible company)
Purpose: To change how the public views OGM interaction from “dangerous” or “uncertain” to “responsible” and “reliable.”
Design: Any gallery exhibiting the 3d bio-art sculptures must implement a mandatory hand-sanitizing and briefing station. The actors here are the gallery owner and the artist.
Assumptions: We assume that the public will follow all instructions and not try to “vandalize” the sculpture by introducing outside contaminants.
Risks: Success creates a safe, educated public; failure is a “success” where the art becomes so popular that the safety protocols are ignored due to high traffic.
Action 3: Peer-Led Biosecurity Audit (Community Strategy that involves the public and synbio community, the artists and the responsible company)
Purpose: To move away from slow federal oversight and use the agility of the SynBio community locally and globally.
Design: A “Safety Buddy” system where a fellow scientist must audit the genetic circuits and the physical mold design before it leaves the lab.
Assumptions: We assume peers will be rigorous and not just let their friends’ projects go on without revising them.
Risks: Success builds a strong self-regulating culture. Failure is a lapse in judgment that leads to a public health scare, potentially getting bio-art banned or detained.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the action:
Action 1
Action 2
Action 3
Enhance Biosecurity
• By preventing incidents
1
2
2
• By helping respond
2
2
1
Foster Lab Safety
• By preventing incident
1
n/a
1
• By helping respond
3
n/a
2
Protect the environment
• By preventing incidents
1
2
2
• By helping respond
2
2
3
Other considerations
• Minimizing costs and burdens to stakeholders
3
1
1
• Feasibility?
2
1
2
• Not impede research
1
1
1
• Promote constructive applications
1
2
1
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the scored framework, I recommend that we prioritize action 1 (Technical Multi-Layered Kill Switches) as the foundation, supported by action 3 (Community Peer Led Biosecurity Audit).
The technical multi-layered kill switches are the only way to ensure the biology is ethical by design; if the bacteria can’t survive outside the mold, the “risk” is effectively zero. However, I’m trading off some technical simplicity for absolute peace of mind. On the other hand, the peer led biosecurity audit is important because it builds the “social tissue” of responsibility among us students. We don’t need more laws; we need better engineers and technicians who check each other’s work. Lastly, my biggest uncertainty is the mutation rate of the kill switches, which is why the community audit must be a recurring process, with constant feedback loops and not a one-time thing.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
This project made me realize that when we make biology “interactive” and “eye-catching,” we might lower people’s guard. However, a concern that arose was about the ethical autonomy of the biological parts of 3d bio-art: are we just “enslaving” these bacteria for a 3-second glow? Or are we letting them decide what is best for them? By using Action 3, we ensure that as artists and scientists, we are also managers of the life we modify, treating it with the respect and conscience it deserves.
Week 2 HW: DNA read, write & edit
Week 2 Lecture Prep
Homework Questions from Professor Jacobson:
1) Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of polymerase is 1 in 106 compared to the ~3.2 billion bp of the human genome. This means that the polymerase makes 3200 errors each time it replicates. Biology manages this discrepancy through DNA repair mechanisms, such as real-time proofreading and post-replication mismatch repair (MutS Repair system).
2) How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
There are more than exponential ways to code an average protein (~1036 bp) due to genetic code redundancy. In practice, many codes do not work because the sequences can fold into minimum free energy secondary structures (like hairpins) that interfere with the system, or they may trigger specific RNA cleavage rules that degrade the message.
Homework Questions from Dr. LeProust:
1) What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligonucleotide synthesis currently is solid-phase phosphoramidite chemistry. This method builds DNA chains through a repeating four-step cycle: coupling with phosphoramidite, capping unreacting sites, oxidation and deblocking. These steps are iterated n times and are usually performed on a solid support like, for example, a silicon chip.
2) Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult to make oligos longer than 200 nucleotides via direct synthesis because of the cumulative error rate. Even though each coupling step has a very high efficiency (>99%), the total yield rapidly decreases as nucleotide length increases. Even a 1% error rate per step would result in almost no full-length, error-free product. Additionally, side reactions and incomplete deprotection create truncated sequences that are difficult to purify from the target molecule.
3) Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct synthesis of a 2000bp gene is impossible because the chemical method cannot maintain the necessary precision over thousands of steps. Instead, scientists use hierarchical assembly. They synthesize many smaller oligos and then “glue” them together using enzymatic methods like Polymerase Cycling Assembly (PCA) or Gibson Assembly to reach the full bp length.
Homework Question from George Church:
1) [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in all animals are phenylalanine, valine, threonine, tryptophan, isoleucine, methionine, histidine, arginine, leucine, and lysine (Wu, 2014). Since lysine is already an essential amino acid, the “Lysine Contingency” in Jurassic Park is redundant because animals (dinosaurs included) naturally lack the metabolic pathways to produce it and would need to obtain it from their environment. A more effective approach that they could have used is to make them dependent on synthetic amino acids that don’t exist in nature with the help of synthetic biology.
Wu, G. Dietary requirements of synthesizable amino acids by animals: a paradigm shift in protein nutrition. J Animal Sci Biotechnol 5, 34 (2014). https://doi.org/10.1186/2049-1891-5-34
AI citation: I used Gemini to understand better the lecture materials and evaluate the feasibility of the “Lysine Contingency”.
Homework 02
Part 1: Benchling & In-silico Gel Art
I successfully made a Benchling account and imported the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
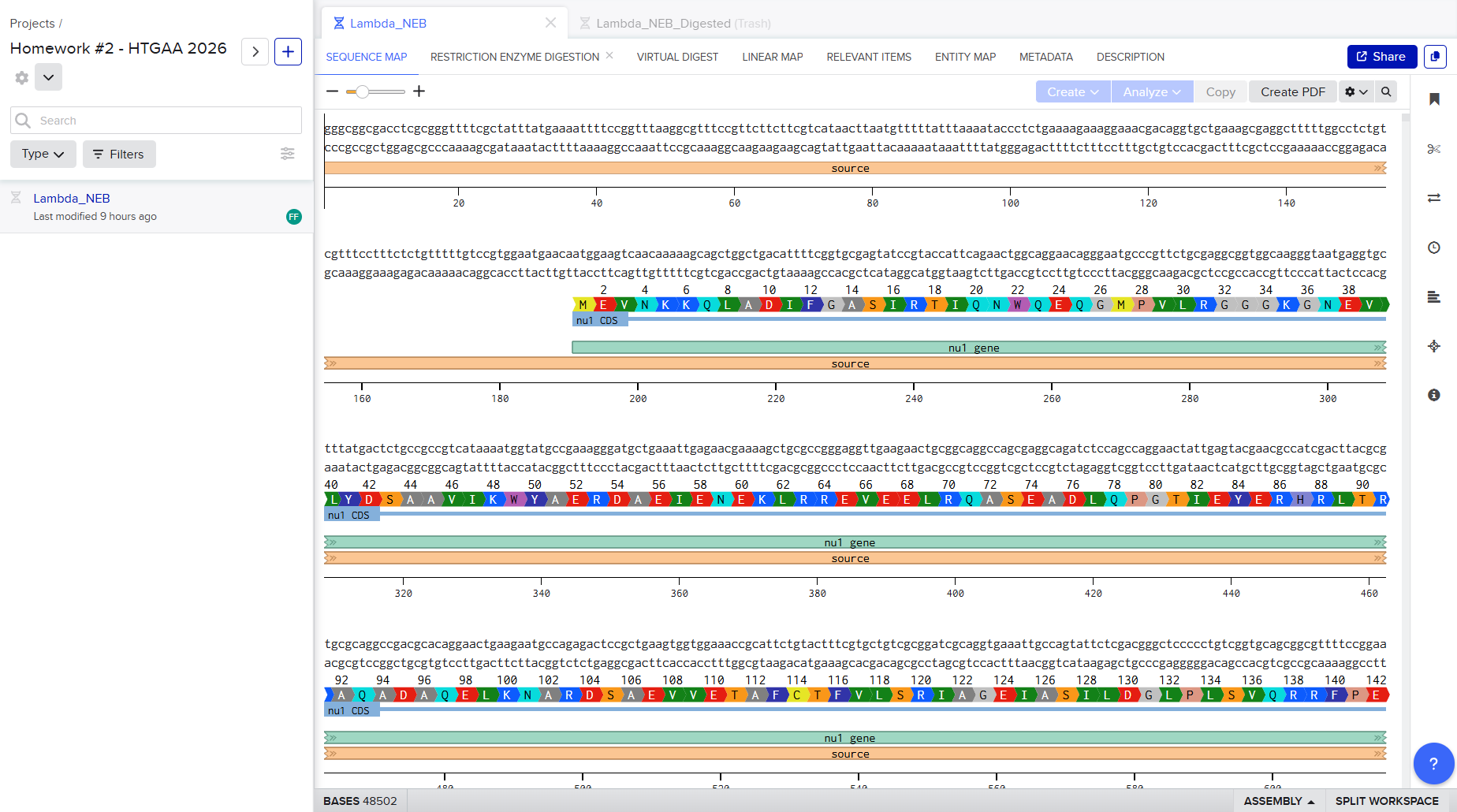
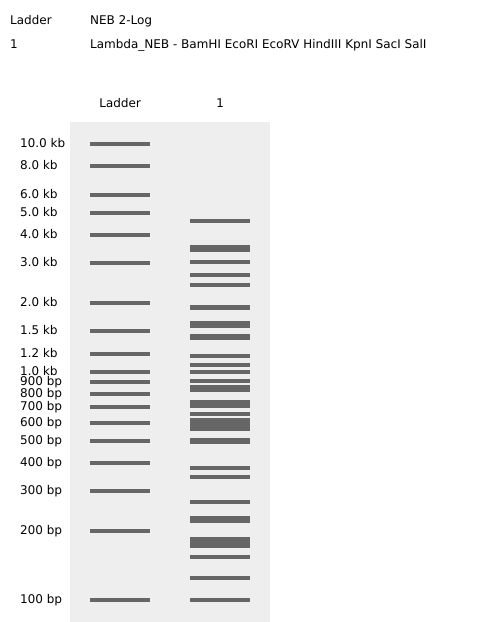
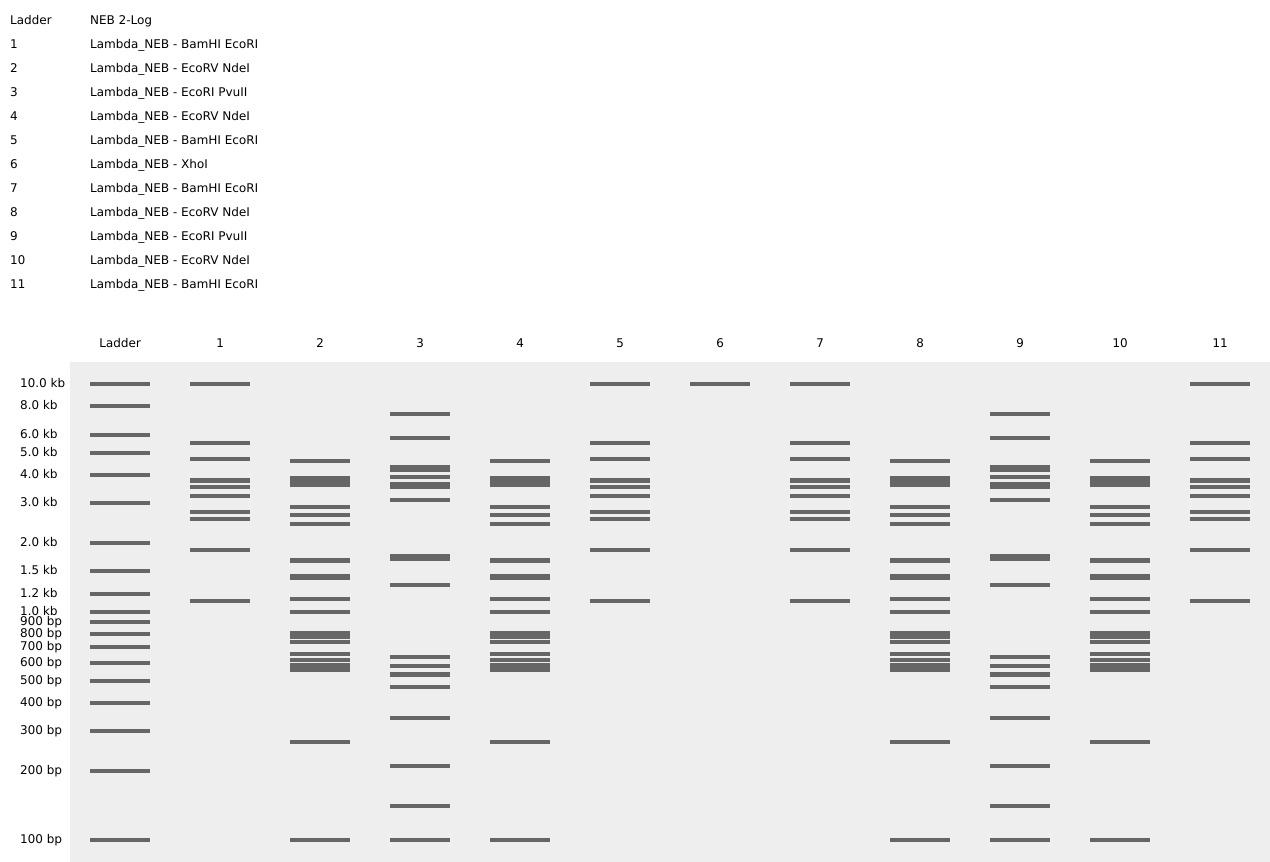
Restriction Enzyme Digestion Simulation using enzymes EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI using Benchling.
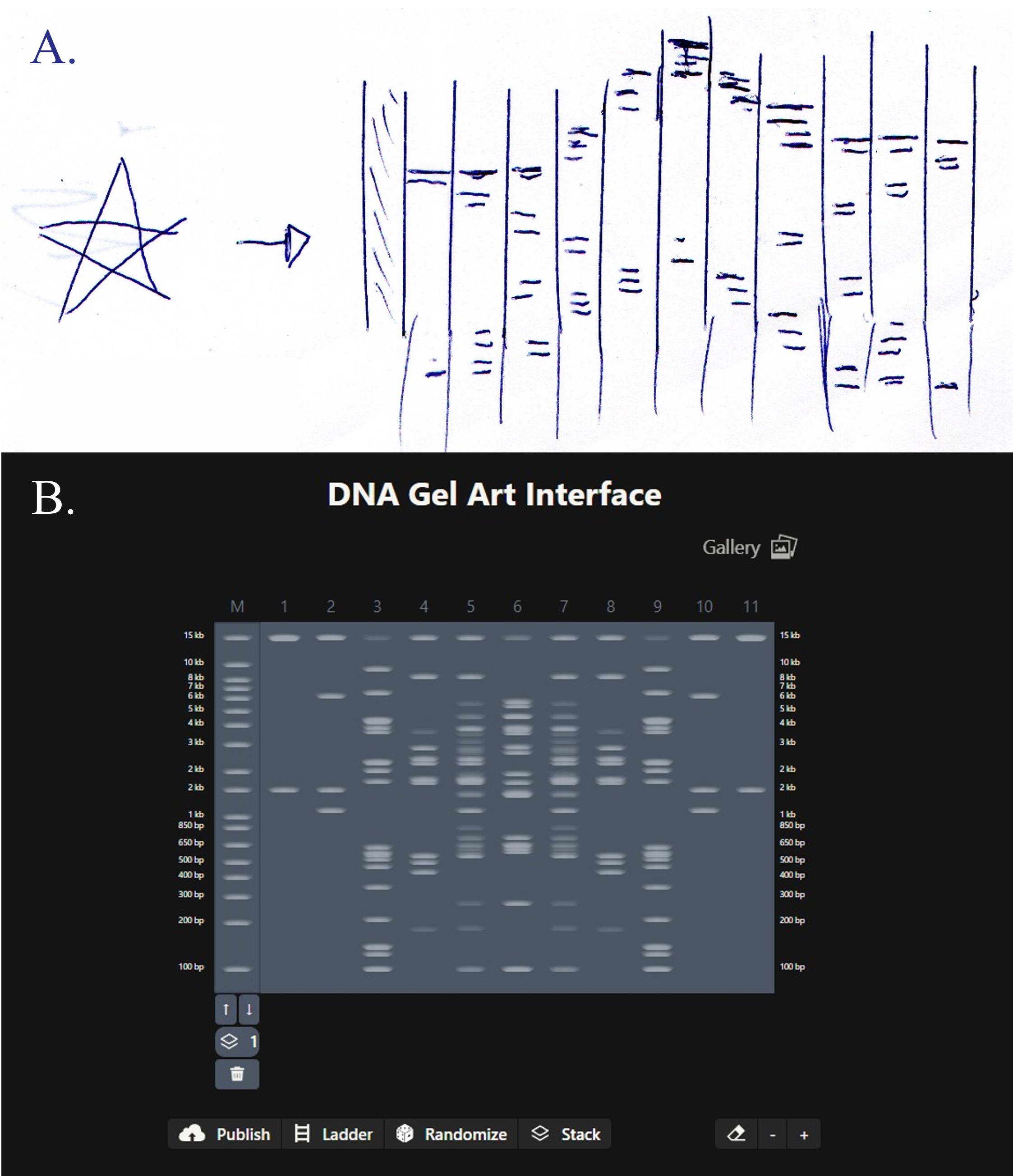
For my in-silico Gel Art I wanted to initially make a star! Sadly, after using Ronan’s website to visualize my idea, I realized that it would be a bit complicated using the listed Restriction Enzymes.
Here is a rough initial sketch for the star and my attempt to do it on Ronan’s website tool
So, I ended up making some tulips instead! You can check out my design on Ronan’s website too!
Here is a picture of the tulips design using Benchling!
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I skipped this one since I do not have Lab access.
Part 3: DNA Design Challenge
3.1. Choose your protein.
I have chosen the Chitinase enzyme from the bacterium Bacillus thuringiensis (NCBI Accession: WCH14858.1).
I found this protein interesting because of its potential in environmental conservation and biotechnology. This enzyme is capable of degrading chitin, which is a primary component of fungal cell walls and insect exoskeletons. Based on the literature, the chitinase protein is particularly efficient due to its modular structure, which typically includes a catalytic domain and chitin-binding domains that enhance its hydrolytic activity (1). Because of that, this protein becomes a very powerful tool for biological control: it can act synergistically with Cry proteins to perforate the peritrophic matrix of insect pests, increasing the efficiency of biopesticides. Additionally, I selected this specific protein because Bacillus thuringiensis is a safe organism to handle in a Level 1 biosafety laboratory (BSL-1), making it a practical and efficient candidate for recombinant protein production in E. coli.
Chitinase protein DNA sequence atgttaaacaagttcaaatttttttgttgtattttagtaatgttcttacttctaccgttatcccctttccaagcacaagcagcaaacaatttaggttcaaaattactcgttggatactggcataattttgataacggtactggcattattaaattaaaagacgtttcaccaaaatgggatgtaatcaatgtatcttttggtgaaactggtggtgatcgttccactgttgaattttctcctgtgtatggtacagatgcagaattcaaatcagatatttcttatttaaaaagtaaaggaaagaaaatagttctttcaataggtggacaaaatggggtcgttttacttcctgacaatgccgctaaggatcgttttattaattccatacaatctctgatcgataaatacggttttgacggaatagatattgaccttgaatcaggtatttacttaaacggaaatgacactaacttcaaaaacccaactacccctcaaatcgtaaatcttatttcagctattcgaacaatctcagatcattatggtccagattttctattaagcatggcccctgaaacagcttatgttcaaggcggatatagcgcatatggaagcatatggggtgcatatttaccaattatttatggagtgaaagataaactaacatacattcacgttcaacactacaacgctggtagcgggattggaatggacggtaataactacaatcaaggtactgcagactacgaggtcgctatggcagatatgctcttacatggttttcctgtaggtggtaatgcaaataacattttcccagctcttcgttcagatcaagtcatgattgggcttccagcagcaccagcggcagctccaagtggtggatacatttcgccaactgaaatgaaaaaagctttaaattatatcattaaaggagttccattcggaggaaagtataaactttctaaccagagtggctatcctgcattccgcggcctaatgtcttggtctattaattgggatgcaaaaaacaactttgaattctctagtaactatagaacatattttgatggtctttccttgcaaaaataa
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
I need to optimize codon usage because, although the genetic code is redundant, different organisms have distinct ‘codon biases.’ Since I am using a sequence from Bacillus thuringiensis, I have optimized it for Escherichia coli K-12 using Benchling’s Codon Optimization tool to ensure that the host cell can translate it efficiently. I chose the K-12 strain specifically because it is the gold standard in synthetic biology laboratories since it is a safe, non-pathogenic, and well-characterized model that guarantees reliable folding for my chitinase enzyme.
Chitinase protein DNA sequence Codon-Optimization ATGCTGAACAAATTTAAATTTTTTTGCTGCATTCTGGTGATGTTTCTGCTGCTGCCGCTGAGCCCGTTTCAGGCGCAGGCGGCGAACAACCTGGGCAGCAAACTGCTGGTGGGCTATTGGCATAACTTTGATAACGGCACCGGCATTATTAAACTGAAAGATGTGAGCCCGAAATGGGATGTGATTAACGTGAGCTTTGGCGAAACCGGCGGCGATCGCAGCACCGTGGAATTTAGCCCGGTGTATGGCACCGATGCGGAATTTAAAAGCGATATTAGCTATCTGAAAAGCAAAGGCAAAAAAATTGTGCTGAGCATTGGCGGCCAGAACGGCGTGGTGCTGCTGCCGGATAACGCGGCGAAAGATCGCTTTATTAACAGCATTCAGAGCCTGATTGATAAATATGGCTTTGATGGCATTGATATTGATCTGGAAAGCGGCATTTATCTGAACGGCAACGATACCAACTTTAAAAACCCGACCACCCCGCAGATTGTGAACCTGATTAGCGCGATTCGCACCATTAGCGATCATTATGGCCCGGATTTTCTGCTGAGCATGGCGCCGGAAACCGCGTATGTGCAGGGCGGCTATAGCGCGTATGGCAGCATTTGGGGCGCGTATCTGCCGATTATTTATGGCGTGAAAGATAAACTGACCTATATTCATGTGCAGCATTATAACGCGGGCAGCGGCATTGGCATGGATGGCAACAACTATAACCAGGGCACCGCGGATTATGAAGTGGCGATGGCGGATATGCTGCTGCATGGCTTTCCGGTGGGCGGCAACGCGAACAACATTTTTCCGGCGCTGCGCAGCGATCAGGTGATGATTGGCCTGCCGGCGGCGCCGGCGGCGGCGCCGAGCGGCGGCTATATTAGCCCGACCGAAATGAAAAAAGCGCTGAACTATATTATTAAAGGCGTGCCGTTTGGCGGCAAATATAAACTGAGCAACCAGAGCGGCTATCCGGCGTTTCGCGGCCTGATGAGCTGGAGCATTAACTGGGATGCGAAAAACAACTTTGAATTTAGCAGCAACTATCGCACCTATTTTGATGGCCTGAGCCTGCAGAAATAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To produce chitinase from my designed sequence, I can use either cell-dependent or cell-free methods. In a cell-dependent approach, I would insert the DNA into a host like E. coli K-12, where the cell’s own machinery handles the work: RNA polymerase transcribes the DNA into mRNA, and then ribosomes translate that message into the final enzyme. On the other hand, cell-free protein synthesis allows me to skip the living cell entirely by using just the necessary biological “parts” (like enzymes and ribosomes) in a tube. This last approach is a much faster way to prototype the protein without keeping bacteria alive, although I really have a space in my heart for bacterial cultures.
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
From what I’ve understood, a single gene can produce different proteins through mechanisms like alternative splicing, where the cell mixes and matches different sections of the message (exons) to create several versions of a protein from the same DNA template. In bacteria like Bacillus thuringiensis, they also use polycistronic operons, which group several related genes under a single promoter. This allows the bacteria to produce a whole set of coordinated enzymes all at once.
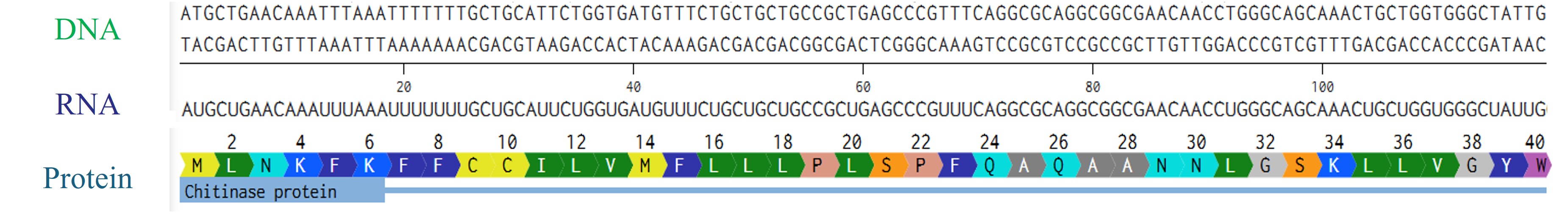
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!!
Rearranged snapshot of Chitinase protein information flow from DNA to RNA to protein. Captured from Fabri’s Benchling and arranged in PowerPoint
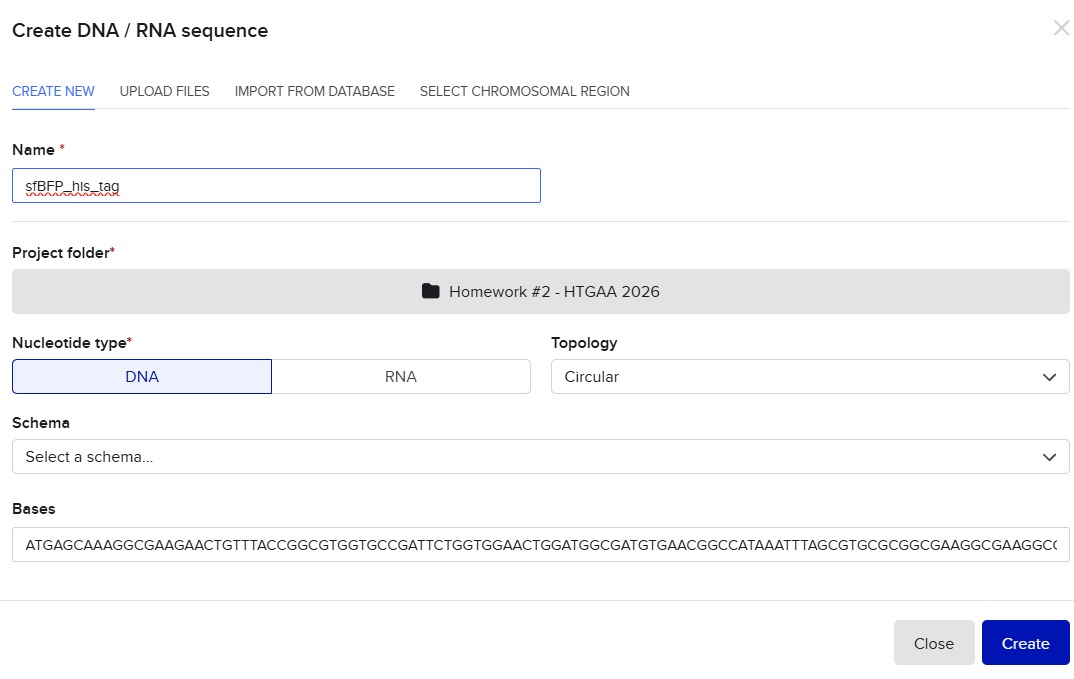
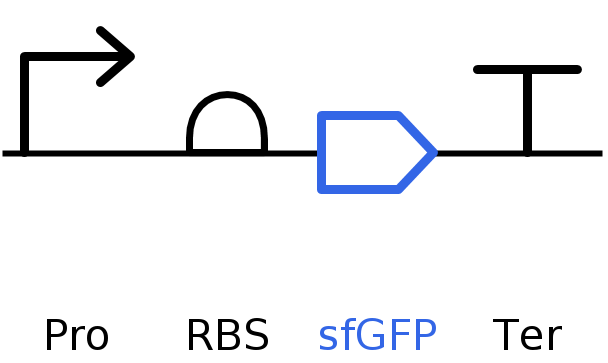
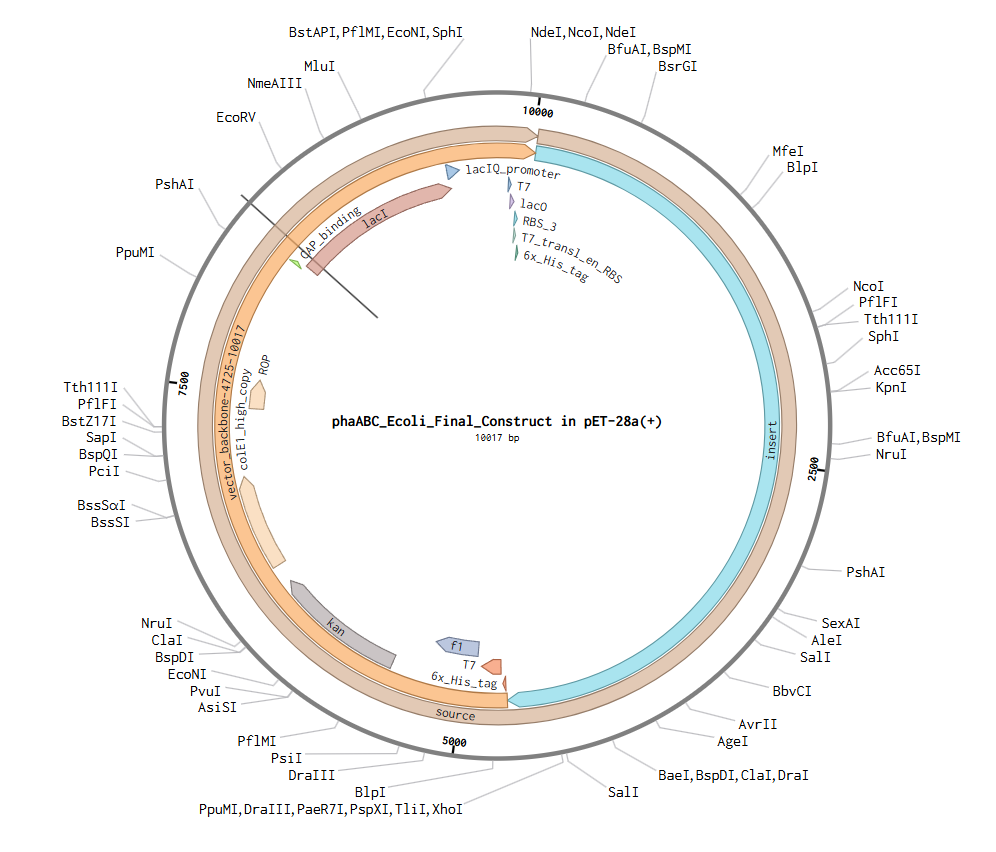
I’ll make a sequence that will make E. coli glow fluorescent blue under UV light by always expressing sfBFP (a blue fluorescent protein):
Screenshot of the creation of the sfBFP sequence in Benchling
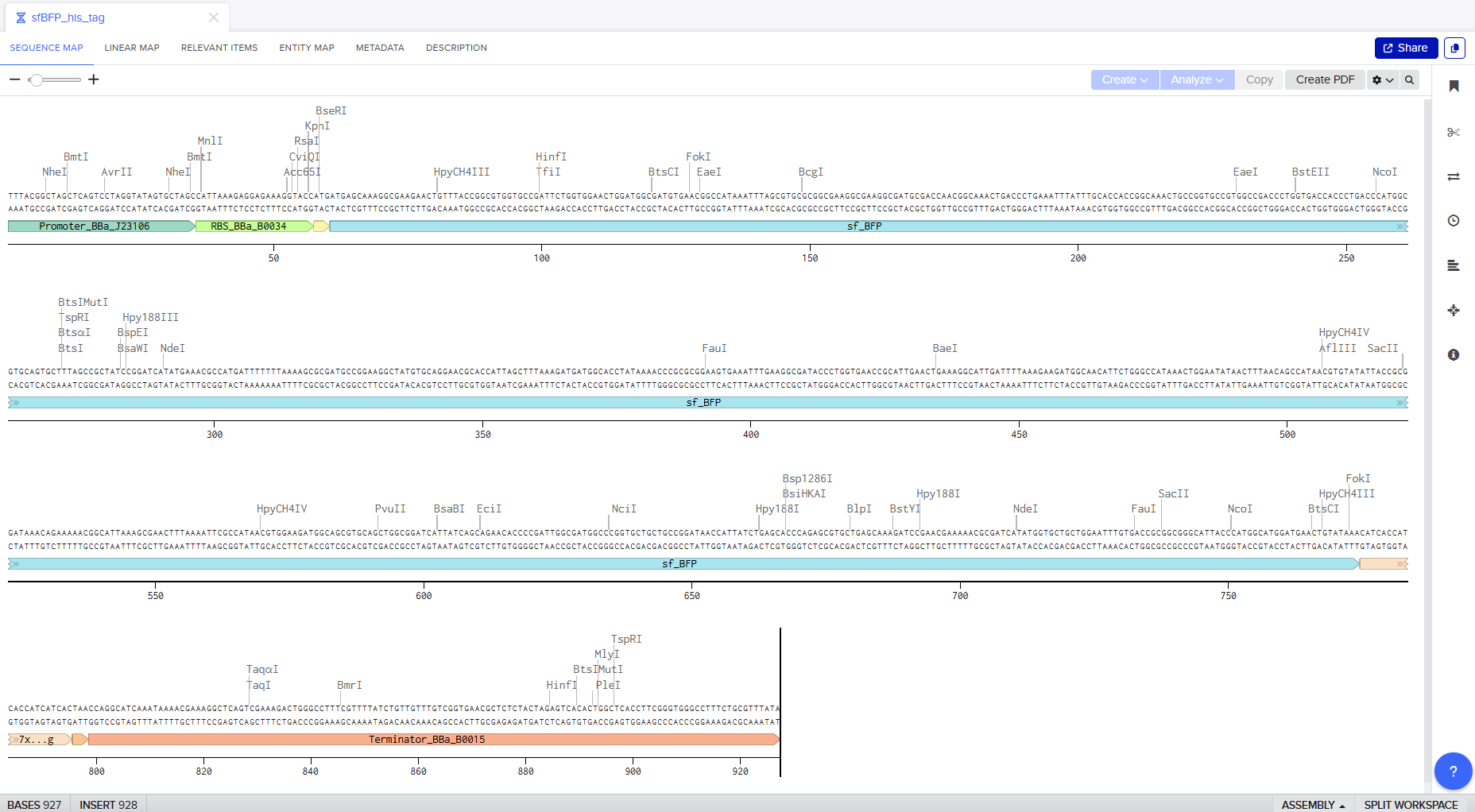
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
Screenshot of the whole sequence with its annotations!
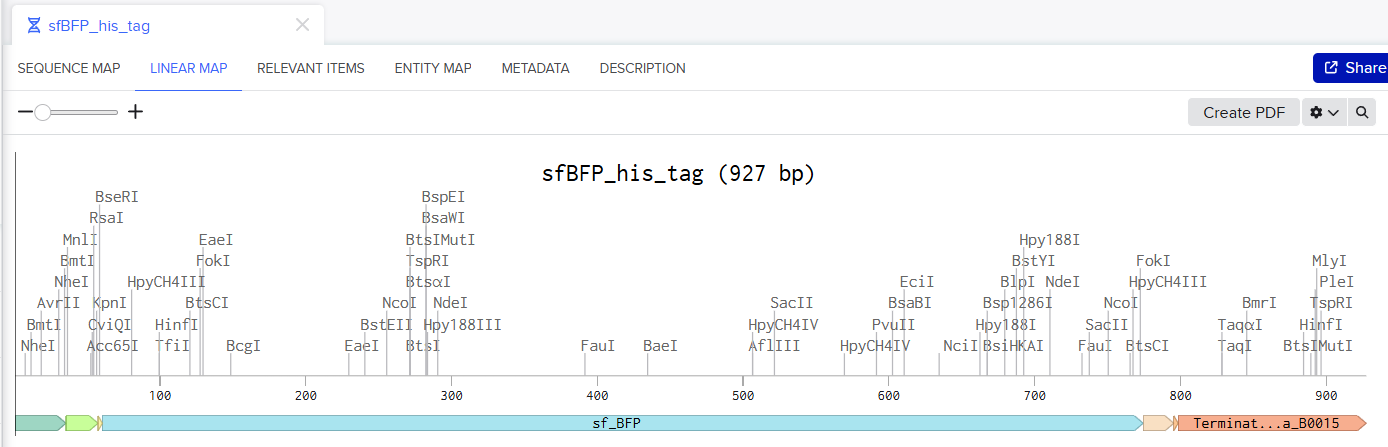
Screenshot of the Linear map of Constitutive sfBFP DNA and here is the Benchling Link
SBOL of the Linear map of Constitutive sfBFP DNA.
4.3. On Twist, Select The “Genes” Option
4.4. Select “Clonal Genes” option
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.5. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
Screenshot of my uploaded sfBFP FASTA file in Twist
4.6. Choose Your Vector
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
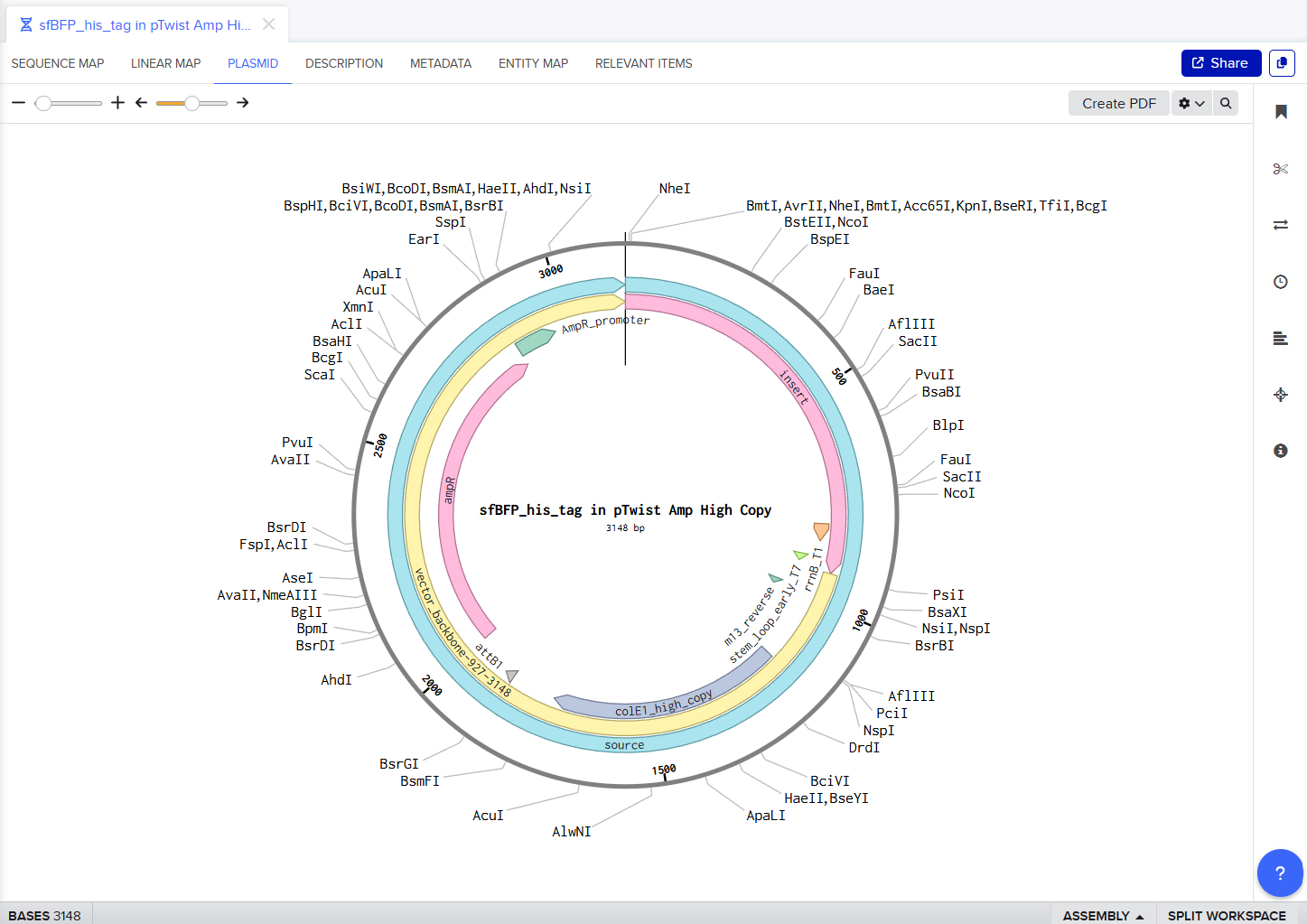
Screenshot of sfBFP with pTwist Amp High Copy vector
My Twist Ready Plasmid!!
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I want to sequence eDNA from river water samples collected at different points in different regions, especially near my hometown. Rivers collect DNA from fish, amphibians, and even terrestrial animals that drink from or live near the water. By sequencing the DNA, I can perform a biodiversity assessment to detect invasive species (like the trout in some Andean rivers) and/or monitor the presence of endangered amphibians without the need for traditional trapping methods.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?\
I would use Illumina (Next-Generation Sequencing) because its massive parallelization would allow me to read millions of sequences from hundreds of species in a single run, which is perfect for complex environmental samples (e.g., in rivers).
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
Illumina’s NGS is second-generation. That’s because it uses synthesis-based sequencing on a solid surface rather than reading single long molecules.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
My input would be filtered river water DNA. Preparation involves metabarcoding (amplifying specific markers like 16S or COI) and adapter ligation to attach fragments to the flow cell.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Illumina’s NGS has many steps but these are the essential ones that make the work itself. First, DNA fragments are attached to a flow cell where they form dense clusters through bridge amplification to ensure the detection signal is strong enough. Next, fluorescently labeled nucleotides are added one by one, and a high-resolution camera records the specific color flash emitted as each base is incorporated into the strand. Finally, the software interprets these light patterns and decodes them into a digital DNA sequence through base calling. (2)
What is the output of your chosen sequencing technology?
The output of Illumina’s NGS is a FASTQ file containing millions of digital reads that identify the species present in the river samples. Once I get the file I can analyze it. with bioinformatics and get results.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would like to synthesize a genetic biosensor designed to detect heavy metal contamination, such as mercury, in river water. By placing this circuit into a safe host like E. coli K-12, the bacteria could “glow” or change color when it senses toxins, acting as a real-time environmental monitor to help protect the river’s biodiversity.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?\
I would love to use Twist Bioscience’s Silicon-based Synthesis to perform the DNA synthesis because of its incredible scalability and its promise of making DNA synthesis better and faster. (3)
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
The steps that Twist follows use silicon chips to print thousands of genes simultaneously, which significantly reduces costs and improves precision. First, the digital sequence is uploaded and ‘printed’ onto a silicon chip; using phosphoramidite chemistry, the machine builds thousands of short DNA strands, known as oligonucleotides, base by base. Second, these short oligos are harvested from the chip and gathered together. Finally, the fragments are enzymatically assembled to form the complete, full-length biosensor circuit, ensuring high precision and scalability.
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The main limits are that very complex designs can significantly increase the turnaround time and the cost of production. Additionally, sequences with difficult content, such as high GC-rich regions, can lower the synthesis success rate.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would like to edit the chitinase genes in native river bacteria to make them more efficient at degrading organic waste. This would help prevent fungal outbreaks and the accumulation of debris, keeping the river ecosystem balanced and clean in a natural way.
(ii) What technology or technologies would you use to perform these DNA edits and why?\
I would use CRISPR-Cas9 because it is the most precise, well-known, and easy-to-design tool for genome engineering in bacteria. The system works by using a guide RNA (gRNA) that leads the Cas9 nuclease to a specific target in the chitinase gene to create a cut. By providing a DNA repair template, I can then insert a more efficient version of the enzyme into the genome.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
This technology edits DNA by acting like a pair of molecular scissors. It follows three main steps: first, the guide RNA identifies and binds to a specific target sequence in the genome. Second, the Cas9 nuclease creates a double-strand break at that exact location. Finally, the cell’s natural repair machinery goes and fixes the break; by providing a DNA repair template, the cell can be tricked into incorporating a new, more efficient chitinase sequence during this repair process.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
I would need to digitally design a specific gRNA that is perfectly complementary to the chitinase gene to avoid off-target cuts. Additionally, the required inputs for the experiment include the Cas9 protein (or a plasmid encoding it), the custom synthetic gRNA, a DNA donor template containing the desired edit, and the target bacterial cells that will be transformed with these components!
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
The biggest limitation of this method is the risk of off-target cuts, where the Cas9 might cut a similar DNA sequence elsewhere in the genome by mistake. Additionally, the efficiency of the edit depends a lot on the cell’s repair mechanism; in some bacteria, the rate of successful “homology-directed repair” can be low, meaning many cells might fail to incorporate the new gene correctly.
References
Martínez-Zavala, S. A., Barboza-Pérez, U. E., Hernández-Guzmán, G., Bideshi, D. K., & Barboza-Corona, J. E. (2020). Chitinases of Bacillus thuringiensis: Phylogeny, Modular Structure, and Applied Potentials. Frontiers in Microbiology, 10, 3032. https://doi.org/10.3389/fmicb.2019.03032
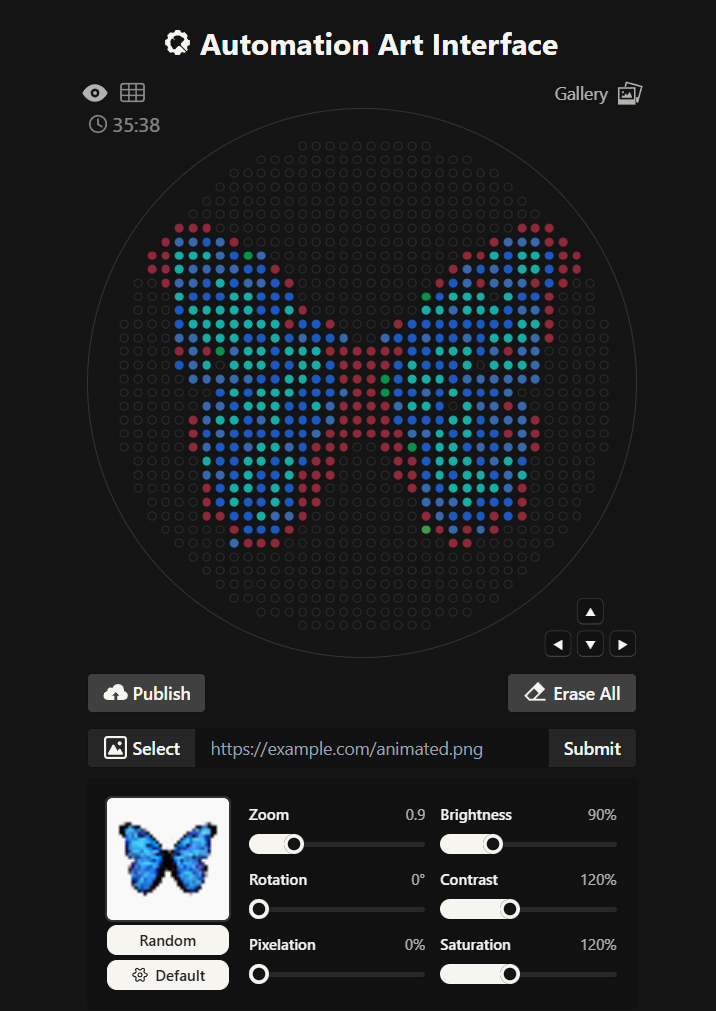
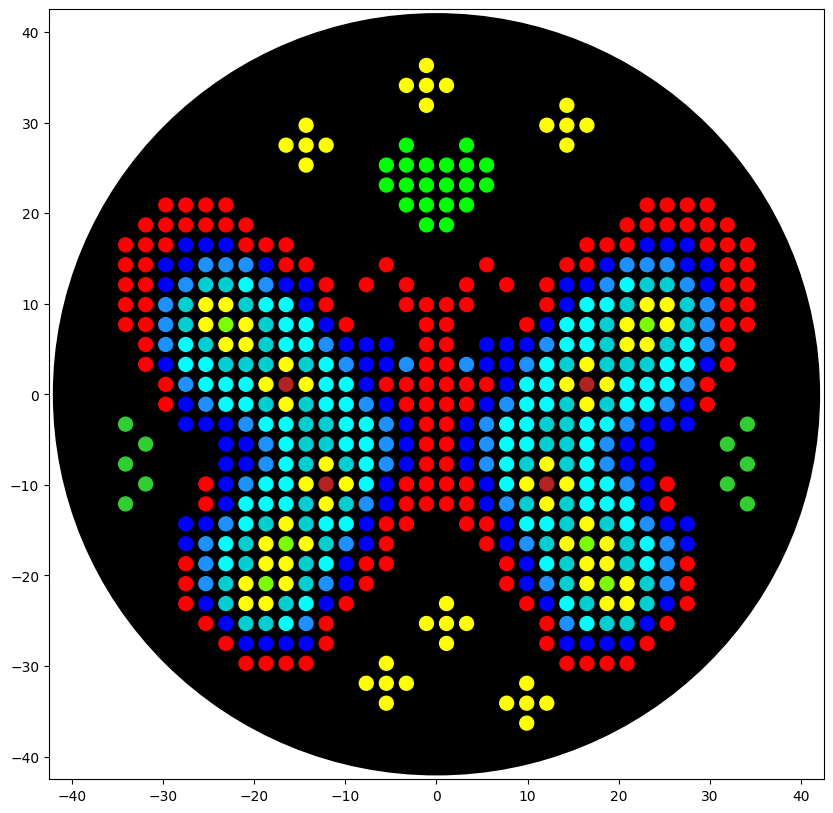
For my first design I made a colorful butterfly! I first used the Opentrons art page to design it by using the upload image option. Initially the design
Here you can see the butterfly image that I uploaded and how it generates on the Opentrons art page side by side!
Then after the image upload, I decided to first move the design a bit lower and also change the colors. Lastly I added some fun details like stars and a heart.
Design process
Here’s the final design!
This is my design: a colorful butterfly! Made using the GUI. You can check it out by yourself here!
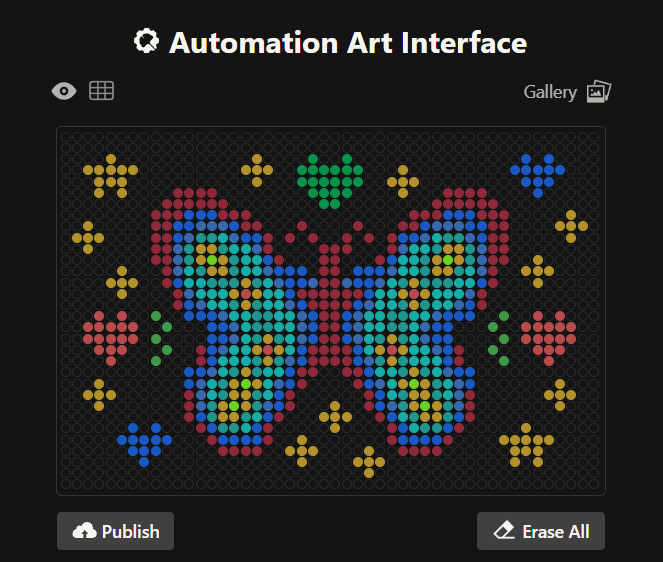
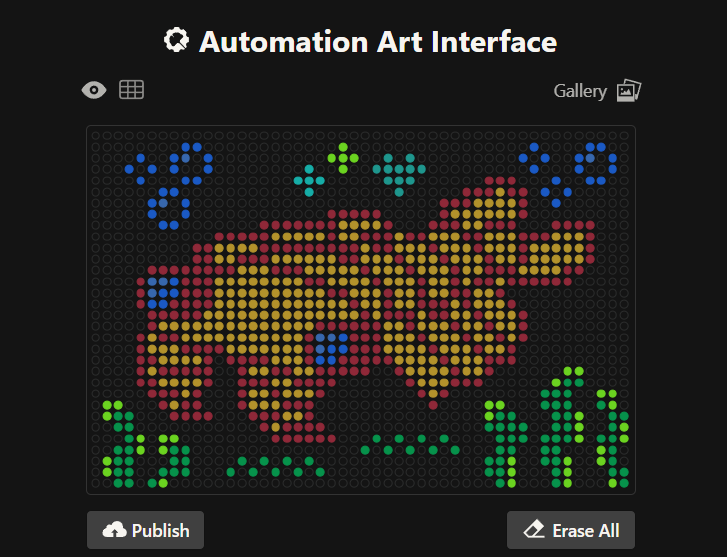
Initially, I made one artistic design on a circular petri dish, but after finding out you could make designs on a rectangular plate, I decided to try it out! I ended up making 2 more designs on rectangular plates.
This is my second design which is a readaptation of the colorful butterfly! Made using the GUI. You can check it out by yourself here!
This is my third design: an anomalocaris! Made using the GUI. You can check it out by yourself here!
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Here’s the Opentrons Lab Simulation in Google Colab for the first design. You can check it out by yourself here!
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Paper: Automation of protein crystallization scale-up via Opentrons-2 liquid handling.
This paper explores the use of the Opentrons OT-2 machine to automate protein crystallization! The researchers developed three Python scripts using the Opentrons Python module to control the robot for mixing and setting up 24-well sitting drop plates using model proteins like lysozyme and a periplasmic protein from Campylobacter jejuni.
The study achieved the desired scale-up goals after minimal trial and error. By automating the liquid handling, the researchers were able to test a wider range of crystallization conditions (reagents, concentrations, and pH) with higher reproducibility than manual pipetting. Although the setup time was around 35 to 40 minutes, it greatly reduces plate variability from person to person. This is a novel application because it makes high-quality structural biology workflows accessible and low-cost, allowing labs to screen protein conditions at a much higher throughput, which is essential for understanding protein function and drug design.
Reference:
DeRoo, J. B., Jones, A. A., Slaughter, C. K., Ahr, T. W., Stroup, S. M., Thompson, G. B., & Snow, C. D. (2025). Automation of protein crystallization scaleup via Opentrons-2 liquid handling. SLAS Technology, 32, 100268. https://doi.org/10.1016/j.slast.2025.100268
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
For instance, my first idea is an interactive 3D bio-art installation that translates a person’s biological data into a living and blooming sculpture. This idea uses genetically engineered bacteria to create a visual representation of a user’s unique microbial/DNA fingerprint. The process starts when a user interacts with a sensor that captures basic biological data, which is then processed by a script to assign specific colors using fluorescent proteins like GFP, RFP, and BFP. In this case, an Opentrons OT-2 acts as a high-precision bio-printer to deposit these living bio-inks into a 3D-printed scaffold made of agar or hydrogel, allowing the sculpture to grow and glow over time to reveal the user’s identity.
Additionally, I will need to design 3D-printed holders with micro-channels and a specialized needle adapter so the OT-2 can deposit the bacteria without breaking the hydrogel/agar structure. I will use capacitive touch sensors to generate the initial data that determines the bacterial/DNA distribution throughout the sculpture. Moreover, I plan to use cloud laboratories like Ginkgo Nebula to synthesize the custom DNA circuits needed to ensure the bacteria express the exact colors and intensity required for the piece.
Here’s a rough python pseudocode for this 3D sculpture idea.
fromopentronsimportprotocol_api# This script translates user data into a 3D bacterial patterndefrun(protocol:protocol_api.ProtocolContext):# Load the custom 3D printed lattice and the bio-inks (bacteria)sculpture_lattice=protocol.load_labware('custom_3d_lattice','1')bio_inks=protocol.load_labware('opentrons_24_tuberack_eppendorf_1.5ml_safelock_snapcap','2')p20=protocol.load_instrument('p20_single_gen2','right')# Logic: If user data indicates Trait X, use Blue Fluorescent Proteinuser_trait="high_diversity"# Example data from sensorifuser_trait=="high_diversity":# Deposit Blue bacteria in the outer ring of the latticeforwellinsculpture_lattice.rows()[0]:p20.pick_up_tip()p20.transfer(10,bio_inks['A1'],well,new_tip='never')p20.drop_tip()# Move in Z-axis to create the 3D effectp20.move_to(sculpture_lattice.wells()[0].top(z=10))
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
As explained in this week’s recitation, add a slide in your Node’s section of this slide deck with an idea you have for an Individual Final Project. Be sure to put your name on your slide!
Here are my three individual final project ideas!
An interactive 3D bio-art sculpture
A river-sensing automated robot system
Chlorella vulgaris in silico optimization and automation
Week 4 HW: Protein Design - Part I
Homework: Protein Design I
Objective:
Learn basic concepts:
amino acid structure
3D protein visualization
the variety of ML-based design tools
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
At first, I thought that it would be a simple math conversion, but after a quick internet search, I realized that not every type of meat has the same amount of protein. According to Barr et al. (2025), 100 g of cooked red meat contains ~28–36 grams of protein, and 100 g of cooked white meat contains ~23–31 grams of protein. Because of this, I decided to use 30 grams of protein per 100 grams of meat as an approximation for the calculations.
Since we know that there’s 30 grams of protein per 100 grams of meat, there would be 150 grams of protein in a piece of 500 grams of meat. These 150 grams of protein, then, are divided by 100 Daltons (which is equivalent to 100 g/mol AA) and finally converted into AA molecules, which gives us a result of approximately 9.033 x 10^23 amino acids!
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Everything we eat is broken down into universal micro building blocks (amino acids, lipids, sugars). Our body doesn’t use the cow’s proteins directly; it hydrolyzes them and then uses our own genetic “code” to reassemble those building blocks into human proteins. It’s about the information (DNA), not the source of the bricks.
Why are there only 20 natural amino acids?
It’s a balance between functional diversity and translational fidelity. These 20 provide enough chemical groups to build almost any catalytic or structural site. Adding more amino acids would increase the risk of errors during translation without a significant evolutionary “payoff.”
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
They likely came from abiotic synthesis (like the Miller-Urey experiment) using simple precursors ($CH_{4}$, $NH_{3}$, $H_{2}$, $H_{2}O$) and energy sources like lightning or hydrothermal vents. Also, carbonaceous meteorites (like Murchison) have shown that amino acids can form in space via Strecker synthesis.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Since natural L-amino acids form right-handed helices, a helix made of D-amino acids would be left-handed. It’s a direct mirror image dictated by the stereochemistry of the Cα atom.
Can you discover additional helices in proteins?
Yes, besides the standard α-helix, proteins show other geometries like the tighter &3sub10;-helix or the wider π-helix. We also see Polyproline helices in collagen and can even design synthetic foldamers with helical shapes that do not exist in nature.
Why are most molecular helices right-handed?
It’s mostly due to the L-homochirality of life. Because all biological proteins are made of L-amino acids, the steric clashes between side chains and the backbone favor the right-handed twist as the most thermodynamically stable conformation (lowest energy).
Why do β-sheets tend to aggregate?
Because they have “sticky” edges. The backbone hydrogen-bond donors and acceptors are exposed at the edges of the sheet, inviting other β-strands to join.
What is the driving force for β-sheet aggregation?
Mainly inter-strand hydrogen bonding and the hydrophobic effect, as burying nonpolar side chains between sheets is energetically favorable.
Why do many amyloid diseases form β-sheets?
Many amyloids form β-sheets because it’s the “global energy minimum” for many sequences; the cross-β structure is incredibly stable and protease resistant.
Can you use amyloid β-sheets as materials?
Yes, they can be used as nanoscaffolds for tissue engineering or as conductive nanowires because of their extreme mechanical strength and self-assembling properties.
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
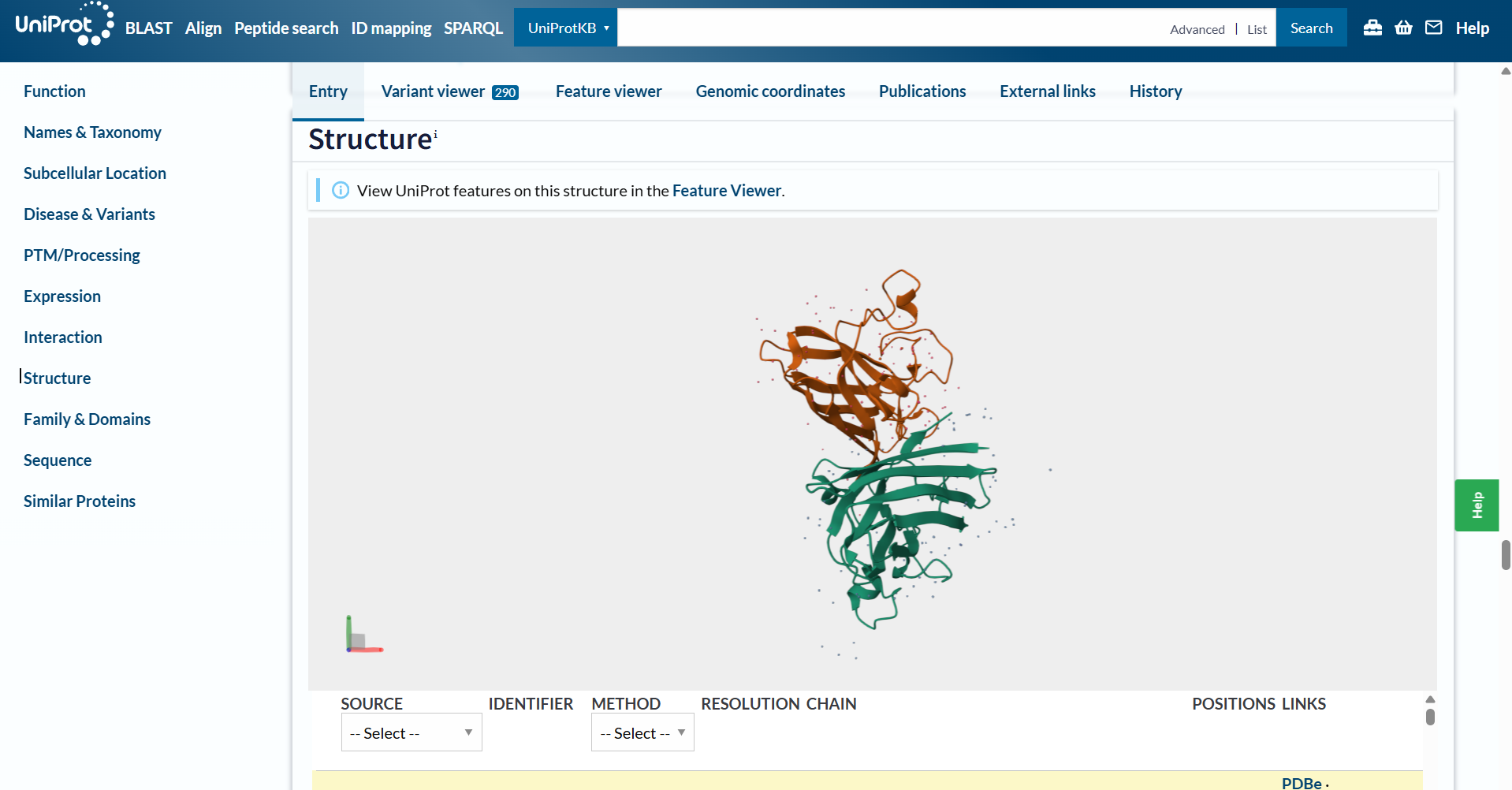
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
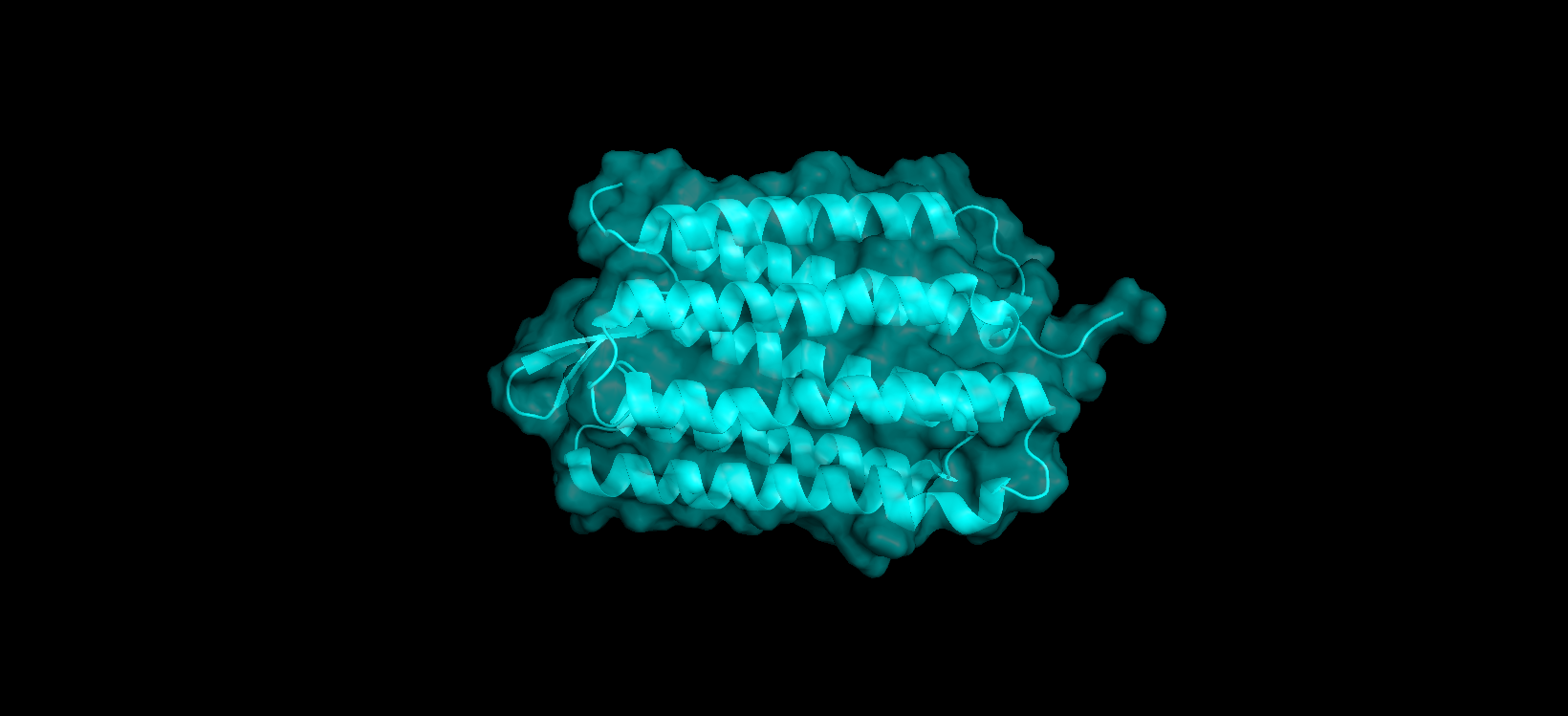
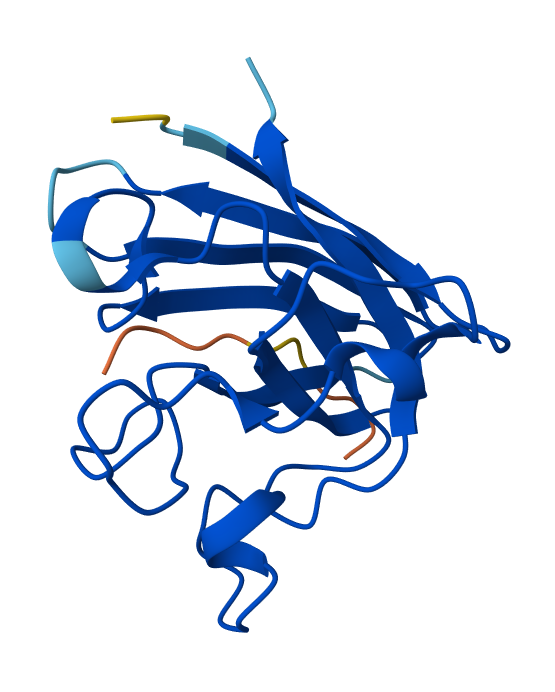
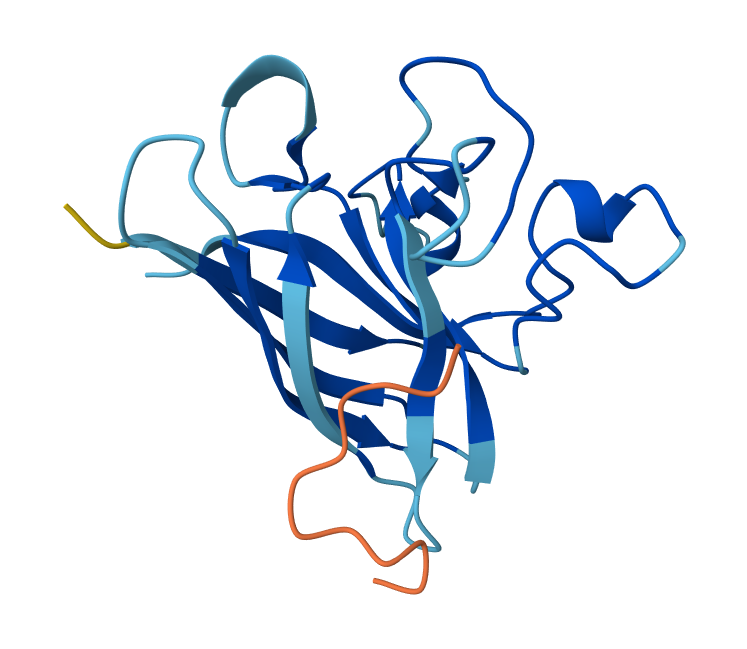
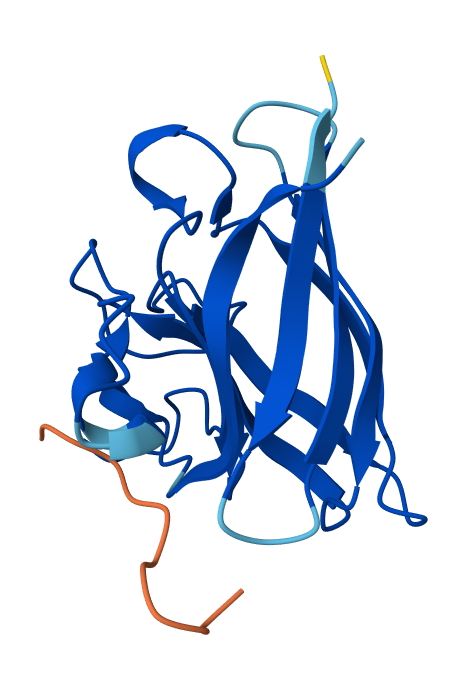
I have selected the bacteriorhodopsin (bR) protein for this part of the homework because of its light conversion cycle and its structure! I am particularly interested in its light-sensitive properties, which offer great potential for applications in bioart. Scientifically, bR is a perfect model for studying single molecule kinetics, as seen in Perrino et al. (2021). Additionally, it provides key insights into membrane protein stability, specifically regarding helical reorganization in the context of membrane protein folding: Insights from simulations with bacteriorhodopsin (BR) fragments (Chatterjee et al., 2024). This combination of biological efficiency and aesthetic potential makes it an ideal choice for my research.
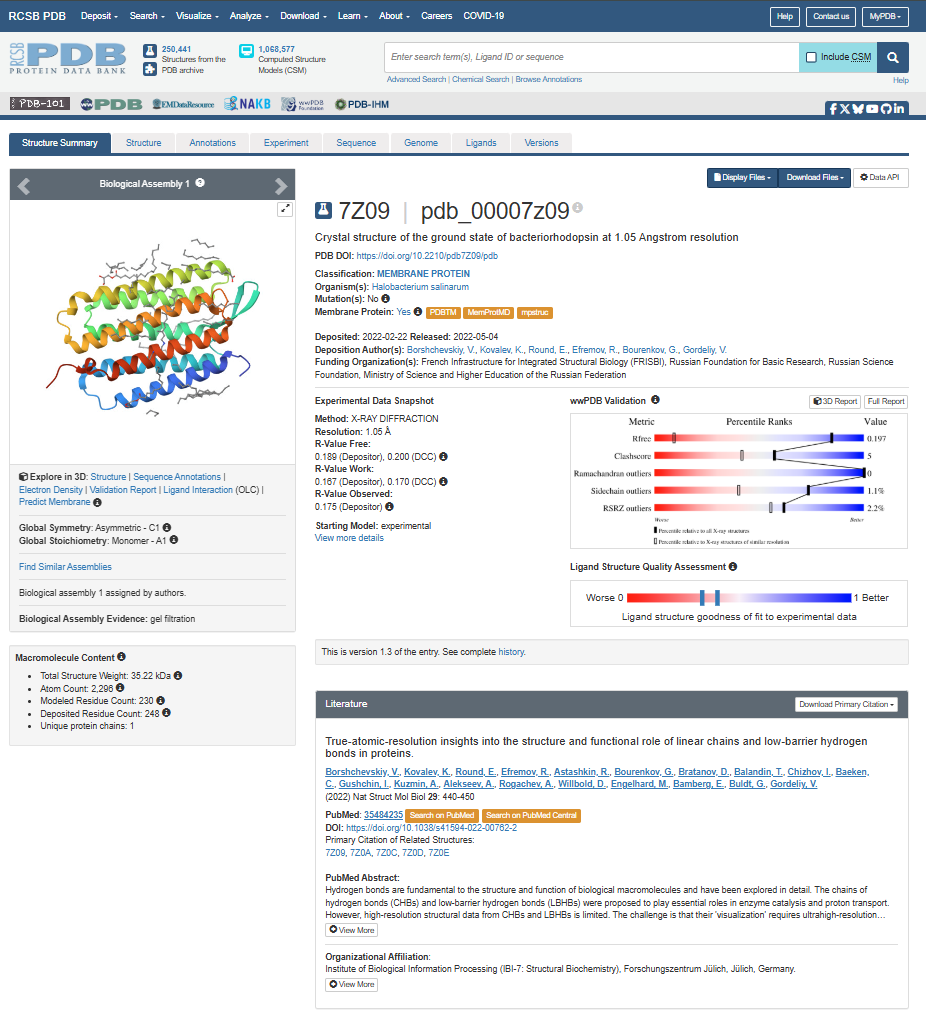
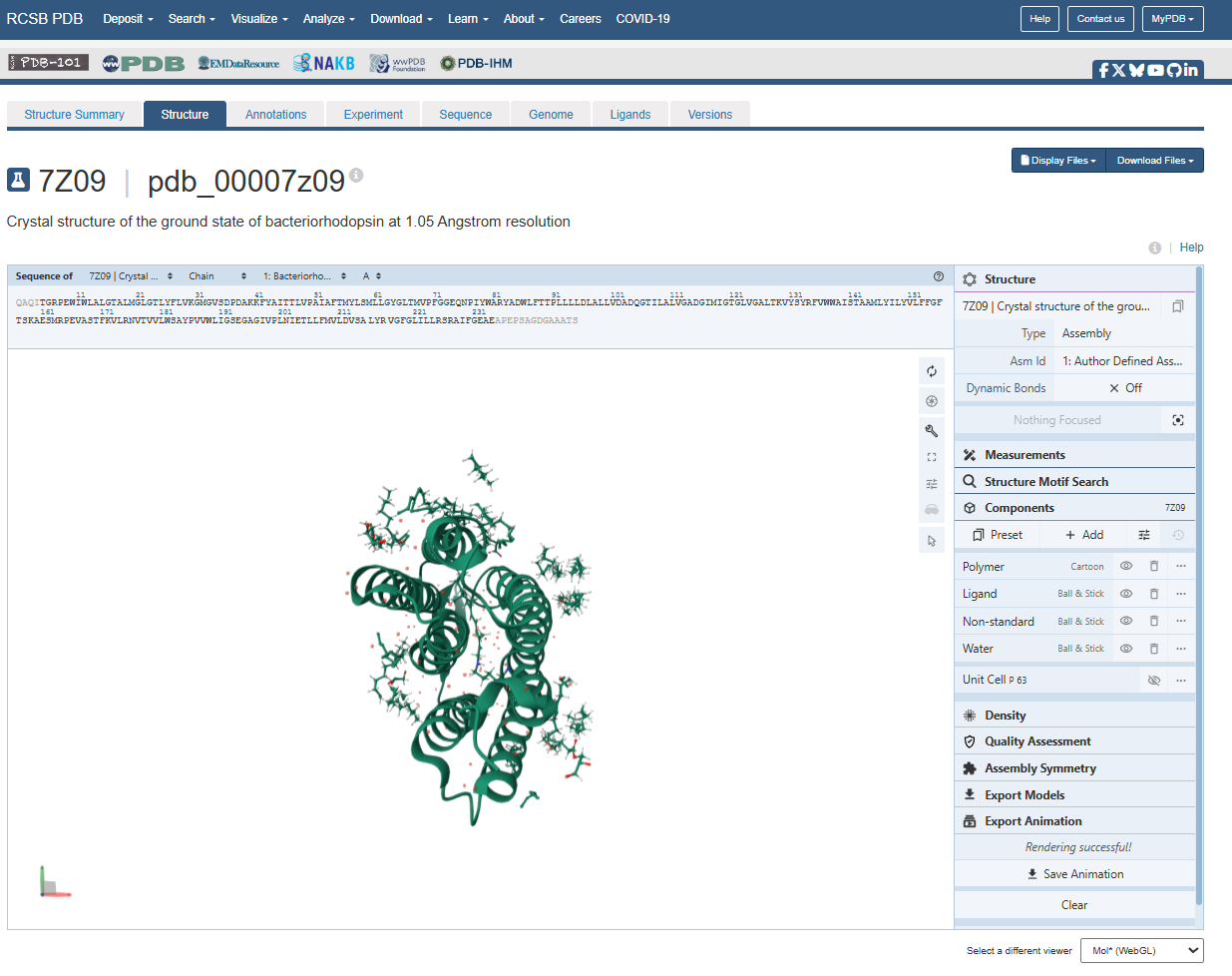
For the next parts of the homework, I will be using the high-resolution crystal structure of the bacteriorhodopsin protein identified by PDB code 7Z09. This specific model was solved using X-ray diffraction and was published recently (2022), representing the protein in its ground state with a resolution of 1.05 Å. I selected this specific entry because of its atomic-level detail that allows for a precise visualization of the retinal chromophore and the internal water networks that are essential for proton pumping.
Identify the amino acid sequence of your protein.
Here’s the bacteriorhodopsin protein sequence I’m using in FASTA format:
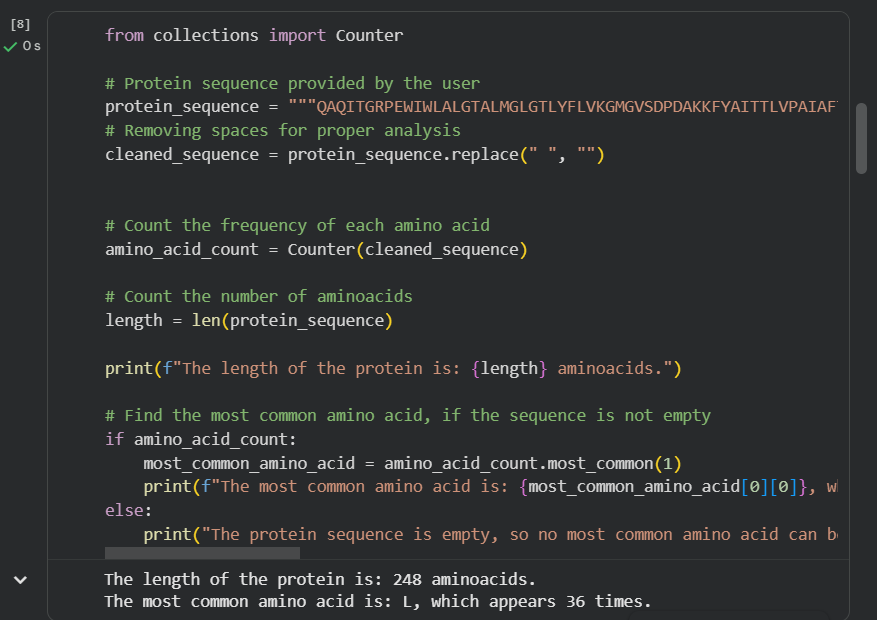
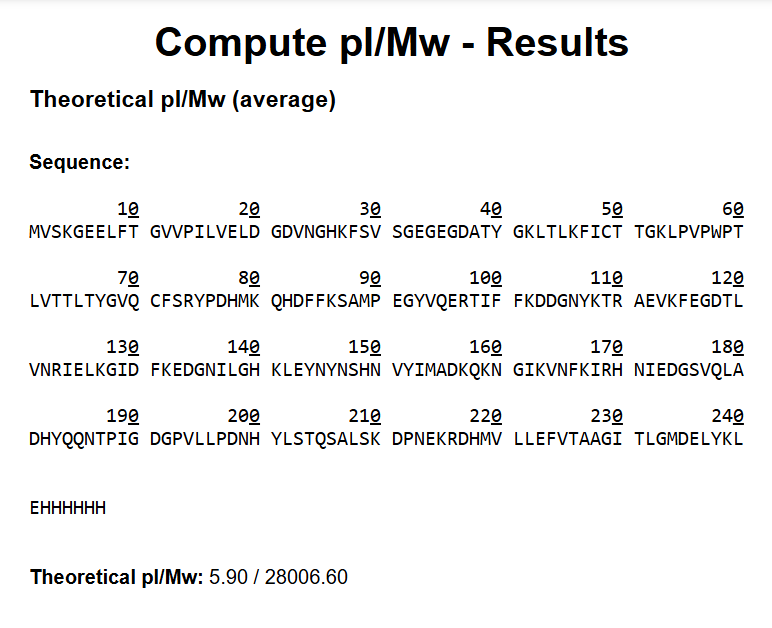
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The bR protein is 238 AA long, and the most frequent amino acid is L (leucine), which appears 36 times in the protein sequence.
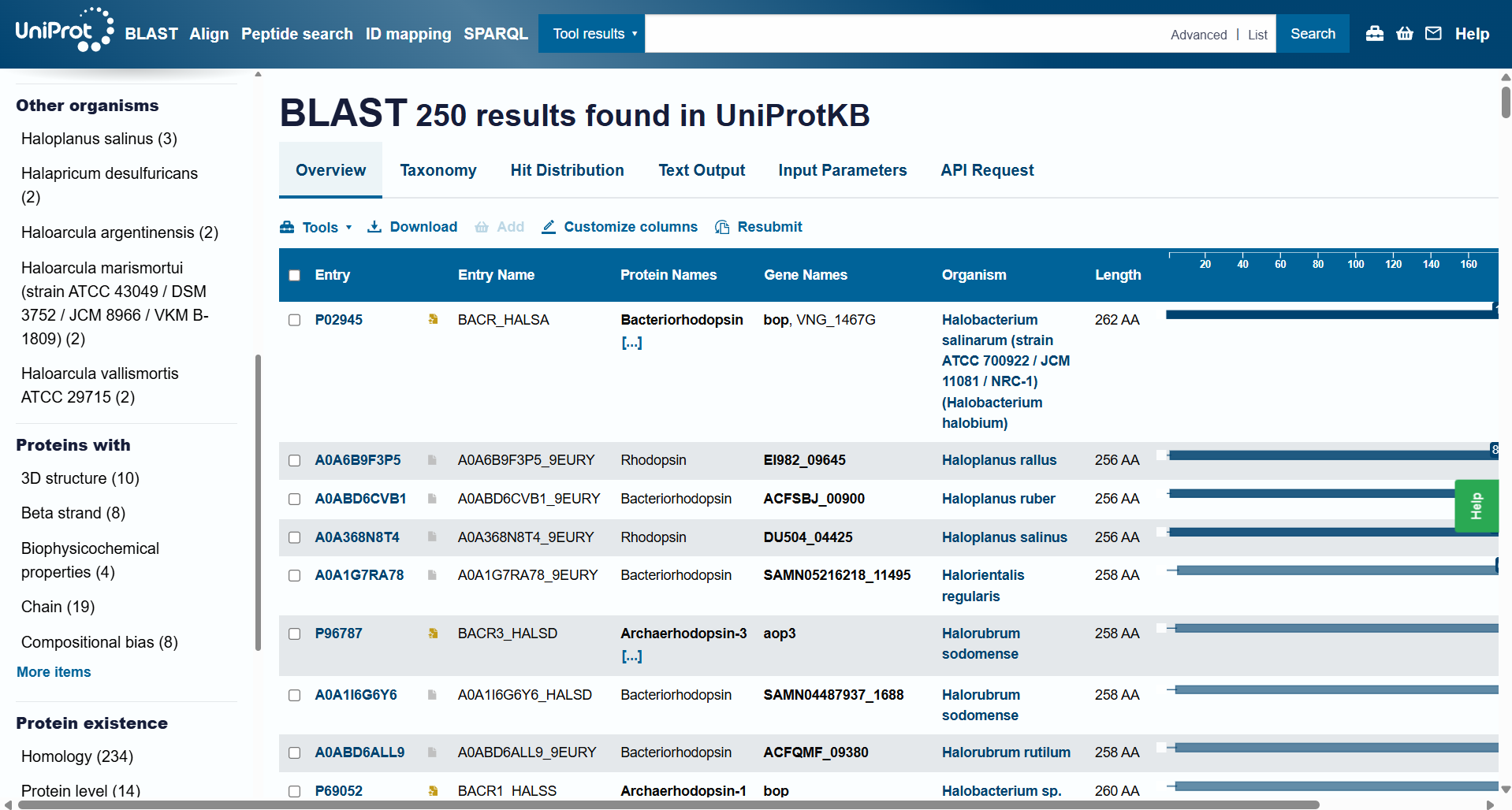
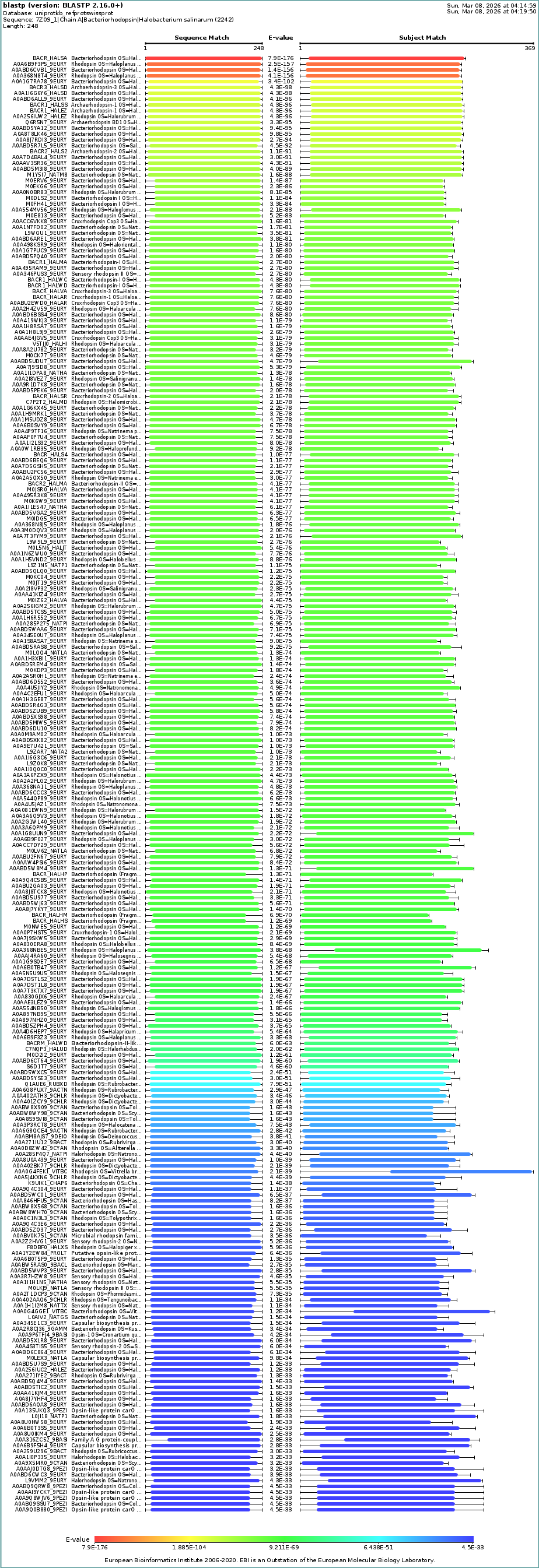
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
For this question, I ran Uniprot’s BLAST tool using the bR protein mentioned before. Here’s the Uniprot’s BLAST ID.
The BLAST tool identified 250 homologs for the protein I selected: 234 of these sequences are inferred through homology, 14 have been experimentally validated at the protein level, and 2 are predicted sequences.
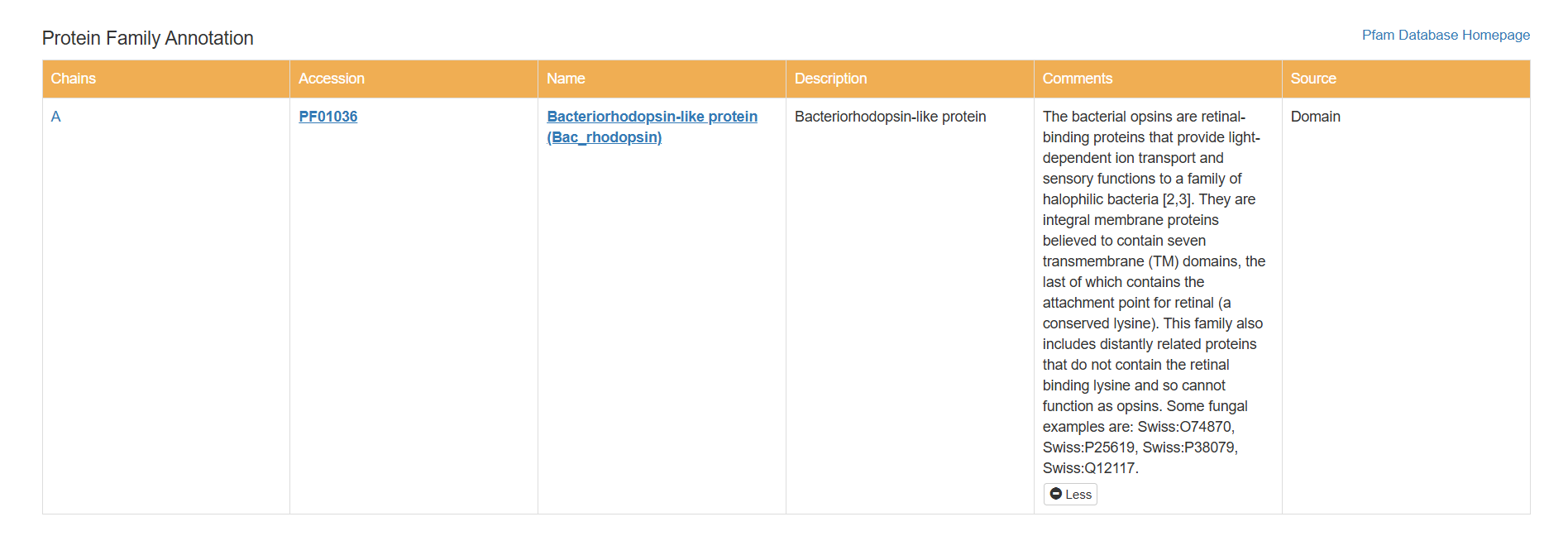
Does your protein belong to any protein family?
Based on my results, my protein belongs to the microbial rhodopsin family, specifically within archaeal-type rhodopsins. Additionally, according to the Pfam PDB annotation (Accession: PF01036), it is classified as a Bacteriorhodopsin-like protein, which are integral membrane proteins characterized by seven transmembrane (TM) domains that utilize a covalently bound retinal to provide light-dependent ion transport.
BLAST’s taxonomy data shows a dominance of homologs within the Haloferacaceae (41%), Haloarculaceae (25%), Natrialbaceae (17%), and Halobacteriaceae (4%) families. The presence of the protein in a wide variety of genera such as Halorubrum, Haloplanus, Haloarcula, and Halobacterium confirms its role as a highly conserved protein across different halophilic microorganisms.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure page for my selected protein can be found at RCSB PDB: 7Z09.
The protein I chose was deposited on 2022-02-22 and officially released three months later, on 2022-05-04.
It is considered an exceptional quality structure because its resolution is 1.05 Å, which is significantly better (smaller) than the 2.70 Å threshold. At this atomic resolution, the positions of individual atoms and the surrounding water are mapped with a lot of precision.
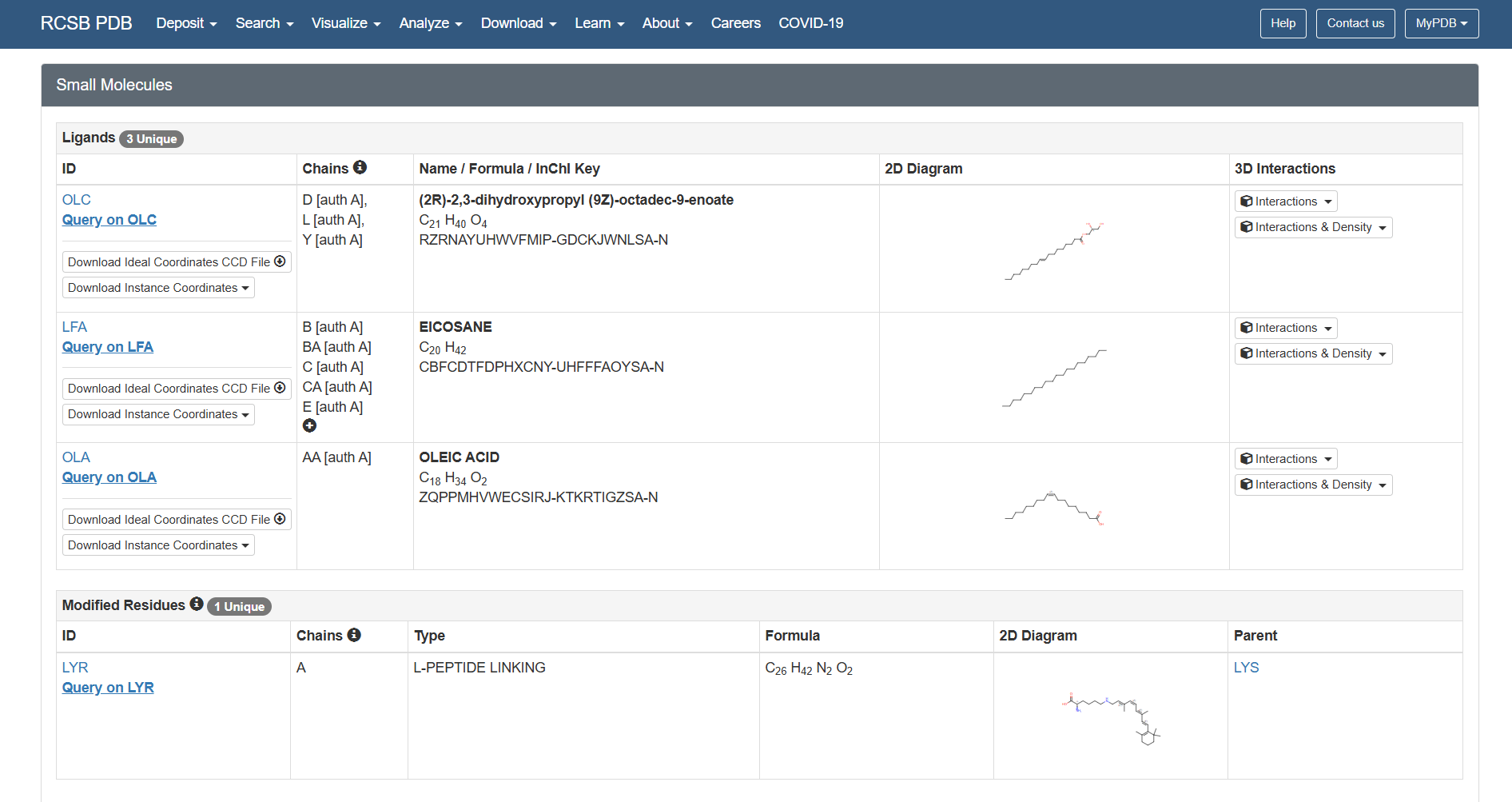
Are there any other molecules in the solved structure apart from protein?
Yes, there are other molecules in the solved structure apart from the bR protein.
Based on the RCSB ligand data, I identified the following molecules: 3 ligands and 1 modified residue.
However, on the structure page we can actually see the 3 ligands, the retinal molecule (LYR), and some water molecules.
The 3 ligands found on the protein are lipids and fatty acids like OLC ((2R)-2,3-dihydroxypropyl (9Z)-octadec-9-enoate), eicosane (OLA), and oleic acid. These ligands represent the lipidic environment that surrounds the protein in its natural state.
The modified residue corresponds to retinal (LYR), which is covalently linked to lysine in the protein chain, and it is the chromophore responsible for absorbing light.
Additionally, there are some water molecules around the bR protein structure that are critical for the proton transport mechanism.
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
So this is my first time using PyMol, it feels intimidating but I hope I get the hang of it!
At first, I uploaded the FASTA bR protein sequence file thinking it would give me the protein structure. But after loading it, all I saw was a very long chain of amino acids. I found that funny for my first experience with PyMol. After that I went back to the PDB page and downloaded the correct .pdb file format.
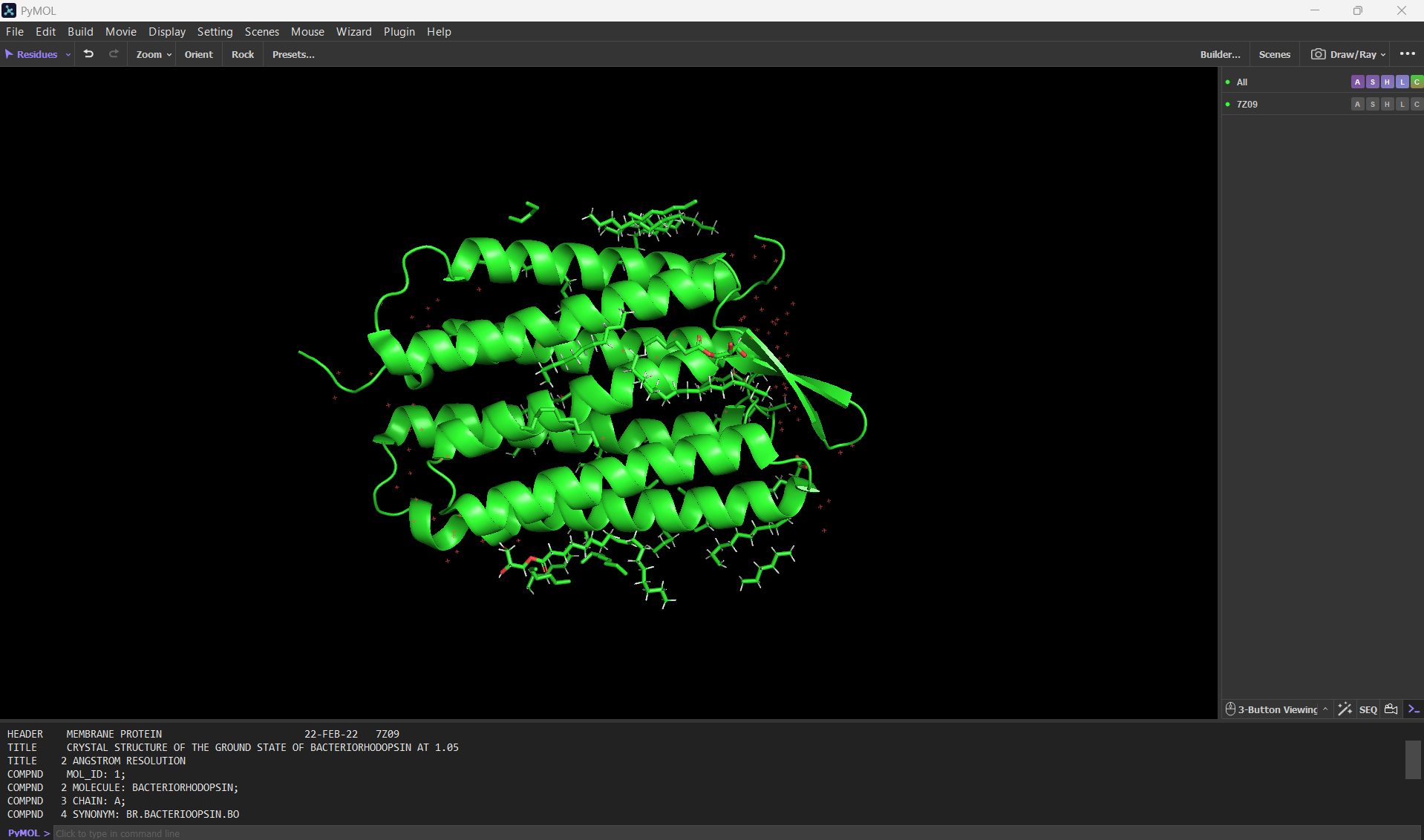
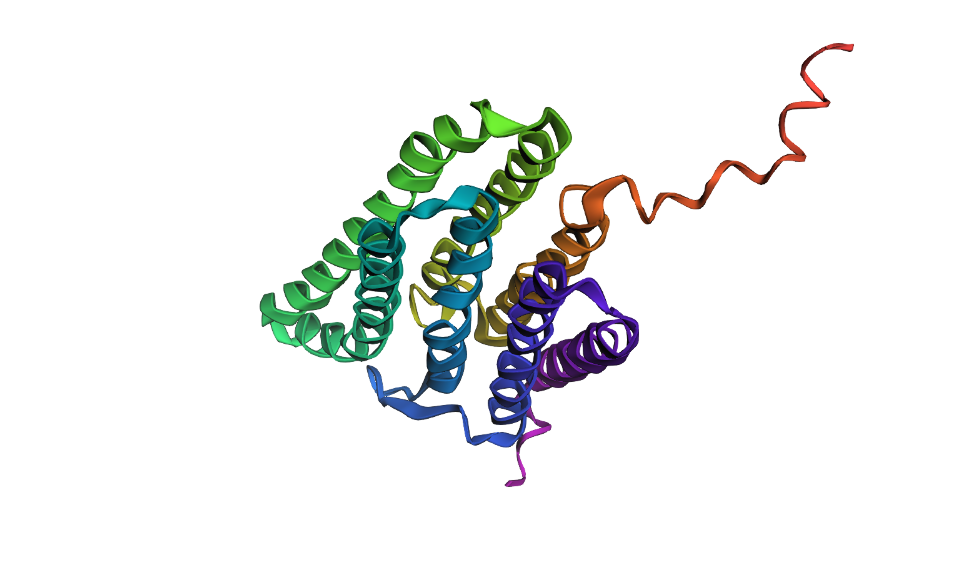
Here’s a screenshot of the bR protein in PyMol using the .pdb file.
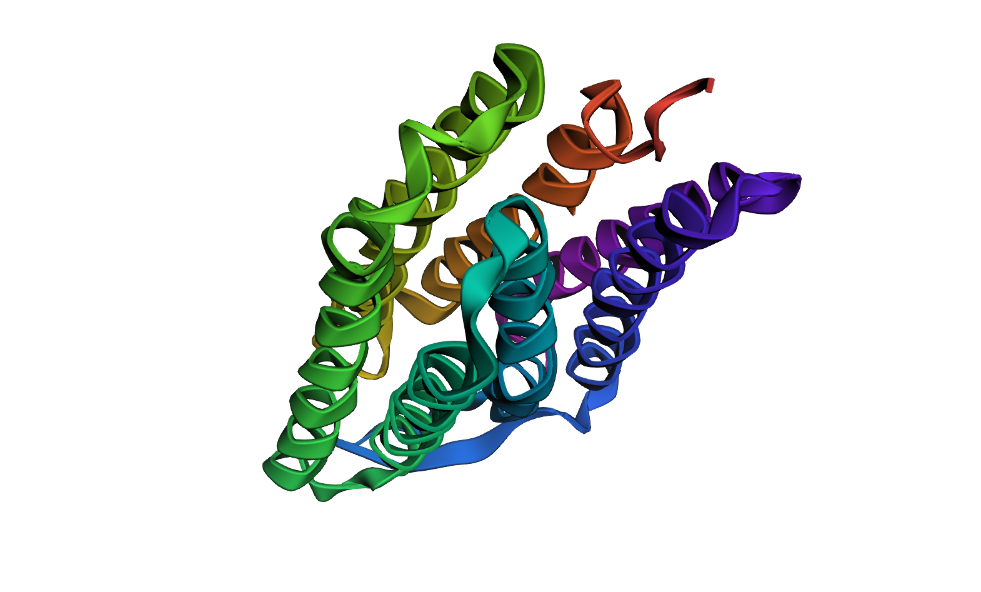
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
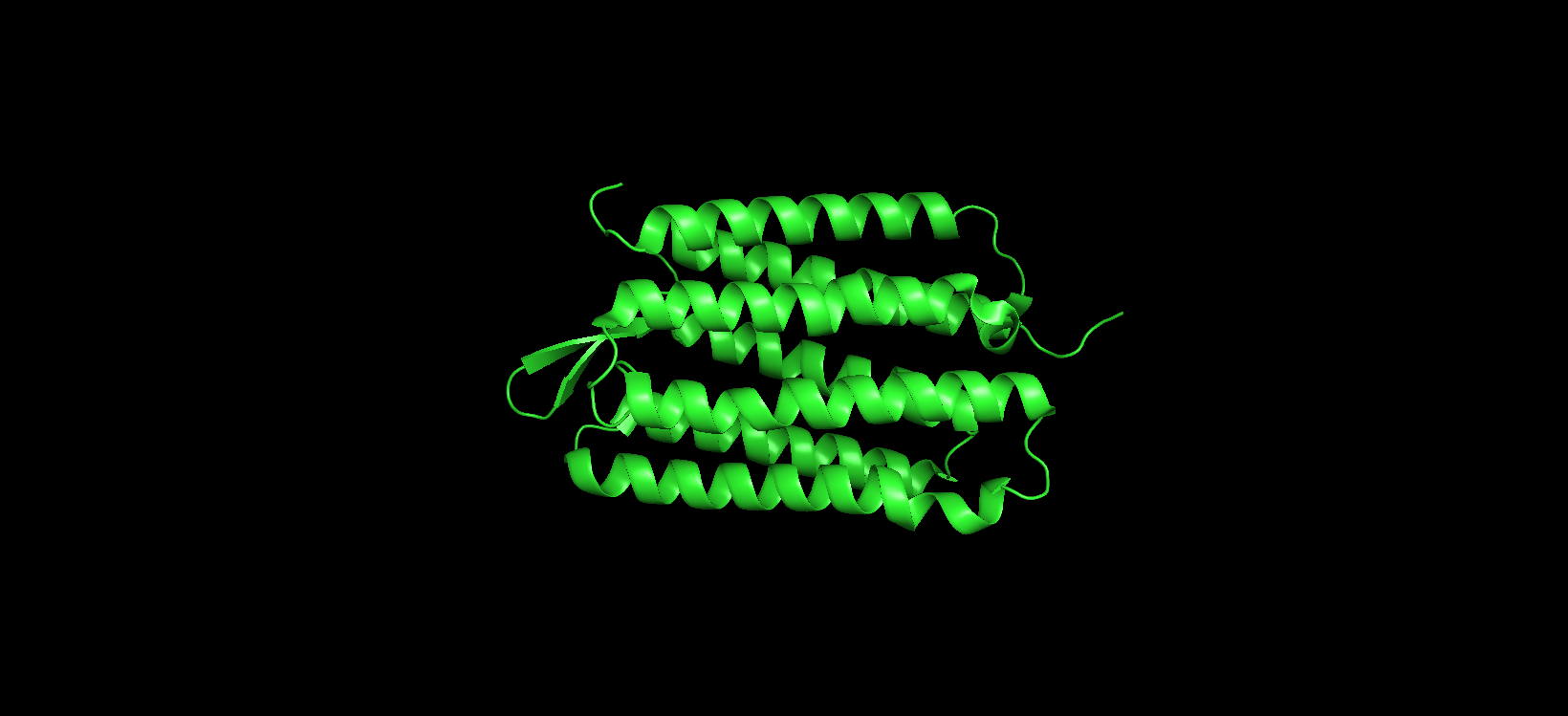
Visualizing the protein as “cartoon”:
Visualizing the protein as “ribbon”:
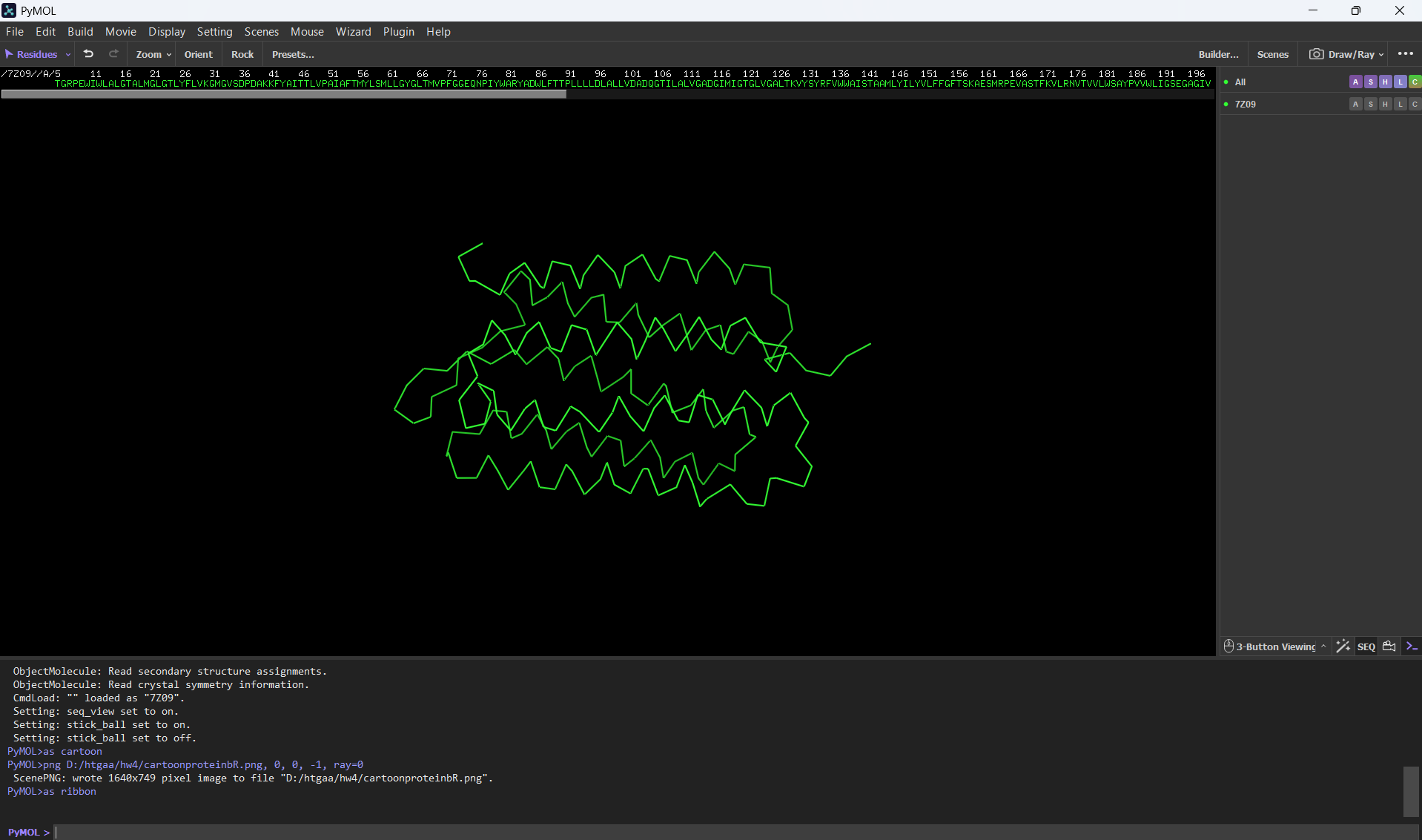
Visualizing the protein as “ball and stick”:
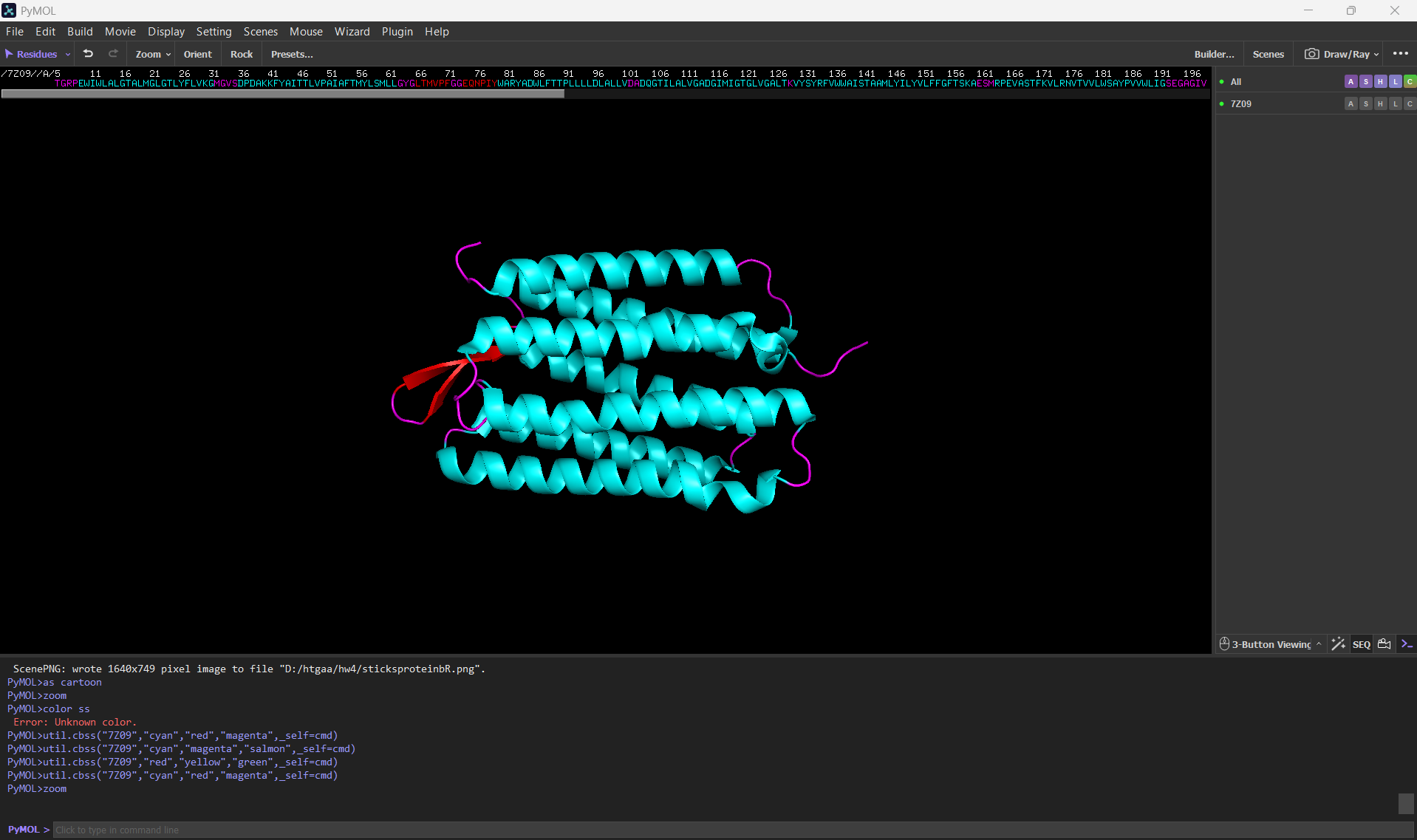
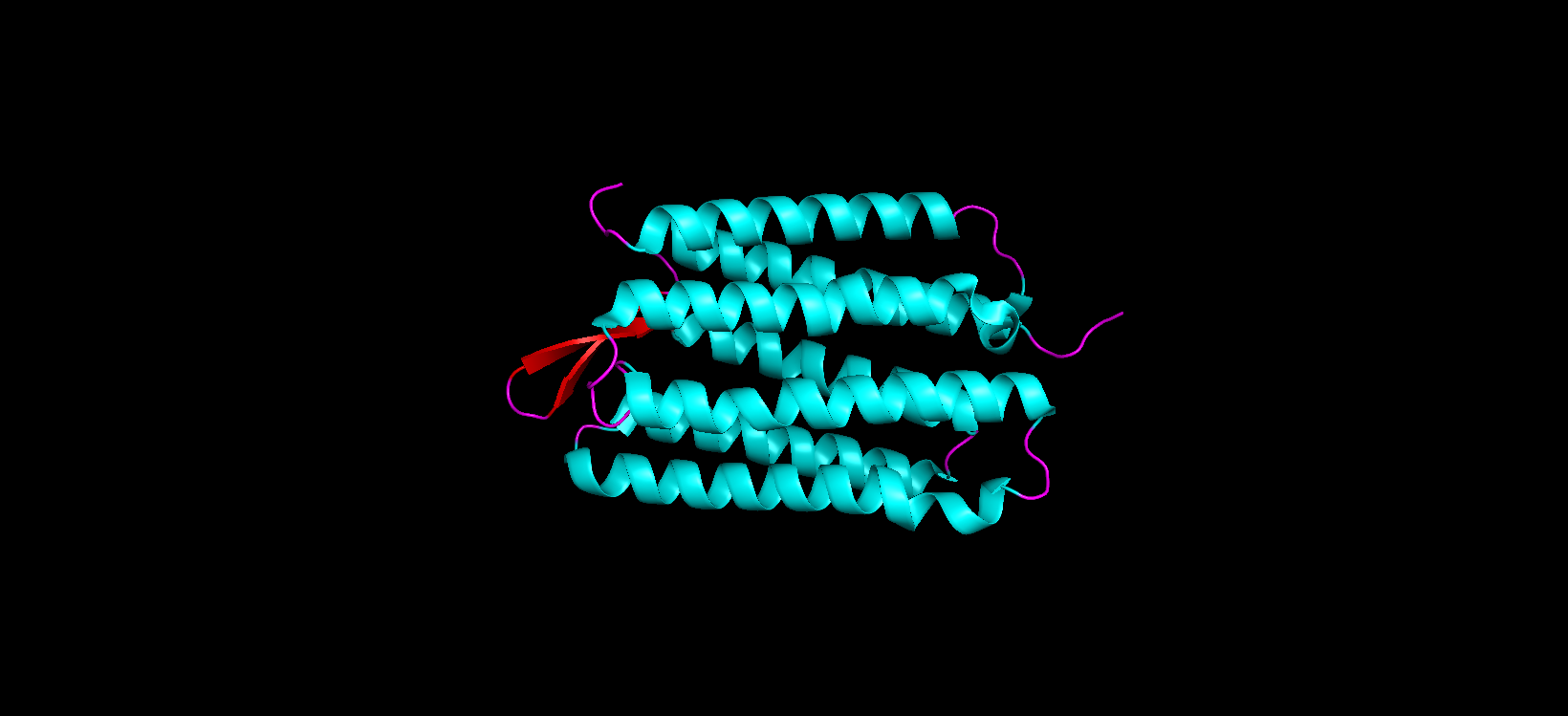
Color the protein by secondary structure. Does it have more helices or sheets?
After coloring the protein according to its secondary structure, I realized there are more helices than sheets. There are 7 alpha helices (colored in cyan), and there are just 2 beta sheets (colored in red) but they are very small. Additionally, PyMol shows that there are 8 loops. which are colored in magenta.
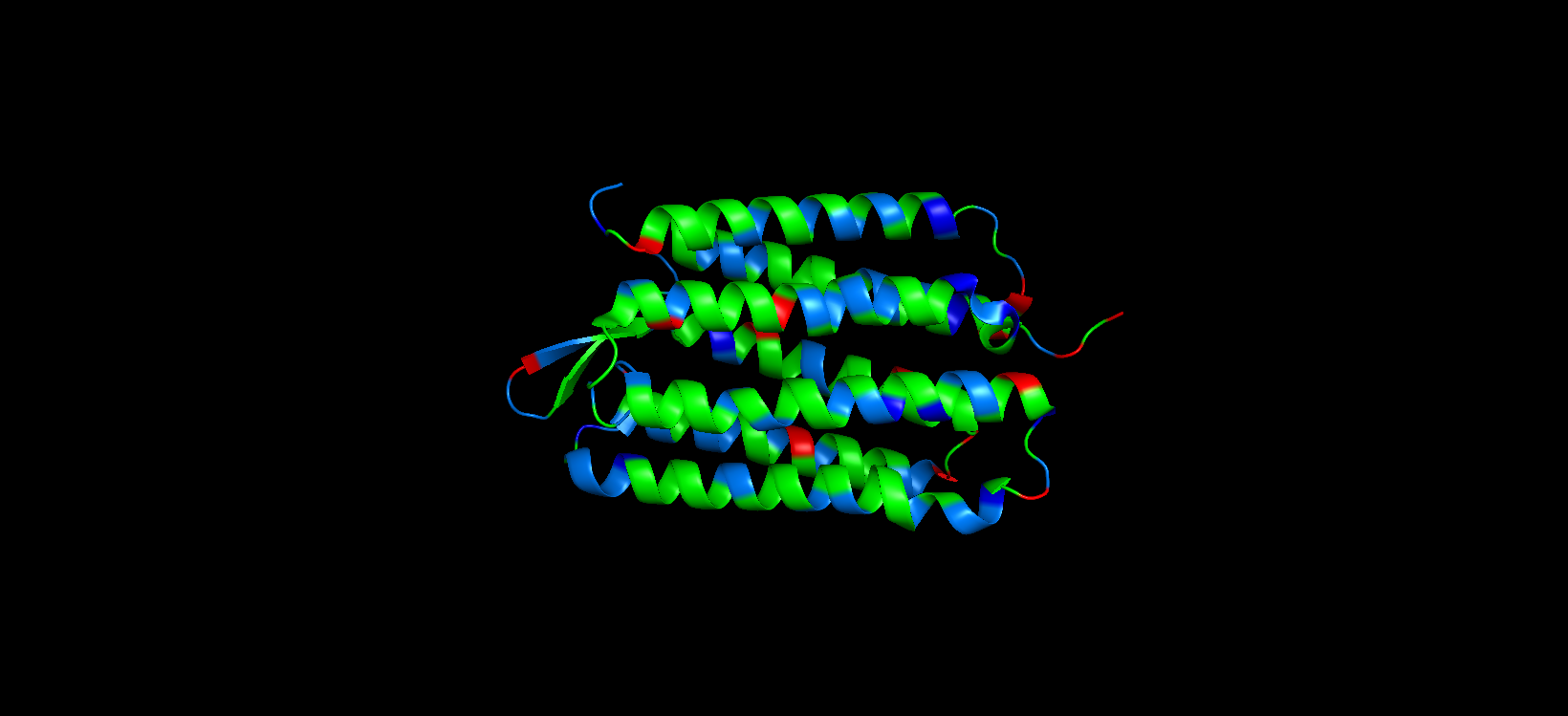
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
For this part, I colored the hydrophilic residues marine, the hydrophobic ones green, and the charged residues red and blue.
After visualizing the colored protein, I recognize that there are more hydrophobic residues than hydrophilic ones, especially along the outer surface of most of the alpha helices, while the hydrophilic residues are mostly in the extremes of the protein, which are mostly exposed to the aqueous environment.
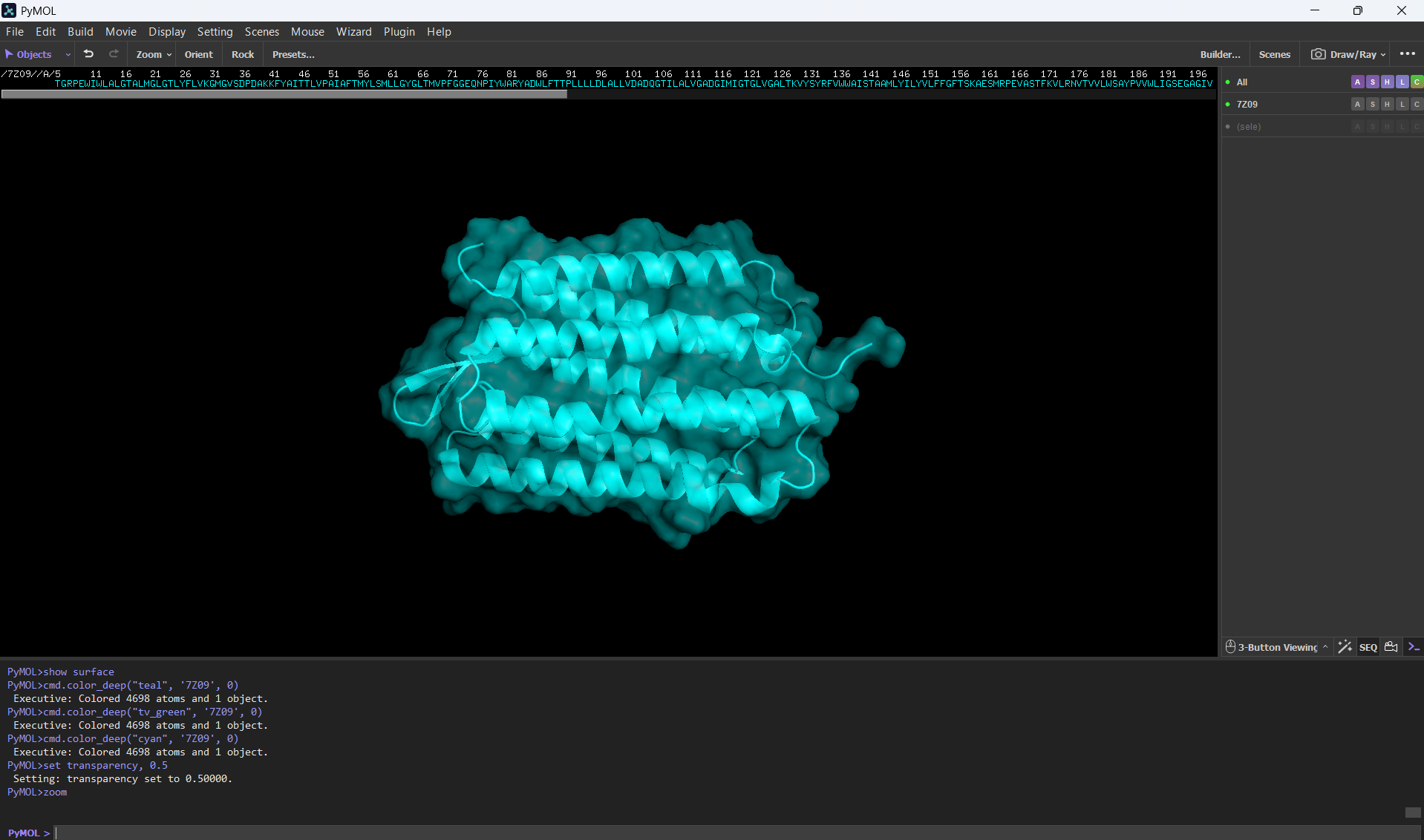
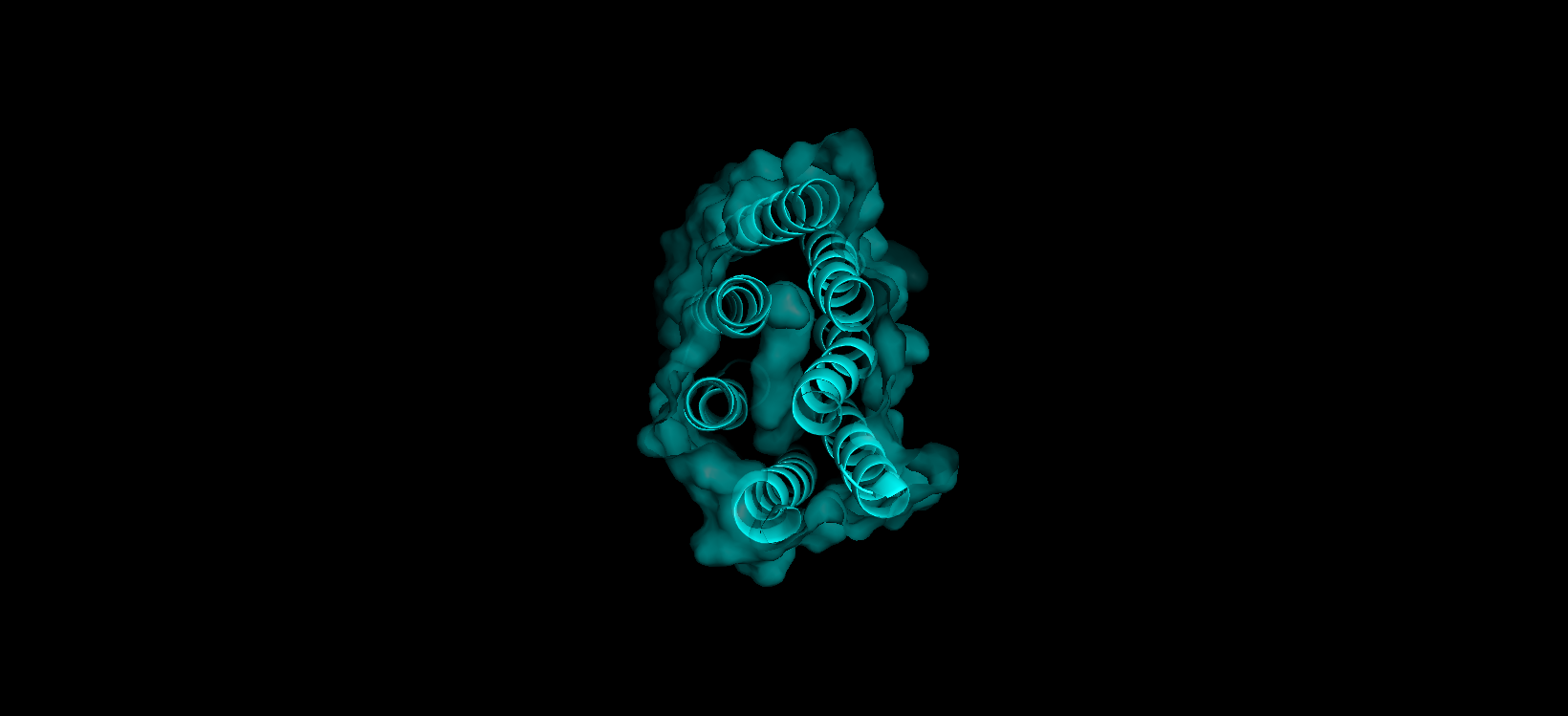
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Here’s a screenshot of the protein visualized by its surface (I set up the transparency to 0.5 to see the inside better):
Here’s a close-up of the protein’s surface:
Finally, here’s another angle of the protein’s surface and its interior:
At first sight, it seems that the protein is very compact and would not have any holes. After using PyMol, I can actually see the central binding pocket that houses the retinal chromophore. Beyond this main site (“hole”), the visualization reveals a continuous internal channel rather than isolated holes along the protein. These results correspond to the bR protein function as a proton pump because of the binding pocket in the middle of it.
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
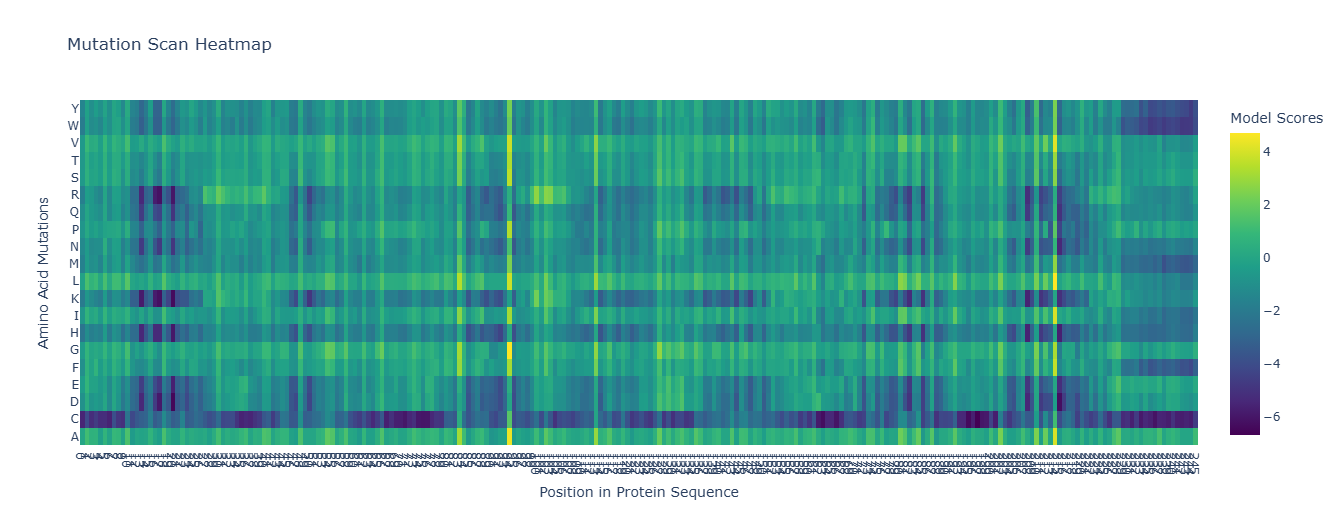
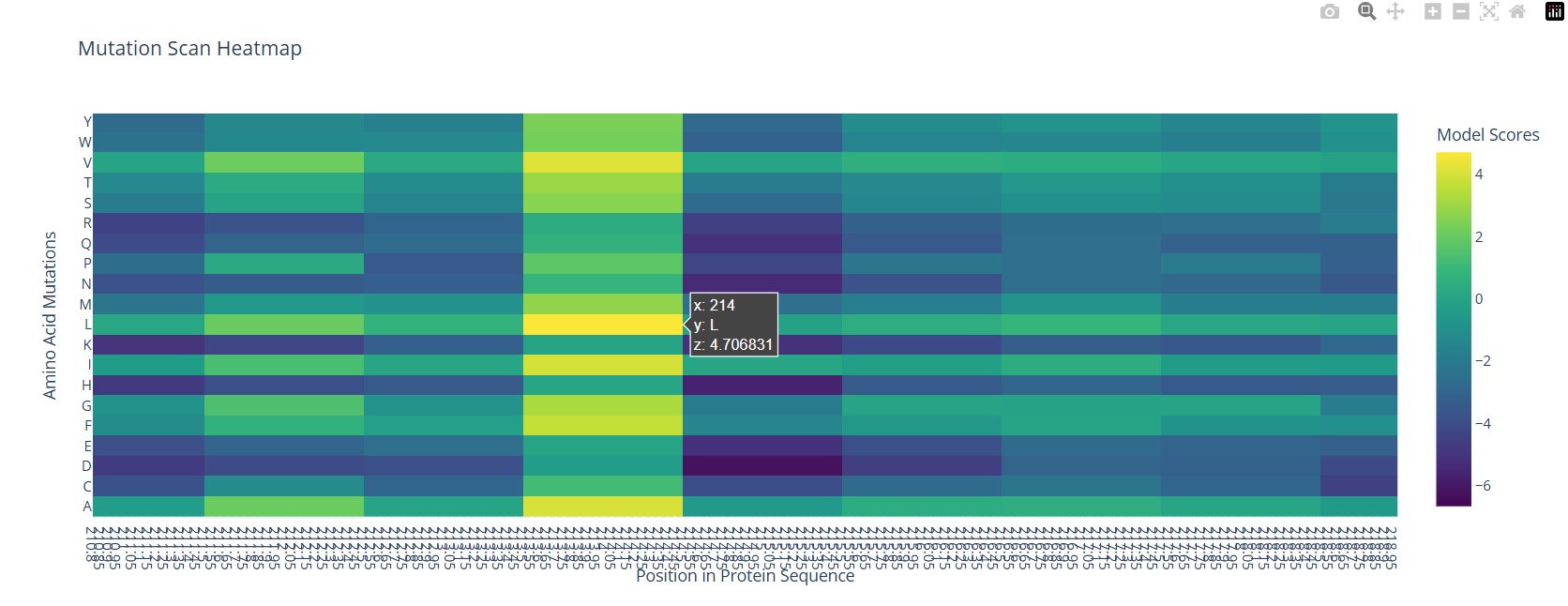
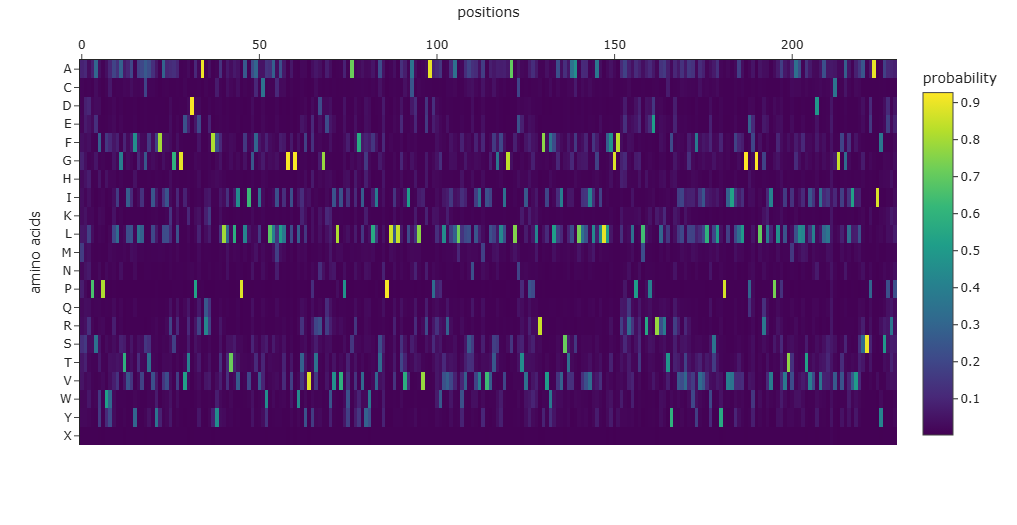
For the unsupervised deep mutational scan, I used the ESM2 8M parameter model!
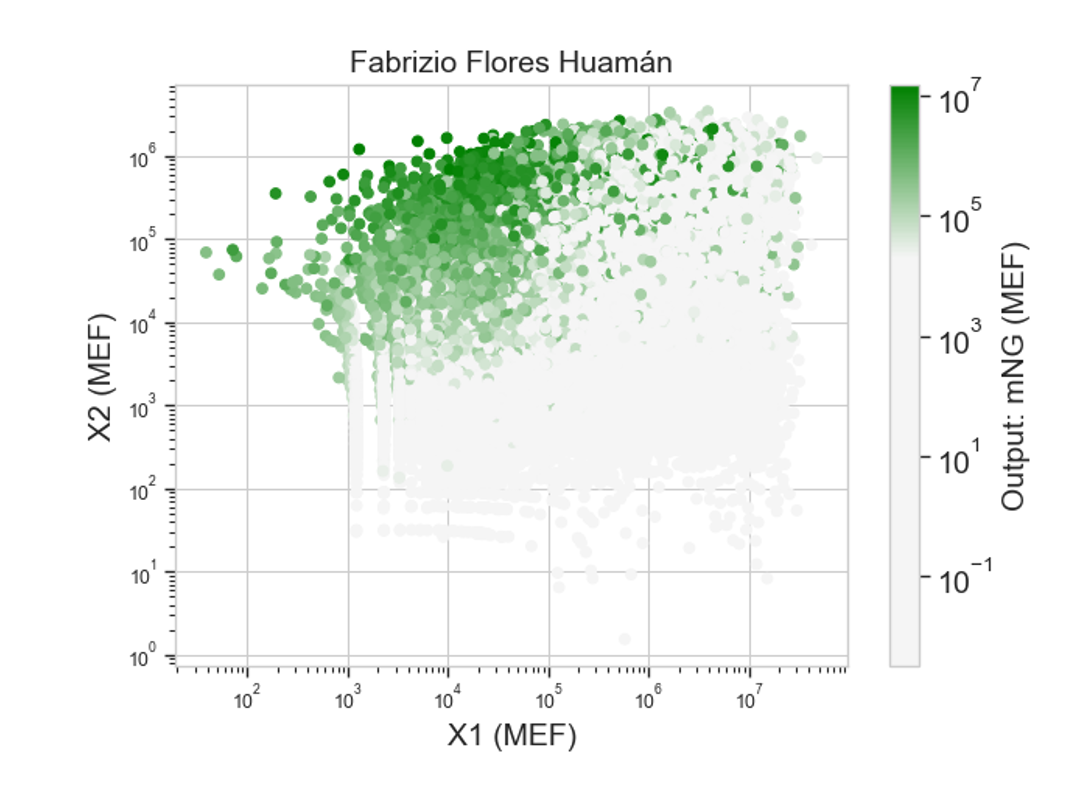
Here’s the heatmap the model produced using the bR protein.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
After looking at the heatmap for a while, I noticed that there were high model scores for two interesting positions: the 94 and the 214 positions showed mostly yellow colors in comparison to the other positions. But between the two, the 214 position had the highest model score.
With the help of the zoom tool, we can clearly identify which residue it corresponds to: leucine (L) with a model score of 4.7. However, this mutation corresponds to the wild-type protein, which indicates that it is a conserved position that bR has perfected in its evolution.
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I compared my ESM-2 unsupervised predictions with experimental data from Jacobson and Perkins (2021). They measured energy changes for mutations in bacteriorhodopsin using single molecule force spectroscopy in a native lipid bilayer. Their results show that even small mutations in the 7-helix core significantly impact protein stability. This correlates with my ESM-2 heatmap, where internal residues like L214 or A94 get the highest scores while destabilizing changes are penalized with dark, low-probability colors. This proves that the model can sense the physical constraints of 7Z09 without needing experimental labels.
Latent Space Analysis
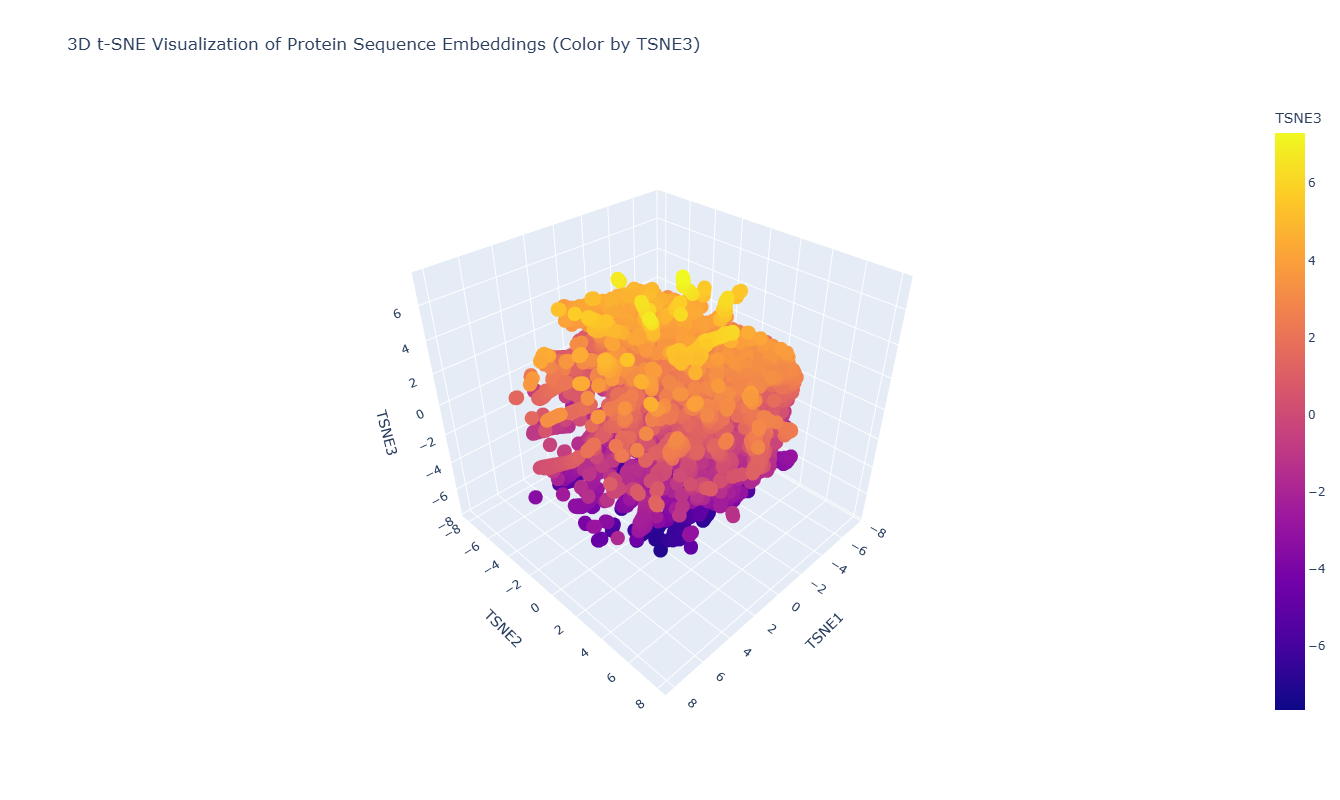
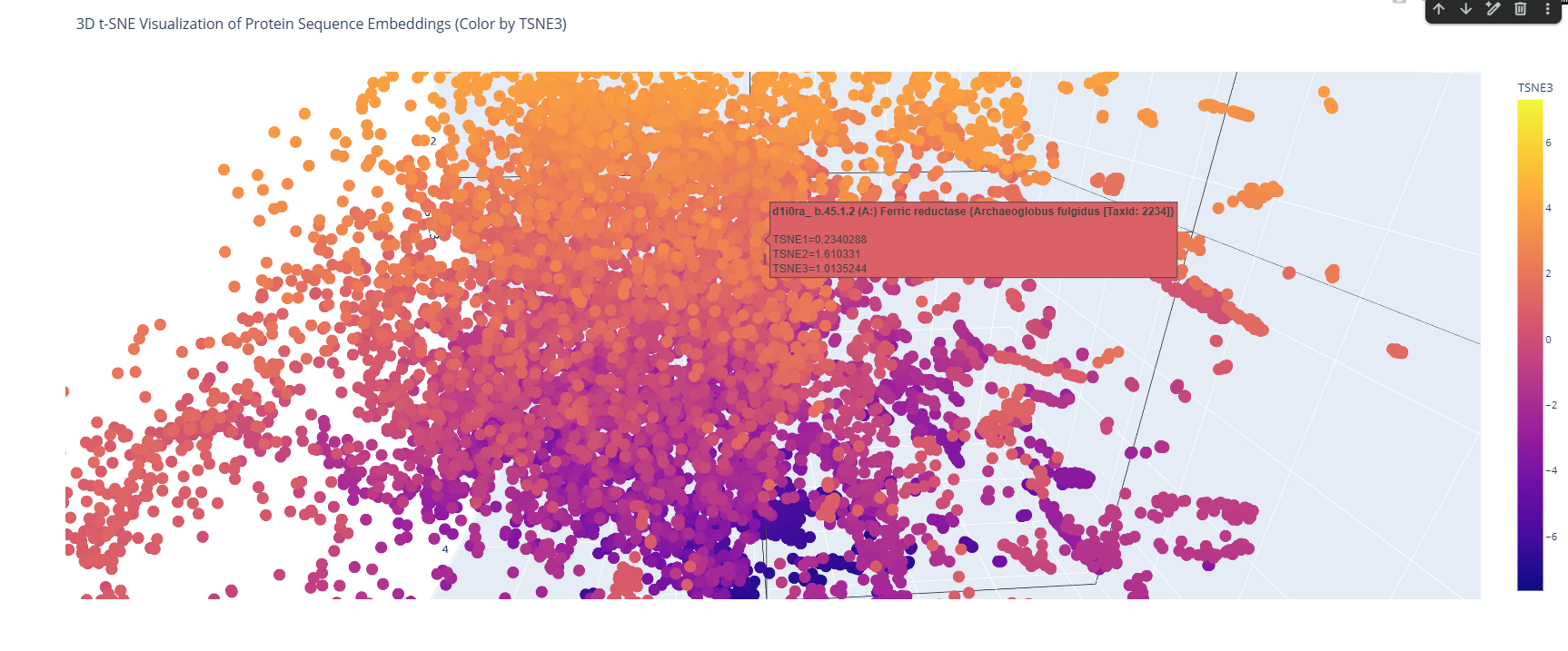
Use the provided sequence dataset to embed proteins in reduced dimensionality.
I used the ESM-2 model to process a protein sequence dataset from ASTRAL SCOP. The model generates high-dimensional mean embeddings for each sequence, which then is reduced to three dimensions using the t-SNE algorithm to visualize the latent space in a 3D plot.
Analyze the different formed neighborhoods: do they approximate similar proteins?
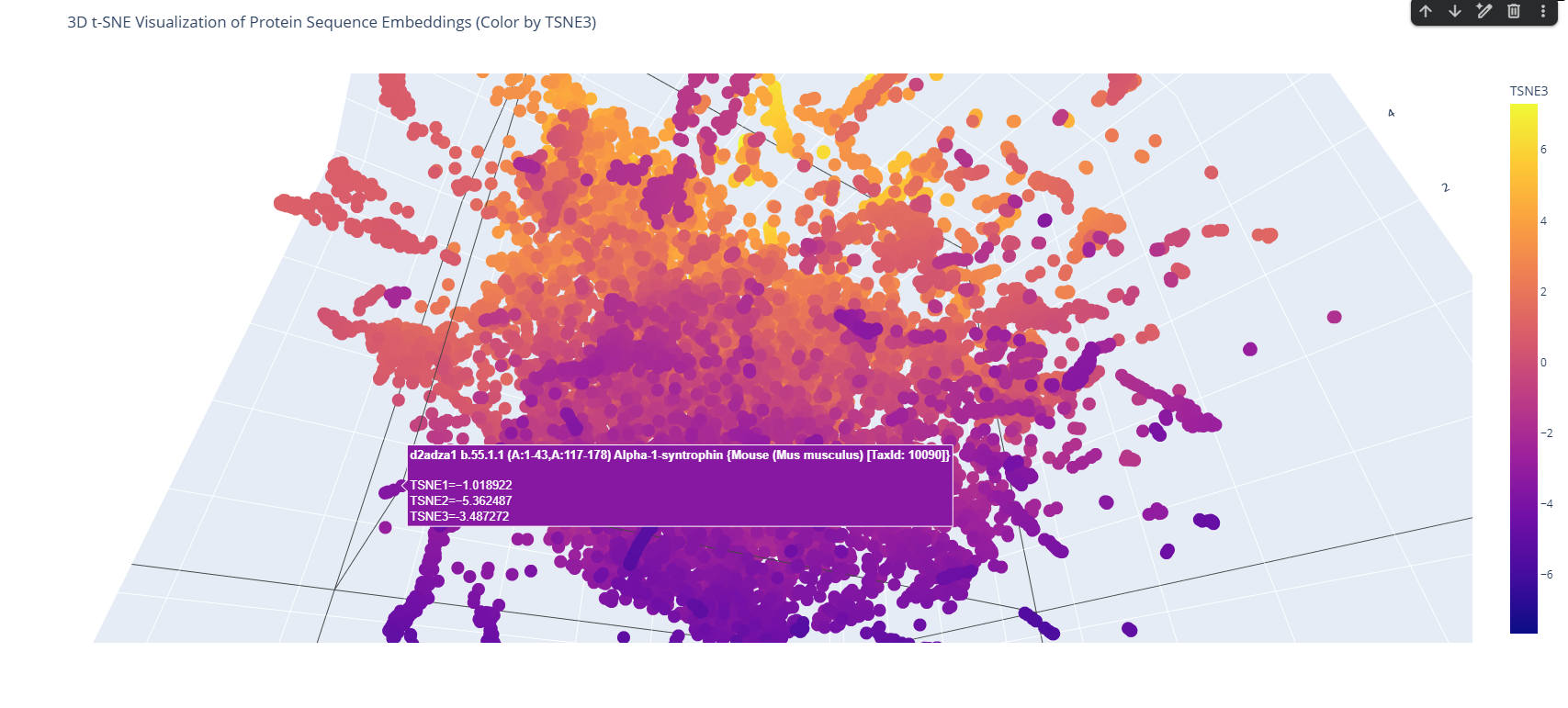
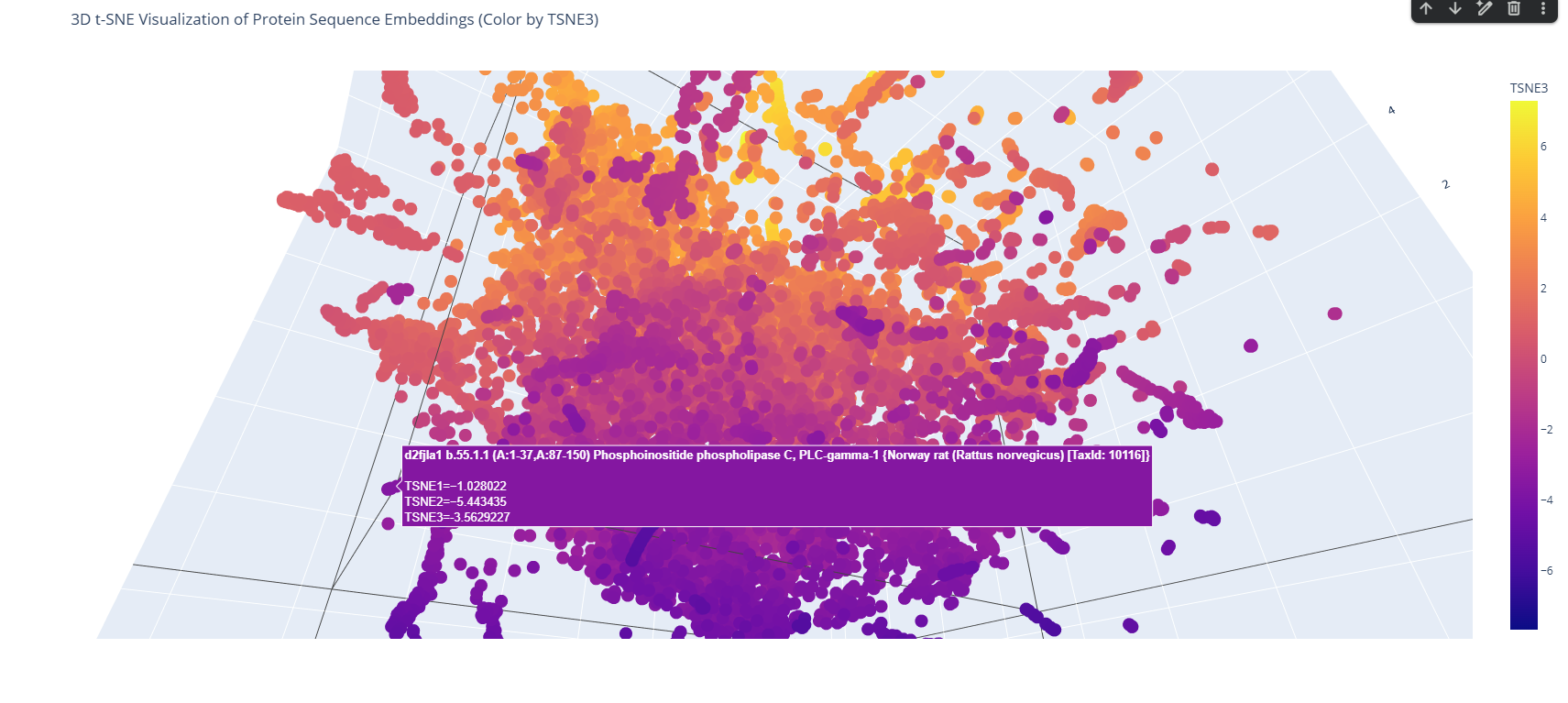
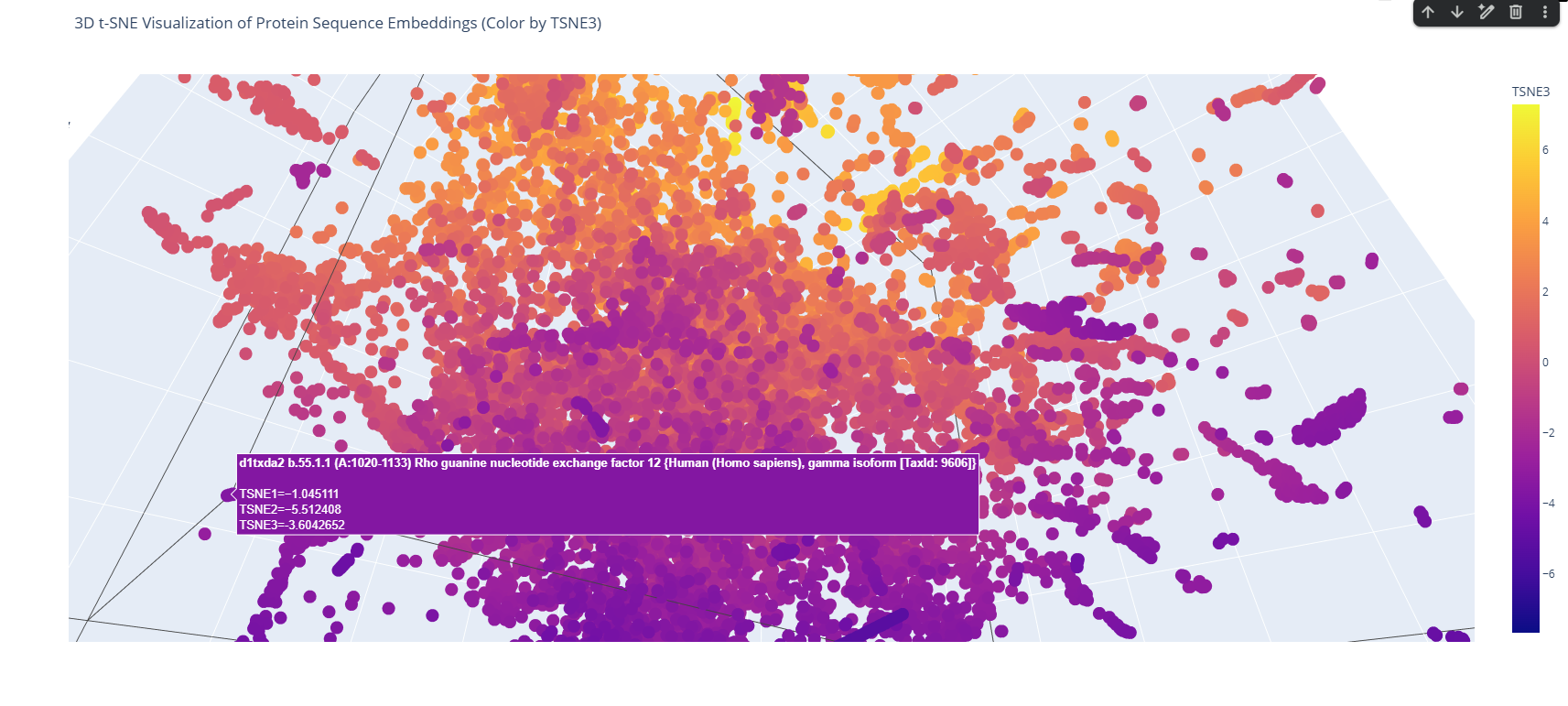
The 3D map reveals distinct neighborhoods where proteins are grouped by their structural characteristics. These neighborhoods approximate similar proteins effectively because of the nature of the SCOP dataset (structural classification). For instance, the following neighborhood contains alpha-1-syntrophin, phosphoinositide phospholipase C, and Rho guanine nucleotide exchange factor 12.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
I place my protein within Class f: Membrane and cell surface proteins and peptides. Its position in the latent space is determined by the specific patterns of its seven transmembrane alpha-helices and its high hydrophobic content. It clusters near other microbial rhodopsins and light-driven pumps, as the model recognizes that these proteins share the same biological logic and structural constraints required to function within a lipid bilayer.
I couldn’t find my protein exactly, but it is near other bacteriorhodopsin-like proteins since my protein’s Tax ID is 2242. So in the 3D plot I would probably find my protein somewhere in the middle.
C2. Protein Folding
Folding a protein
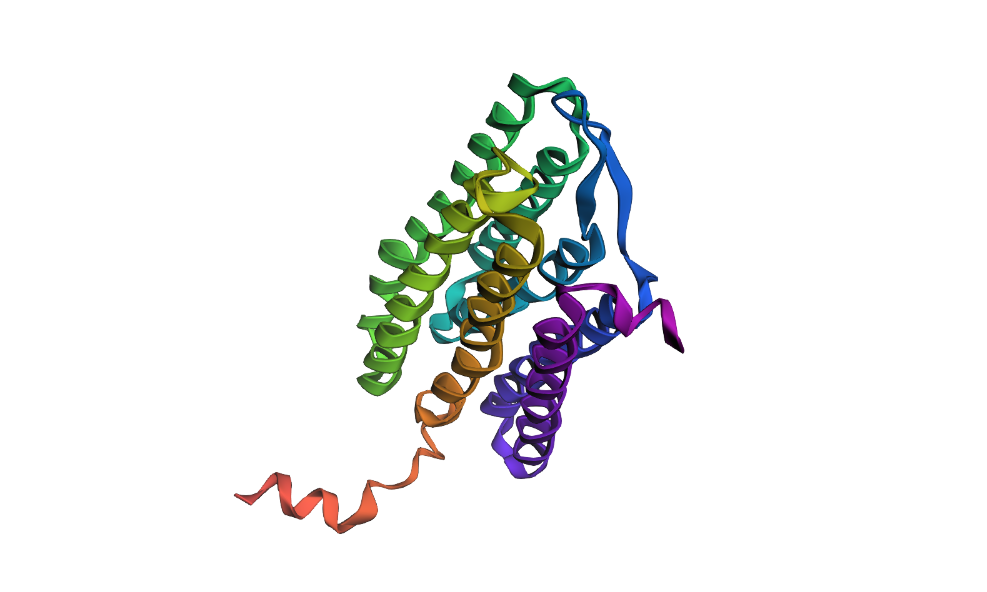
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Here’s my protein folded with ESMFold:
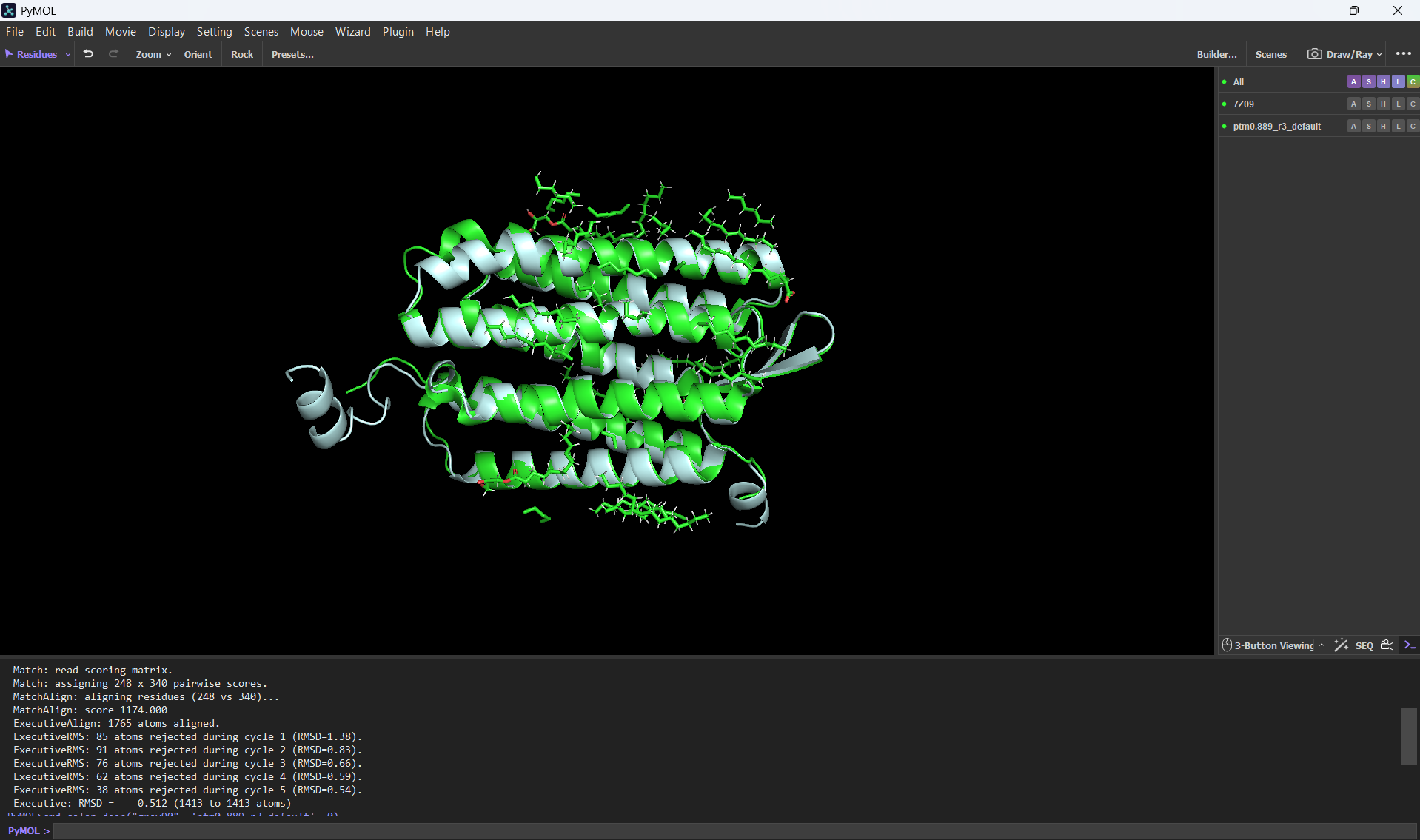
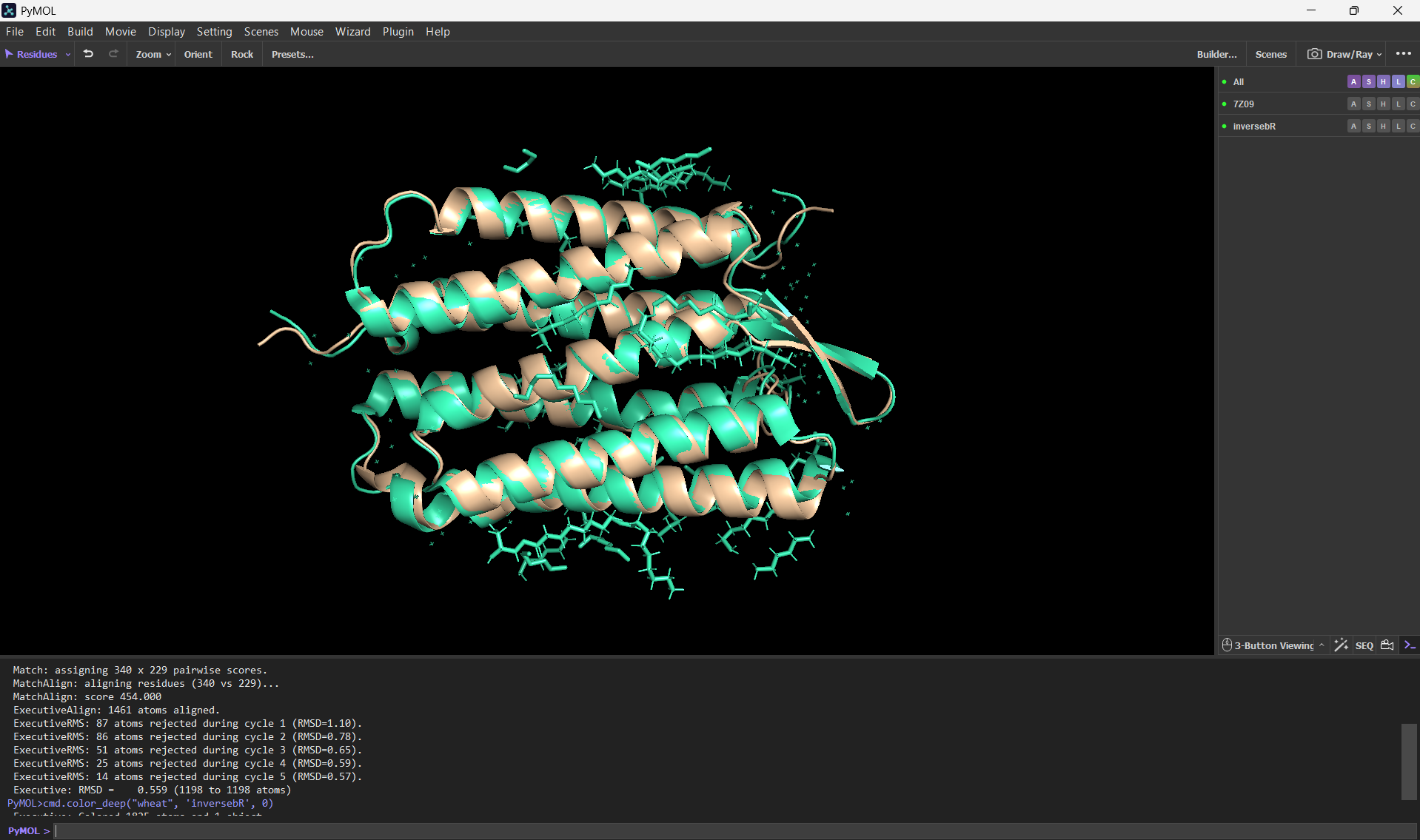
And here’s an alignment of the original 7Z09 bR protein and the ESMFolded one (this was done using the align command on PyMol using the ESMFold .pdb result and the 7Z09.pdb)
Aditionally, here’s the PyMOL command results after doing the alignment.
PyMOL>align ptm0.889_r3_default, 7Z09 Match: read scoring matrix. Match: assigning 248 x 340 pairwise scores. MatchAlign: aligning residues (248 vs 340)... MatchAlign: score 1174.000 ExecutiveAlign: 1765 atoms aligned. ExecutiveRMS: 85 atoms rejected during cycle 1 (RMSD=1.38). ExecutiveRMS: 91 atoms rejected during cycle 2 (RMSD=0.83). ExecutiveRMS: 76 atoms rejected during cycle 3 (RMSD=0.66). ExecutiveRMS: 62 atoms rejected during cycle 4 (RMSD=0.59). ExecutiveRMS: 38 atoms rejected during cycle 5 (RMSD=0.54). Executive: RMSD = 0.512 (1413 to 1413 atoms)
The RMSD was only 0.512 Å over 1413 atoms; this statistic confirms that the ESMFold code can accurately reconstruct the bacteriorhodopsin protein. This means that the language model has deeply learned the structural patterns of the bacteriorhodopsin fold with high accuracy.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To test the resilience of the bR 7Z09 fold, I performed two types of sequence modifications. First, I introduced point mutations in the transmembrane helices.
The structure showed high resilience to minor changes, maintaining its 7-helix architecture with minimal RMSD shifts. However, when I replaced large segments of the helical core with random or polar sequences, the ESMFold prediction collapsed or showed significant unfolding in those regions.
>Large segment mutation using A's and G's QAQITGRPEWIWLALGTALMGLGTLYFLVKGMGVSDPDAKKFYAITTLVPAIAFTMYLSMLLGYGLTMVPFGGEQNPIYWARYADWLFTTPLLLLDLALLVDADQGTILALVGADGIMIGTGLVGALTKVYSYRFVAAGGAAAGGAAAGGAAAGGAAMLYILYVLFFGFTSKAESMRPEVASTFKVLRNVTVVLWSAYPVVWLIGSEGAGIVPLNIETLLFMVLDVSAKVGFGLILLRSRAIFGEAEAPEPSAGDGAAATS
This confirms that while the bacteriorhodopsin fold is structurally robust, its stability is strictly dependent on the conserved hydrophobic patterns that allow the helices to pack correctly within the membrane.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Here are the results after running the ProteinMPNN colab section
The ProteinMPNN analysis shows a 44.1% sequence recovery and a high 0.9490 confidence score, indicating that the model successfully redesigned over half of the residues while maintaining the protein’s evolutionary “grammar.”
The probability heatmap confirms that internal transmembrane positions remain conserved, while external loops allow for significant sequence variability.
Input this sequence into ESMFold and compare the predicted structure to your original.
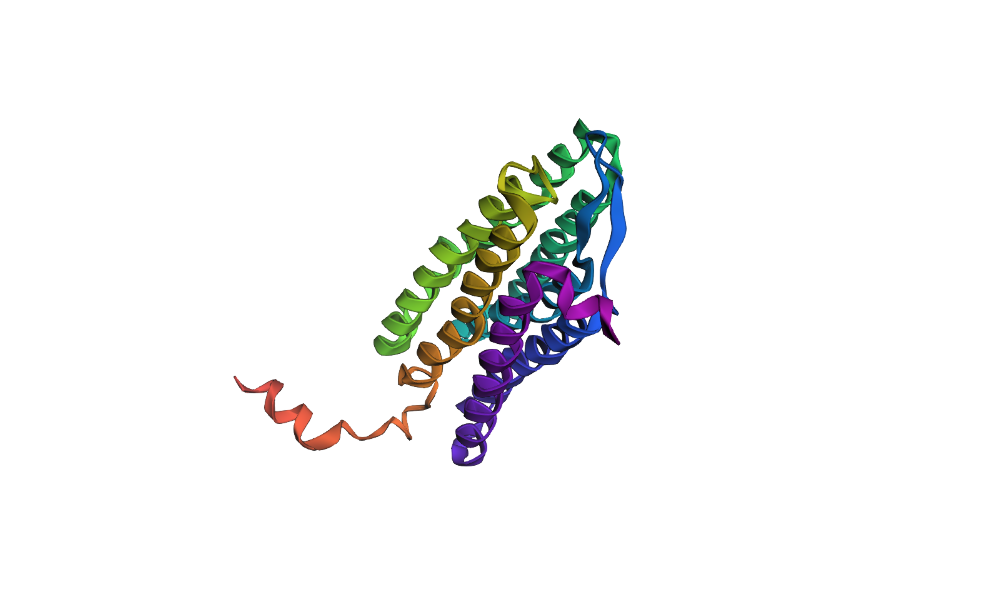
Here’s a screenshot of the protein generated by ProteinMPNN and folded using ESMFold:
Here’s a screenshot of the protein visualized using PyMOL aligned with the original one:
Folding the redesigned sequence with ESMFold resulted in a structure nearly identical to the original 7Z09 backbone, validating that the inverse-folding process preserved the 7-helix architecture. This proves that ProteinMPNN can “hallucinate” valid sequence variants that strictly obey the biophysical and geometrical constraints of the native bacteriorhodopsin fold.
References
Barr, B., Levitt, D. E., & Gollahon, L. (2025). Red Meat Amino acids for Beginners: A narrative review. Nutrients, 17(6), 939. https://doi.org/10.3390/nu17060939
Perrino, A. P., Miyagi, A., & Scheuring, S. (2021). Single molecule kinetics of bacteriorhodopsin by HS-AFM. Nature Communications, 12(1), 7225. https://doi.org/10.1038/s41467-021-27580-2
Chatterjee, H., Mahapatra, A. J., Zacharias, M., & Sengupta, N. (2024). Helical reorganization in the context of membrane protein folding: Insights from simulations with bacteriorhodopsin (BR) fragments. Biochimica Et Biophysica Acta (BBA) - Biomembranes, 1866(5), 184333. https://doi.org/10.1016/j.bbamem.2024.184333
Jacobson, D. R., & Perkins, T. T. (2021). Free-energy changes of bacteriorhodopsin point mutants measured by single-molecule force spectroscopy. Proceedings Of The National Academy Of Sciences, 118(13). https://doi.org/10.1073/pnas.2020083118
Part D. Group Brainstorm on Bacteriophage Engineering
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
We will focus on increasing the structural stability of the L protein to ensure it remains functional under different environmental conditions.
We will also attempt to increase the toxicity of the lysis protein by optimizing its target regions to enhance bacterial cell wall disruption.
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
We will use ESMFold to perform in silico mutational scanning and identify target regions in the L protein.
We propose using Genomic Language Models (GLMs) to design and optimize sequences with higher lytic potential.
Finally, we will use AlphaFold-Multimer to validate the folding and stability of the engineered protein complexes.
Why do you think those tools might help solve your chosen sub-problem?
ESMFold allows for high-speed structural feedback, making it easier to test how mutations affect the 7-helix bundle.
GLMs are essential for capturing the “evolutionary grammar” of toxicity, helping to design proteins that are more aggressive than natural variants.
AlphaFold ensures that our computational designs are biophysically plausible and stable before any potential wet-lab implementation.
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Contextual Gap: There is a lack of specific data regarding the host bacteria’s environment, which might lead to unexpected results in vivo.
Misfolding Risk: The engineered protein might still misfold or aggregate in a real biological system despite having positive simulation results in the pipeline.
Include a schematic of your pipeline.
Here’s a short written schematic of our pipeline:
[Sequence Input] → [ESM-2 Mutational Scan] → [GLM Toxicity Optimization] → [AlphaFold Validation] → [Final Design]
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Week 5 HW: Protein Design - Part II
Homework
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
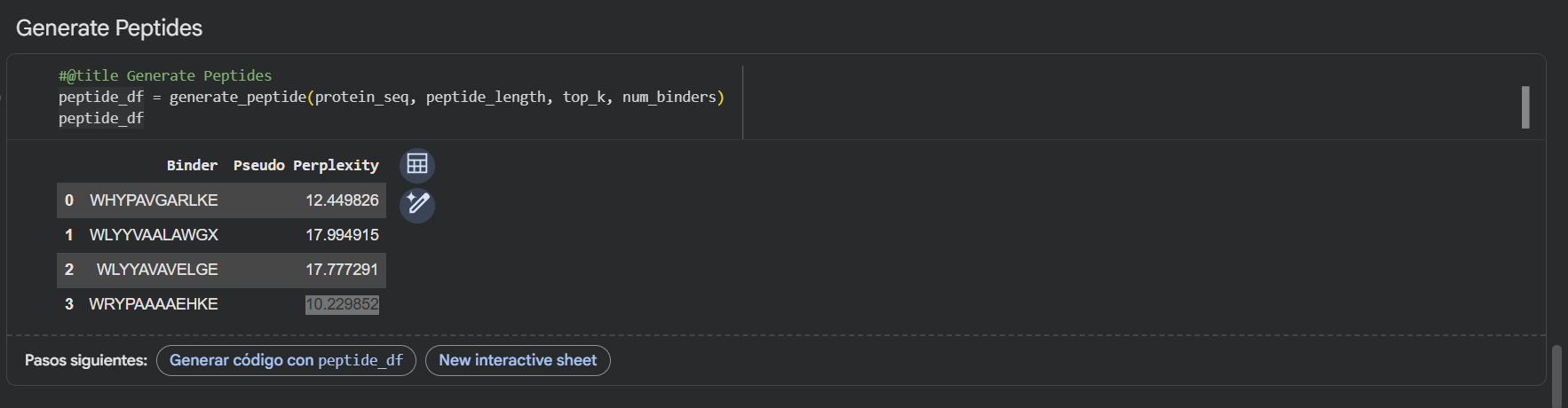
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Using the PepMLM-650M model, I generated four potential binders of length 12 AA’s. The model’s confidence is reflected in the pseudo-perplexity scores, where lower values suggest a more plausible binding interaction.
PepMLM Generated Binder
WRYGVAGVRHWX
WLYPPAVVEHKE
HRYYPTAVRWKX
WHYGVVGLAHKK
Here’s also a screenshot of the binders generated using the PepMLM colab:
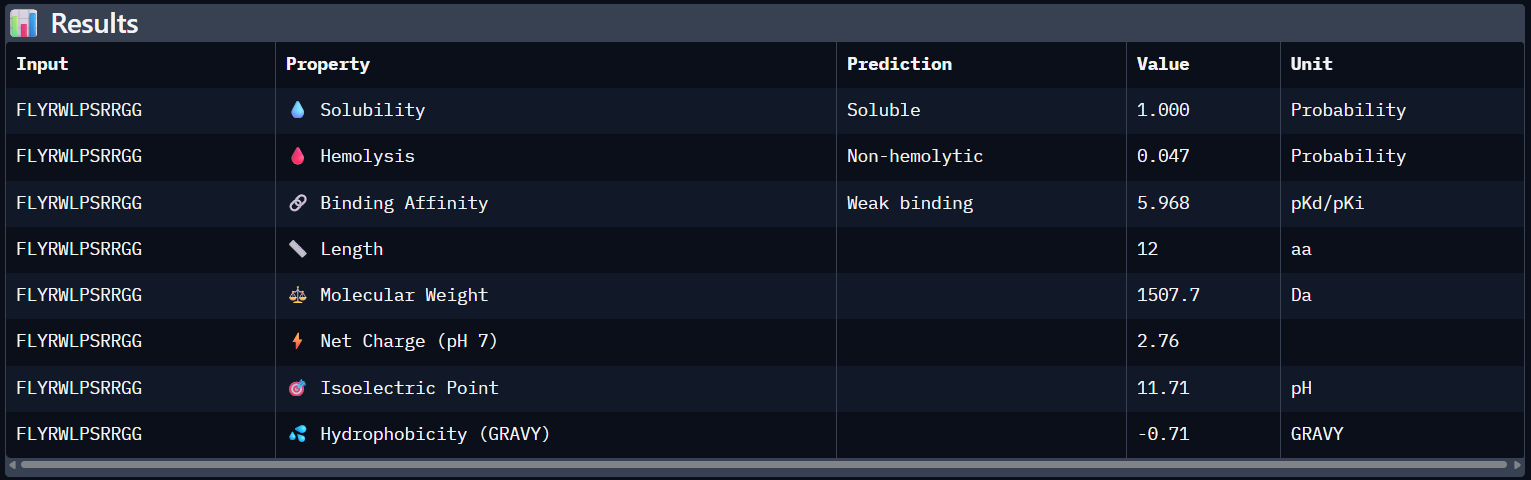
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Here’s the updated list with the known SOD1-binding peptide:
Binder
WRYGVAGVRHWX
WLYPPAVVEHKE
HRYYPTAVRWKX
WHYGVVGLAHKK
FLYRWLPSRRGG
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Here’s the final list of binders with their respective perplexity scores
Binder
Pseudo Perplexity
WRYGVAGVRHWX
13.614870
WLYPPAVVEHKE
20.275341
HRYYPTAVRWKX
10.113044
WHYGVVGLAHKK
12.192786
FLYRWLPSRRGG
-
My top-ranked candidate after looking at the generated binders is the third binder WLYYAVAVELGE (perplexity score: 10.11) because of its low perplexity score. That indicates high model confidence, so it should generate the best results out of the four binders.
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Side note: since AlphaFold doesn’t support the X’s, I decided to use the neutral amino acid Alanine (A)
Peptide N°1: WRYGVAGVRHWX Seed: 1418500094
Peptide N°2: WLYPPAVVEHKE Seed: 1181188013
Peptide N°3: HRYYPTAVRWKX Seed: 826762887
Peptide N°4: WHYGVVGLAHKK Seed: 1427381627
Peptide N°5: FLYRWLPSRRGG Seed: 449653589
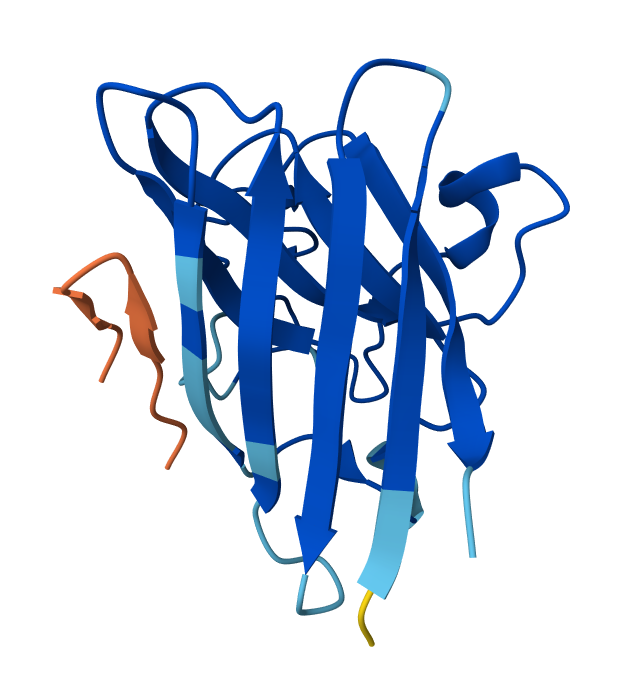
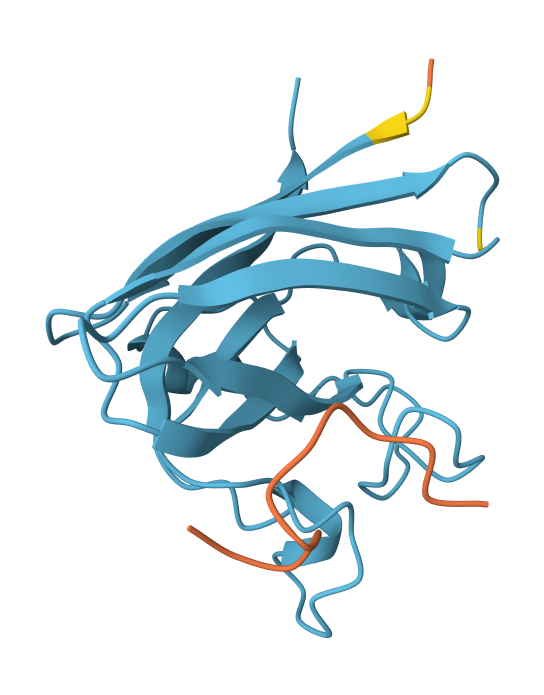
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Binder
Pseudo Perplexity
ipTM score
WRYGVAGVRHWX
13.614870
0.42
WLYPPAVVEHKE
20.275341
0.25
HRYYPTAVRWKX
10.113044
0.41
WHYGVVGLAHKK
12.192786
0.34
FLYRWLPSRRGG
-
0.31
Peptide N°1: This one binds predominantly near the β-barrel region and partially engages the dimer interface. It appears partially buried within a surface groove, suggesting strong structural complementarity.
Peptide N°2: This candidate localizes near the N-terminus, specifically approaching the A4V mutation site. However, it remains mostly surface-bound with lower structural confidence.
Peptide N°3: Similar to the first binder, this peptide anchors itself against the β-barrel, showing a stable orientation that is partially buried against the protein core.
Peptide N°4: It localizes at the edge of the dimer interface, appearing as a surface-bound “cap” rather than a buried ligand.
Peptide N°5 (reference): The known binder shows a moderate ipTM, localizing primarily at the dimer interface of the SOD1 mutant.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The observed ipTM scores are moderate and lower than 0.5, but two of my peptides, mutant_peptide_1 and mutant_peptide_3, significantly exceeded the known reference binder (0.31). This confirms that PepMLM identified novel sequence patterns with better structural affinity for the mutant surface than the reference. While these scores aren’t yet at “drug-level” affinity, they provide a much better starting point for optimization than the current benchmark.
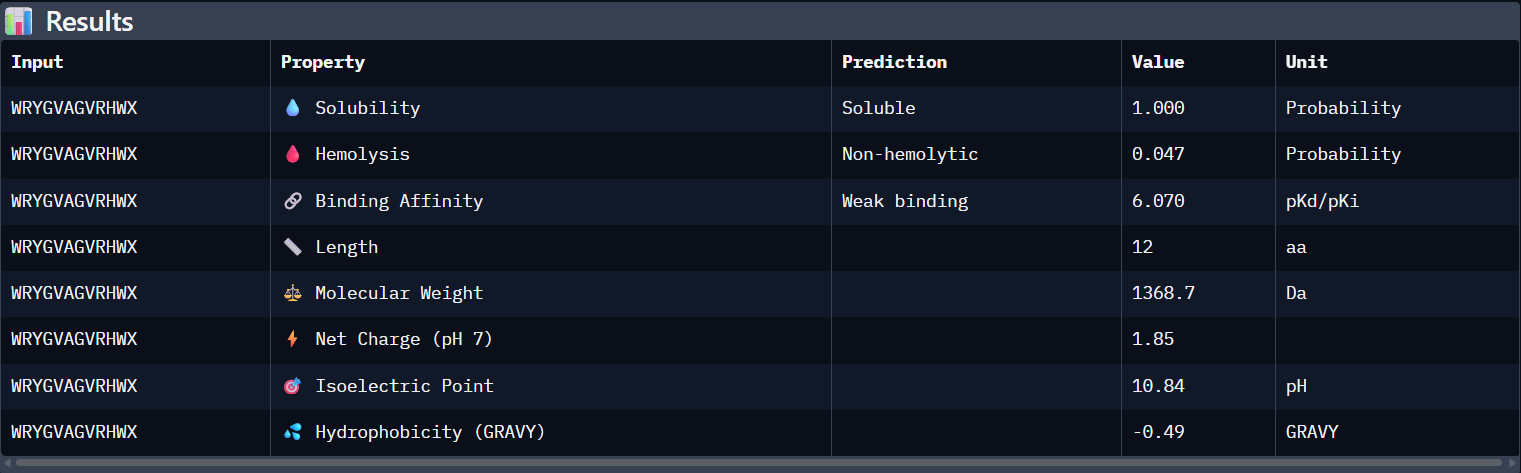
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide!
For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Peptide N°1:
Peptide N°2:
Peptide N°3:
Peptide N°4:
Peptide N°5:
Analysis of the PeptiVerse Results:
Peptide
ipTM (AF3)
Binding Affinity (pKd)
Solubility
Hemolysis (Prob)
Net Charge
WRYGVAGVRHWA
0.42
6.070
Soluble (1.00)
0.047
+1.85
WLYPPAVVEHKE
0.25
5.526
Soluble (1.00)
0.017
-1.14
HRYYPTAVRWKA
0.41
5.471
Soluble (1.00)
0.018
+2.84
WHYGVVGLAHKK
0.34
5.555
Soluble (1.00)
0.025
+1.93
FLYRWLPSRRGG
0.31
5.968
Soluble (1.00)
0.047
+2.76
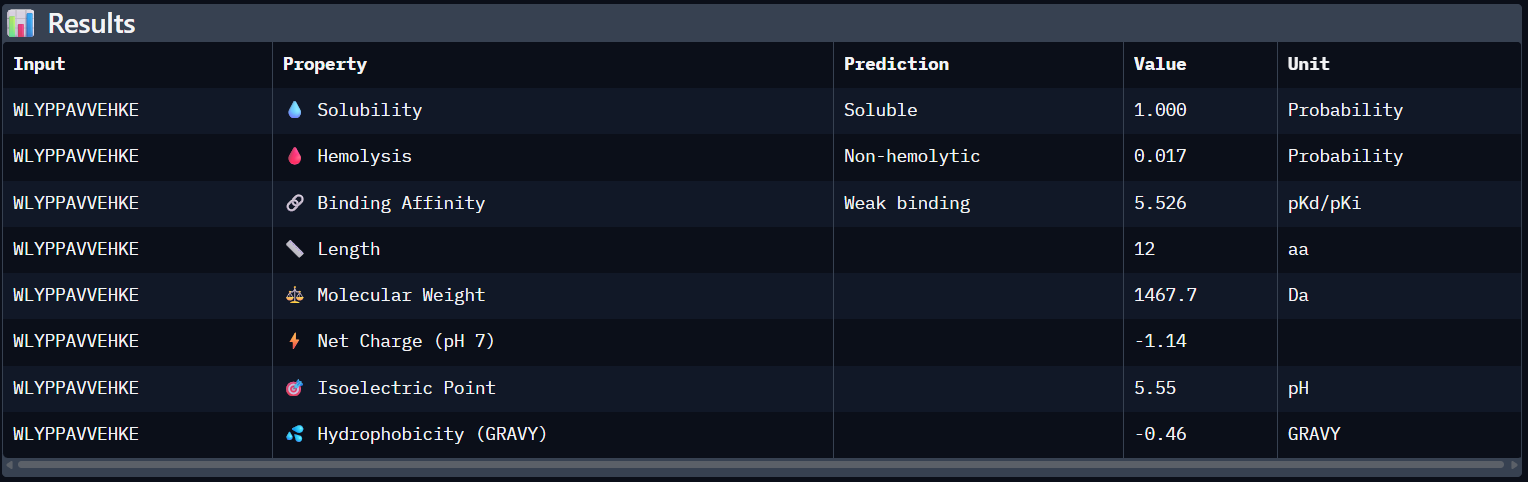
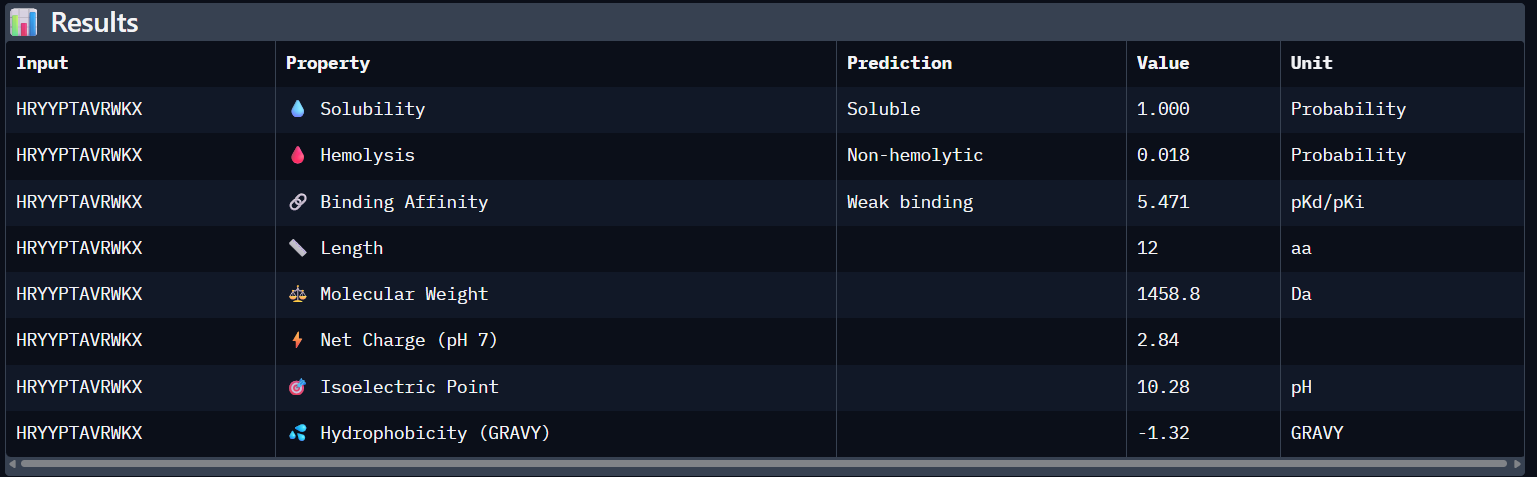
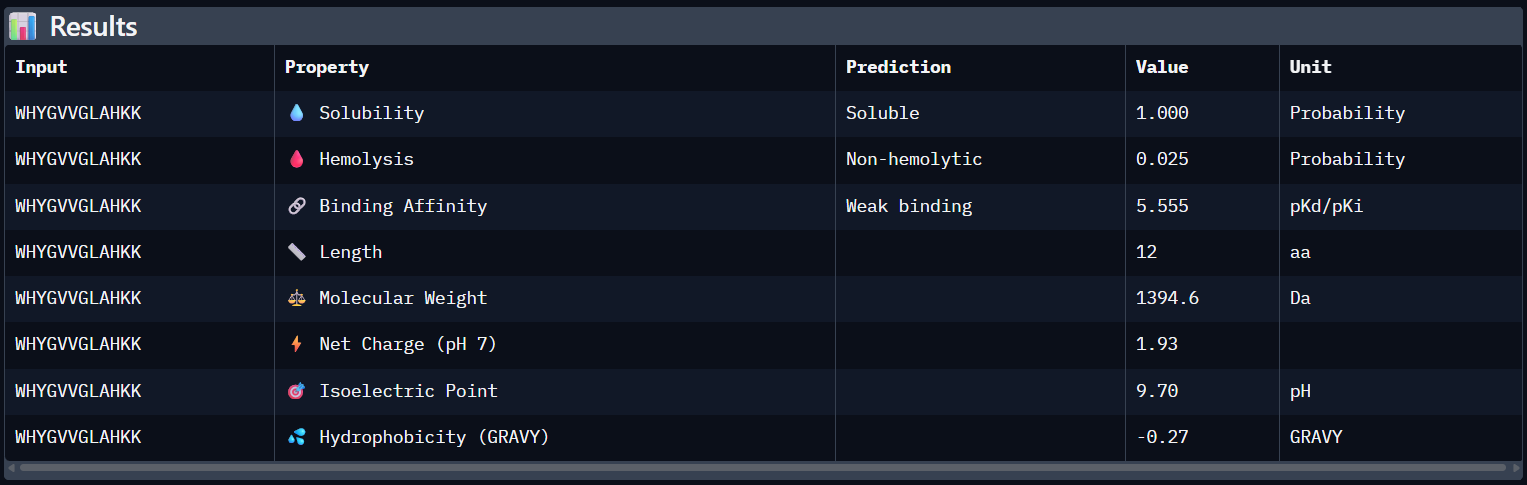
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Comparing AlphaFold3’s structural confidence with PeptiVerse predictions shows that structural docking alone isn’t enough for drug design. My first and third PepMLM binders, WRYGVAGVRHWX and HRYYPTAVRWKX, both exceeded the known reference binder in ipTM score (0.42 and 0.41 vs. 0.31). Crucially, PeptiVerse shows these new designs are less hemolytic than the reference (0.018 vs. 0.047), which is a significant safety improvement.
Choose one peptide you would advance and justify your decision briefly.
I would choose to advance with my third peptide (HRYYPTAVRWKX) for therapeutic development because it strikes the best balance between structural fit and safety. It has the lowest chance of hemolysis (0.018) and is expected to be completely soluble (1.000). Its low pseudo-perplexity (10.11) also reflects high model confidence. Aditionally, the positive net charge (+2.84) should favor its interaction with the mutant SOD1 surface in the cytosolic environment.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
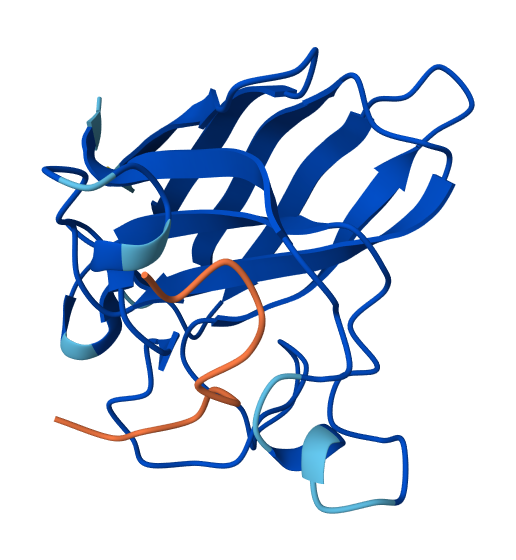
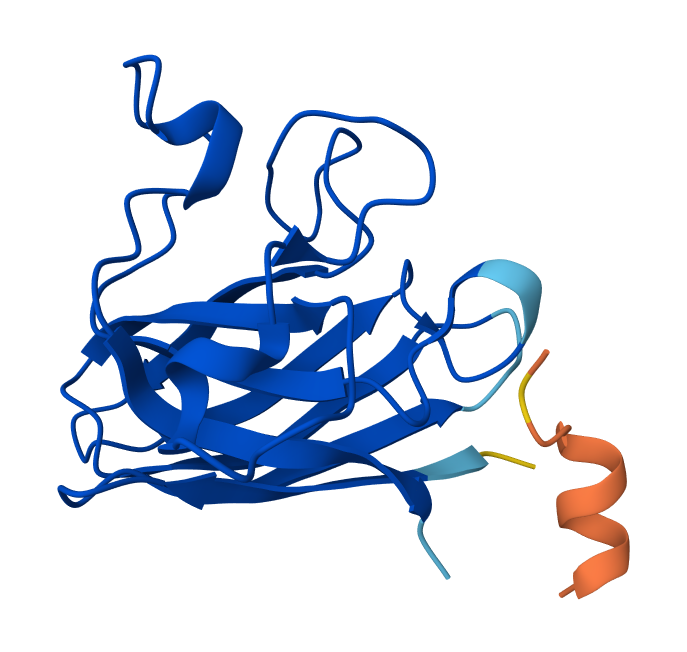
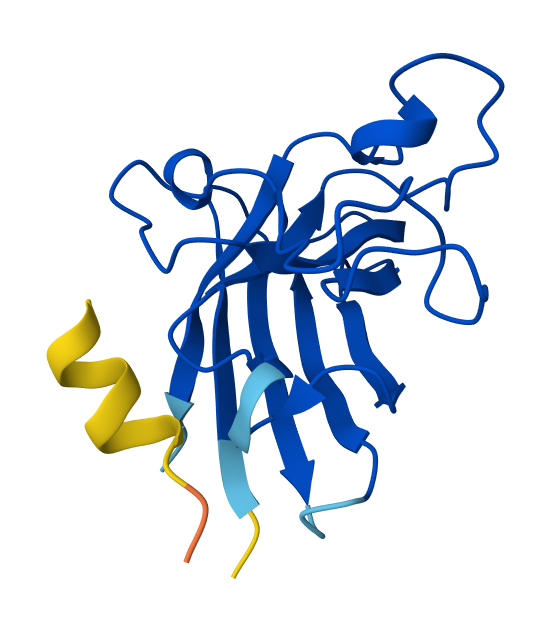
I selected residues 84-90 and 94-102 from two adjacent β-strands (ending at Asp84 and Asp102) to define a broad binding pocket on the β-barrel surface.
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
These are the binders moPPit generated:
RVRTYKRTQKEM
KCYSLKLKKKYY
YEYYKKKTCQKH
Using these parameters:
Additionally, I wanted to see how AlphaFold3 evaluated the new optimized binders, so here are the results:
Optimized Peptide N°1:
Optimized Peptide N°2:
Optimized Peptide N°3:
Binder
AlphaFol3 Seed
ipTM score
RVRTYKRTQKEM
2124434605
0.34
KCYSLKLKKKYY
276492257
0.32
YEYYKKKTCQKH
1326061601
0.45
The third moPPIt-generated peptide is quite interesting since it achieved an ipTM score of 0.45 after evaluating it using AlphaFold3.
This means that by “sculpting” a sequence specifically complemented to the Asp84-Asp102 motif, moPPIt created a high-affinity “molecular staple” that reinforces the SOD1 β-barrel core. This targeted motif approach balances affinity, solubility, and specificity and aims to prevent the structural collapse and toxic aggregation triggered by the A4V mutation, providing a precise lead candidate for clinical development.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a
MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
This homework requires computation that might take you a while to run, so please get started early.
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
Soluble N-terminal domain: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYV
TM domain: LIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
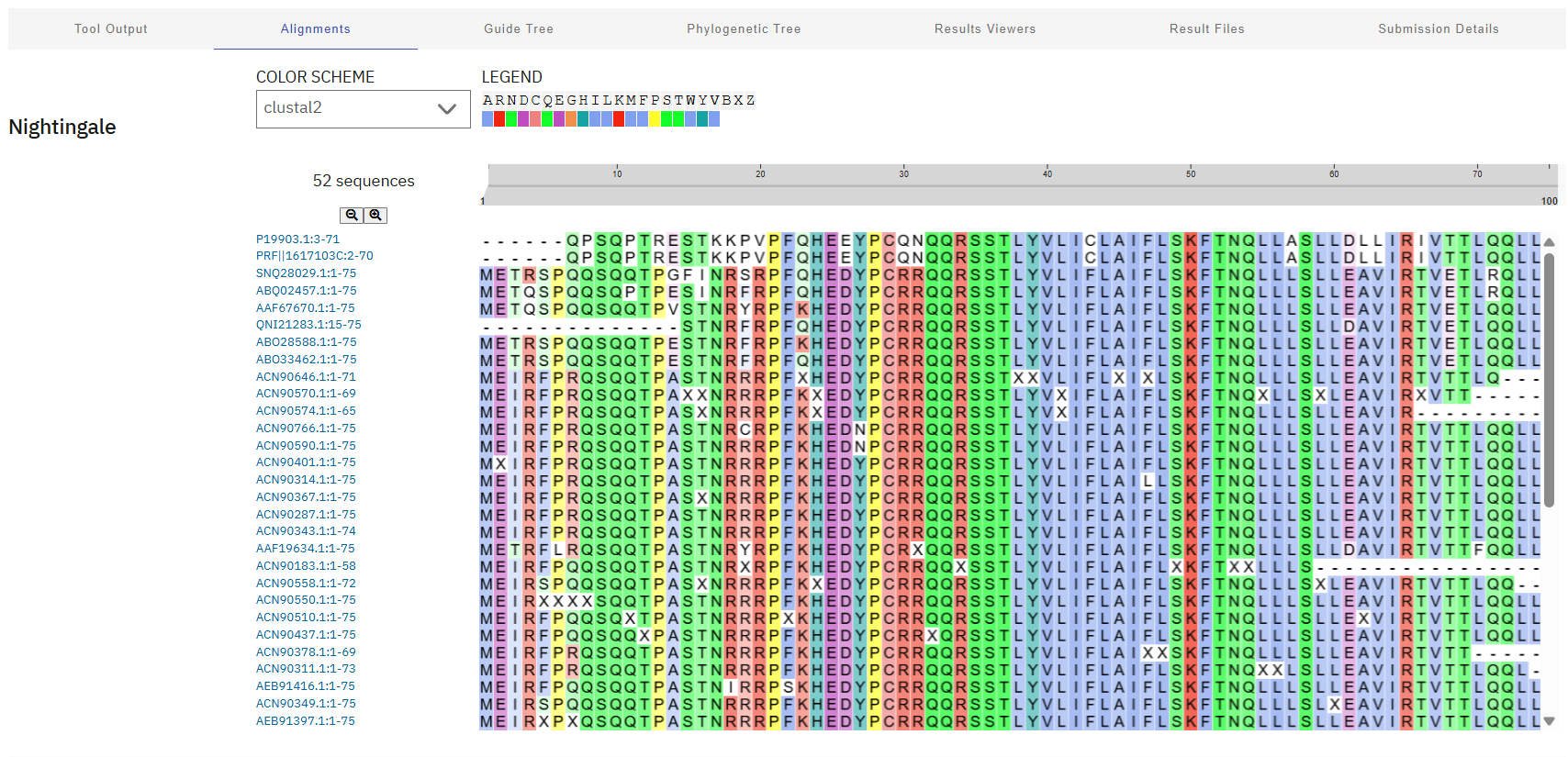
Additionally, here’s a screenshot of the BLAST results for L-protein:
Lastly, these results were aligned using Clustal Omega, revealing a highly conserved “island” (HEDYPCRRQQRSST) at residues 24-38. These sites will be avoided during mutagenesis to preserve the critical interaction with DnaJ and overall biological function of the phage.
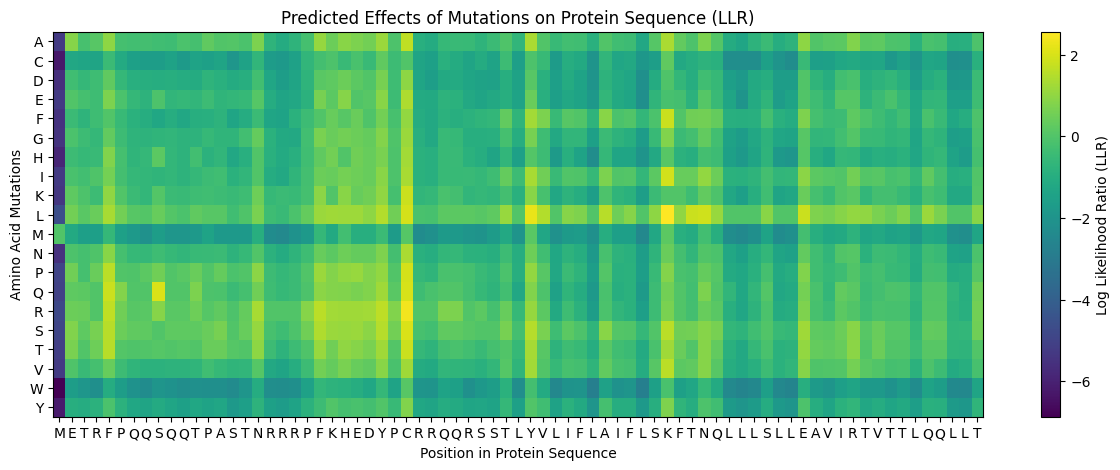
My approach is very straightforward: I combined computational LLR scores with experimental lab data using a copy of the HTGAA Colab. I filtered for mutations that showed “active lysis” (value 1) in the experimental spreadsheet and high positive LLR scores in the notebook.
Step 3: Filtering and Ranking
I used AlphaFold 3 to model the 8-chain assembly. This step was used to rank candidates that showed both positive computational scores and confirmed experimental activity, ensuring they don’t disrupt the pore’s symmetry.
Step 4: Final Mutated Sequences
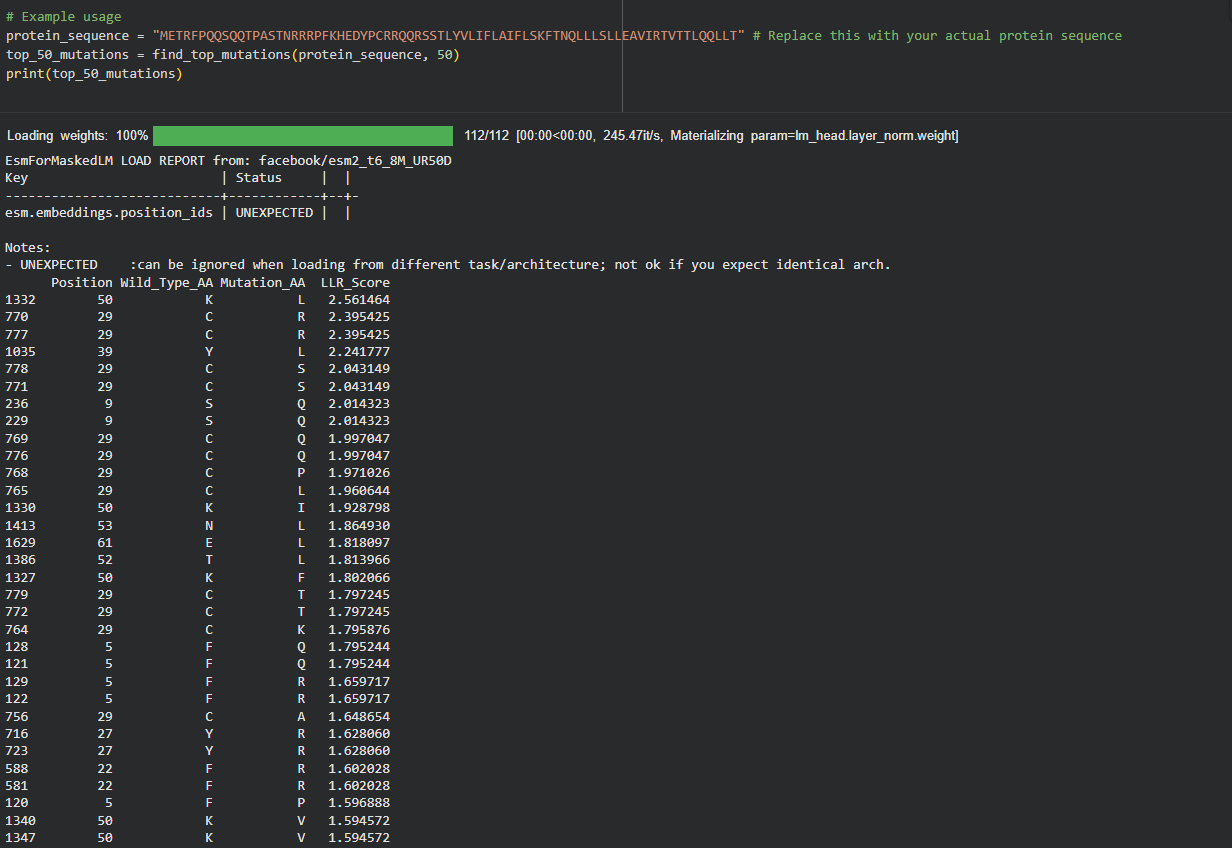
These 5 mutations were selected because they are experimentally proven to maintain lysis (score 1) and show improved or stable computational scores.
Region
Mutation
LLR Score (ESM-2)
Experimental Lysis
Rationale
Soluble
S9Q
2.01
Active (1)
High computational confidence; replaces Serine with Glutamine to stabilize the N-terminal loop.
Soluble
C29R
2.39
Active (1)
One of the top scores; removing this Cysteine likely prevents incorrect disulfide bonding.
TM Domain
Y39L
2.24
Active (1)
High confidence score in the TM interface; optimizes hydrophobicity for membrane entry.
TM Domain
A45L
1.53
Active (1)
Consistent with experimental data; improves the hydrophobic core of the lytic pore.
TM Domain
N53L
1.86
Active (1)
Replaces a polar Asparagine with Leucine, significantly improving helix-helix packing in the multimer.
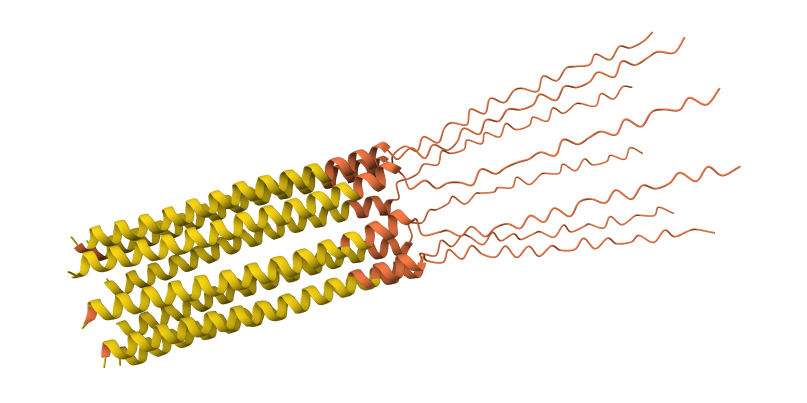
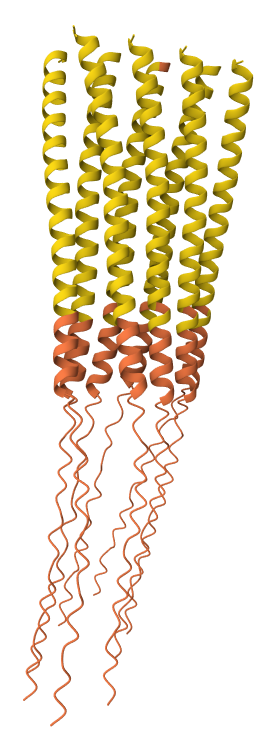
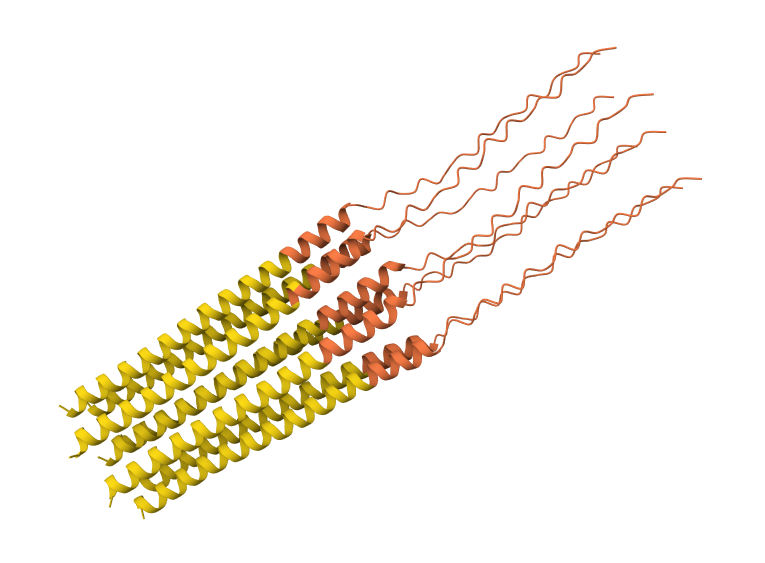
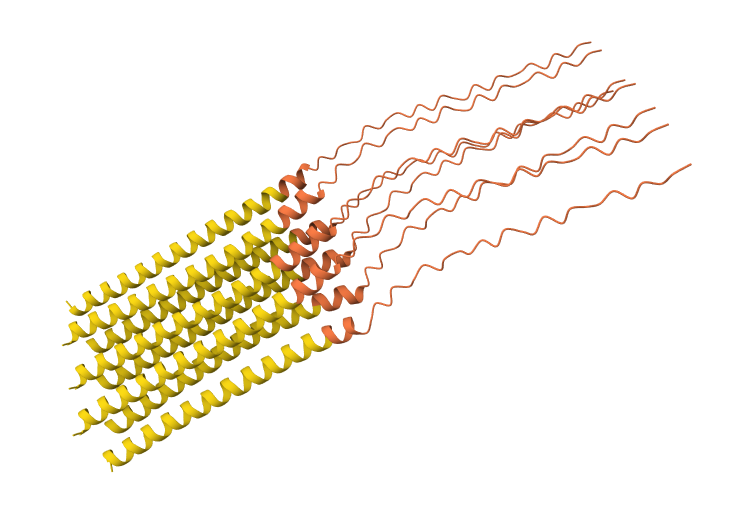
S9Q mutation 8-chain assembly:
C29R mutation 8-chain assembly:
Y39L mutation 8-chain assembly:
A45L mutation 8-chain assembly:
N53L mutation 8-chain assembly:
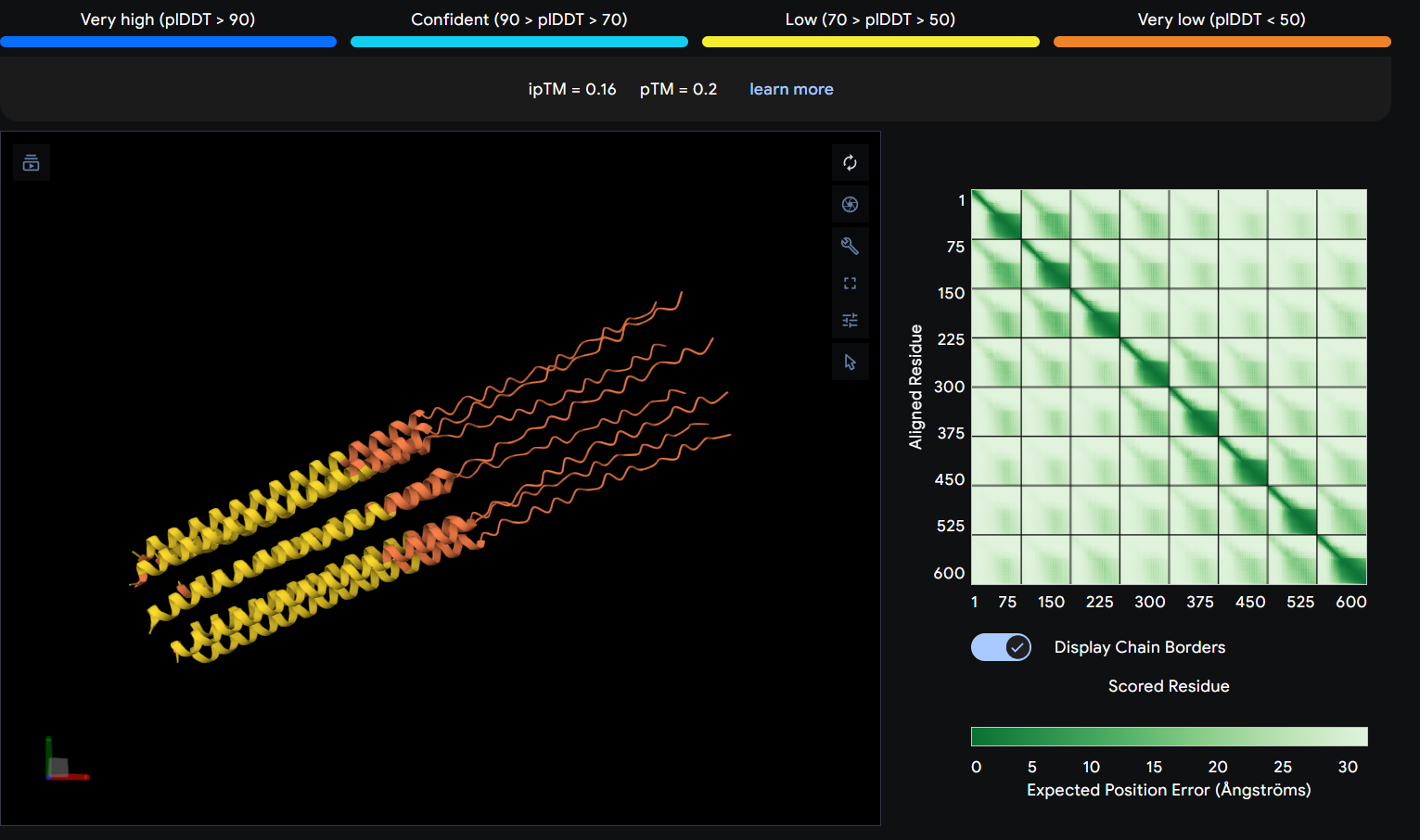
While AF3 structures were used to visualize the multimeric orientation, the ipTM scores remained low (~0.17) across all mutations. This is expected given the small, intrinsically disordered nature of the L-protein and the high flexibility required for its lytic function, which challenges standard multimeric confidence metrics.
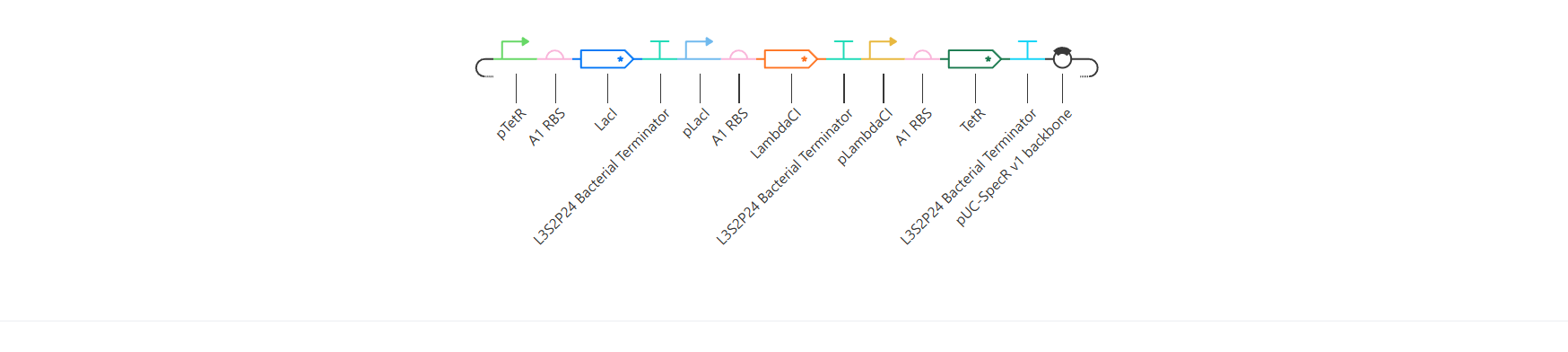
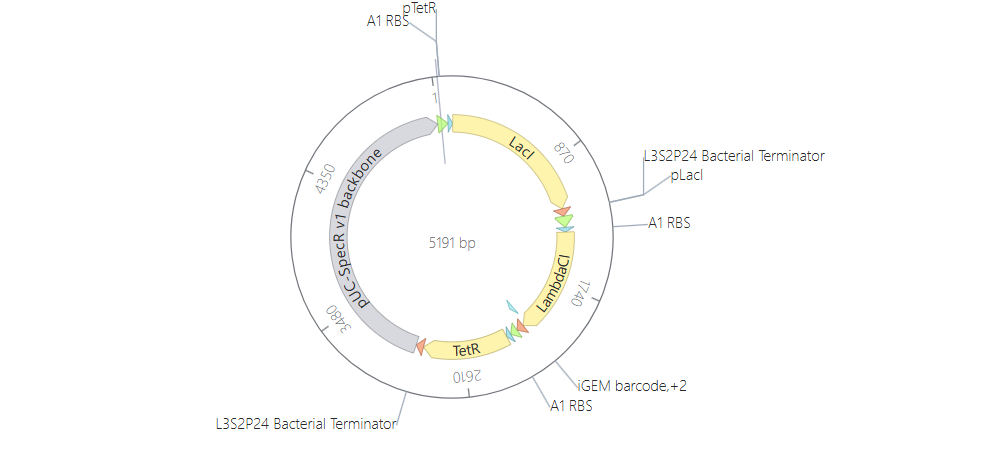
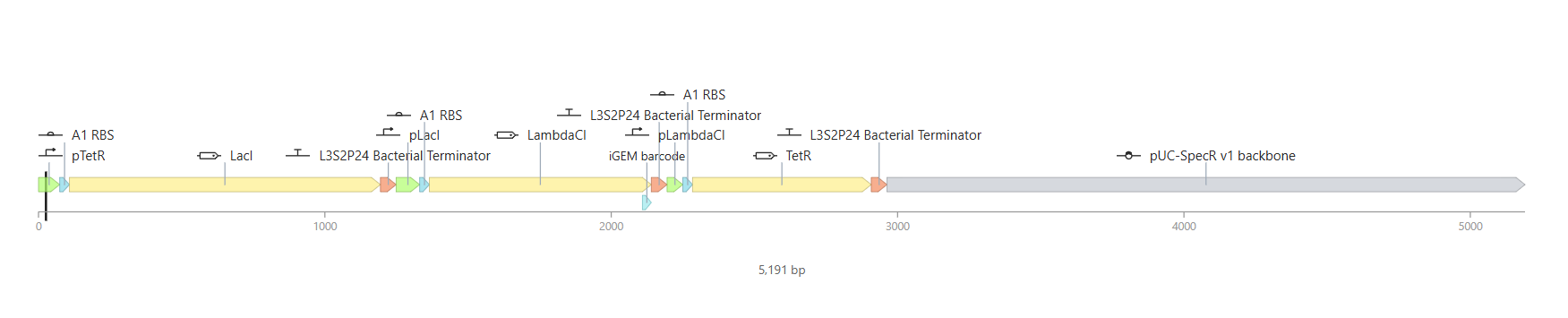
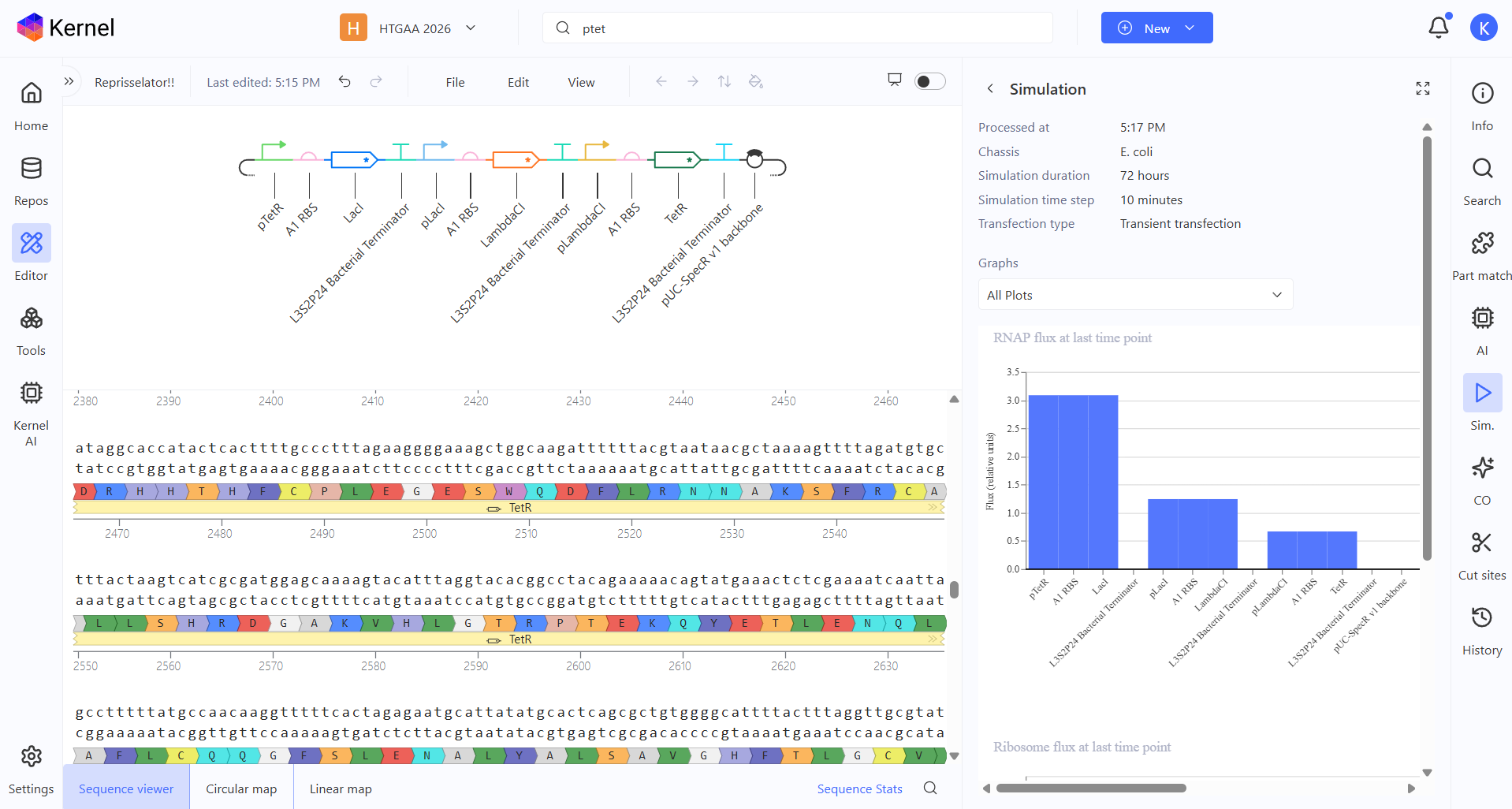
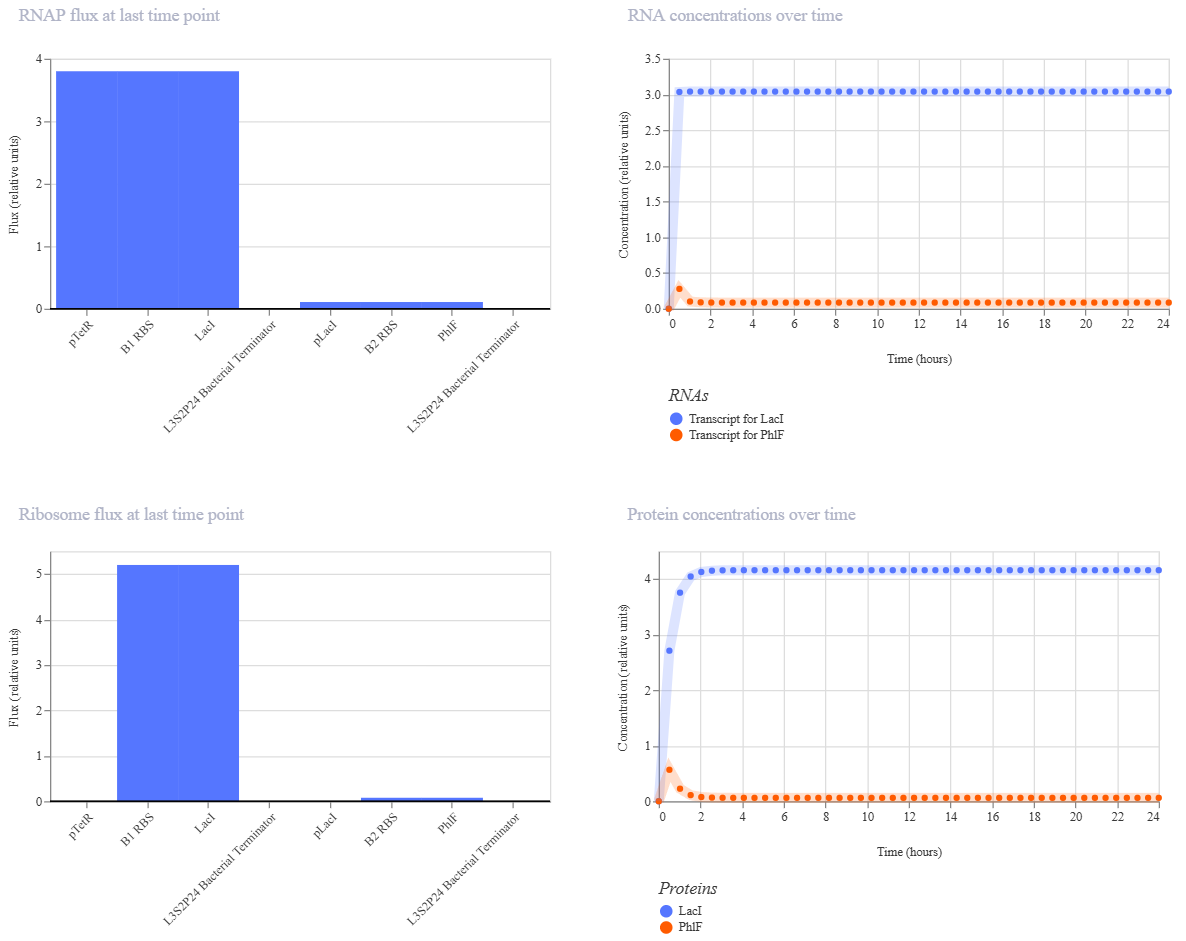
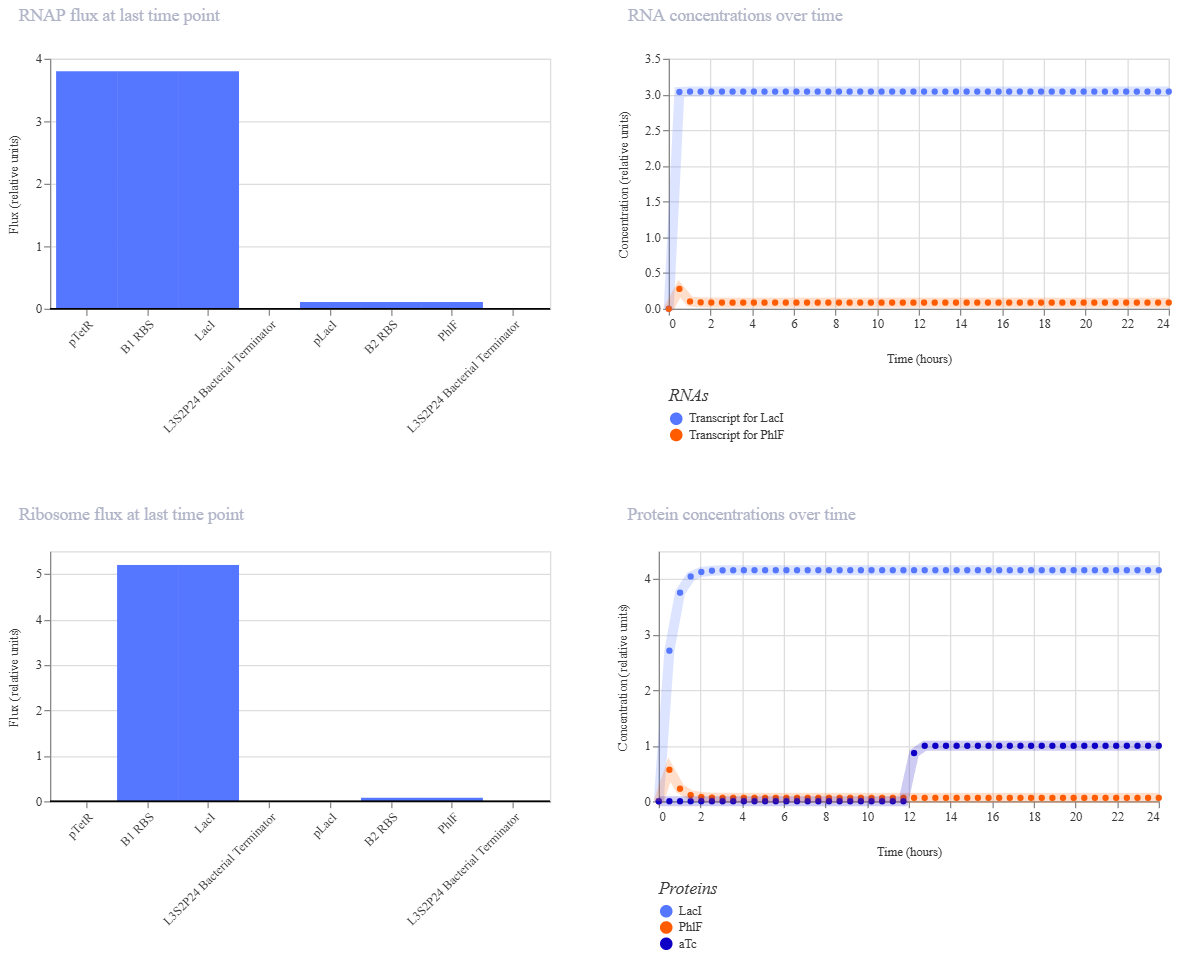
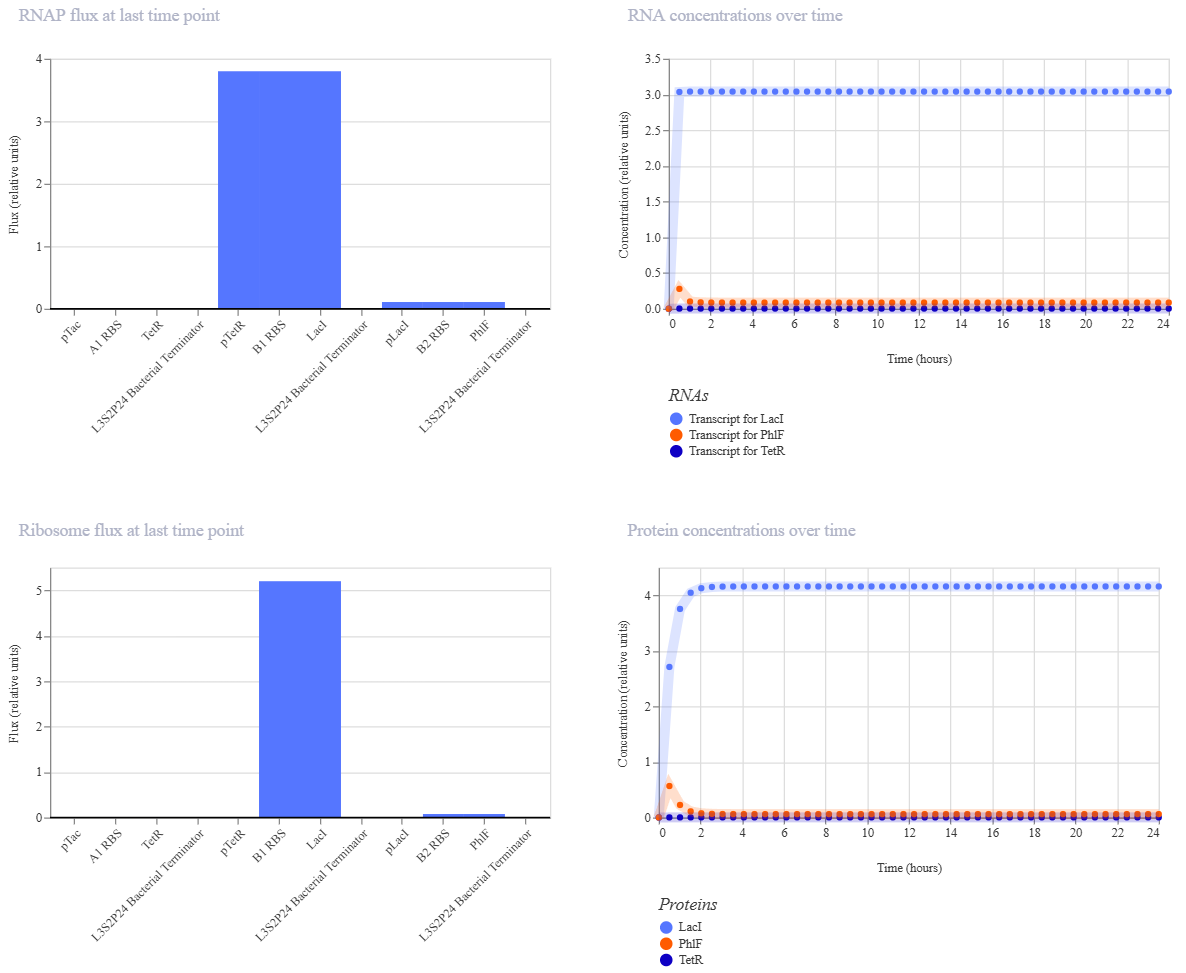
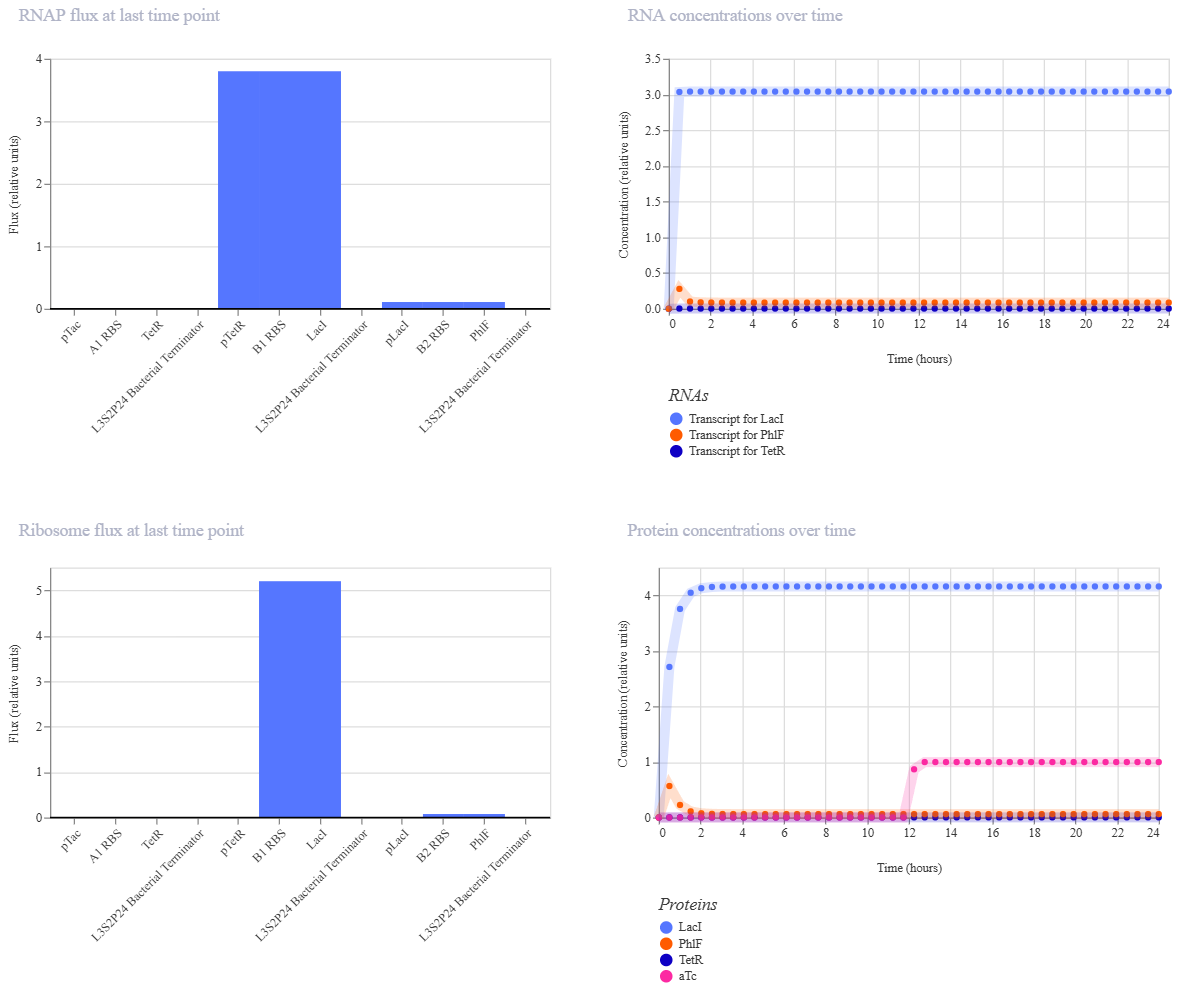
Week 6 HW: Genetic Circuits Part I
Homework
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Some components in the Phusion High-Fideñity PCR Master Mix include the Phusion DNA Polymerase, which is the enzyme that actually builds the new DNA strands with high accuracy. It also contains dNTPs, which are the building blocks (A, T, C, and G) used to synthesize the DNA. There are also buffer salts and magnesium ions (Mg^2+) that provide the right chemical environment for the enzyme to stay stable and work efficiently.
What are some factors that determine primer annealing temperature during PCR?
The biggest factor is the melting temperature (T_m) of the primers, which is mostly determined by their length and GC content since G-C pairs have stronger bonds than A-T pairs. The concentration of salts in the PCR buffer and the concentration of the primers themselves also play a huge role. Usually the annealing temperature is set about 2 to 5°C below the lower T_m of the primer pair to make sure they bind specifically to the template.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
For instance, PCR uses a thermocycler and specific primers to synthesize millions of new copies of a specific DNA segment which is great for adding mutations or overhangs for assembly. While restriction digests use enzymes to cut existing DNA at specific recognition sites, which is a much simpler “cut and paste” protocol.
PCR is preferable when you need to create a lot of DNA from a tiny sample or when you need to change the sequence (like our color mutations). Digests are better when you already have the DNA and just need to move a large, pre-existing block without the risk of mutation errors from a polymerase.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
I’d have to verify that the ends of my DNA fragments have overlapping sequences of about 20 to 40 base pairs that are identical to the fragment they are being joined to. In the lab protocol, it is verified by designing primers with 5’ overhangs that match the other fragment. I’d also need to check the concentration via Nanodrop to ensure I have enough DNA and run a diagnostic gel to confirm the fragments are the correct size before mixing them.
How does the plasmid DNA enter the E. coli cells during transformation?
The DNA enters through temporary pores created in the bacterial cell wall. In heat shock we use an abrupt temperature change to stress the membrane and open these pores, while in electroporation we use a high-voltage pulse to achieve the same thing. Once the pores are open, the plasmid moves into the cell by simple diffusion, and then we give the cells SOC media so they can recover and close those pores back up.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a really cool method that uses Type IIS restriction enzymes and T4 DNA ligase to assemble multiple fragments in a single reaction. Unlike standard enzymes, these cut outside of their recognition sites, which allows for “scarless” assembly because the recognition sequences are removed during the process. You design the fragments so that they create unique 4-base pair overhangs that guide the pieces together in the correct order. This is super efficient for building complex circuits because you can put many fragments together at once in one tube. It relies on a cycle of different temperatures to keep the cutting and pasting going until the final circular plasmid is formed.
Here’s a diagram of the first step of Golden Gate Assembly from Snapgene
Model this assembly method with Benchling or Asimov Kernel!
Assignment: Asimov Kernel
Create a Repository for your work
I created my repository under the name HTGAA 2026 - Fabrizio Flores in Asimov Kernel!
Create a blank Notebook entry to document the homework and save it to that Repository
I created a blank notebook entry!
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Bacterial Demos repo Exploration
After finding the Bacterial Demos Repo on Asimov Kernel, I started looking at all the constructs, starting with the Repressilator one! There are 3 promoters with different properties, biological logic gates and more complex circuits like the self-regulating one or the Multiplexer.
Repressilator demo
Comparing promoters demo
J23117 promoter demo
Multiplexer demo
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Repressilator recreation
So first, I created an empty Construct to start my Repressilator recreation!
Then I searched the Repressilator parts one by one and started adding them up on my empty Construct.
Here you can see the construct, the linear map and the circular map better!
After the Represillator construction, I ran the simulation and got these results:
And here are the results from the Repressilator Construct found in the Bacterial Demos repository:
At first, my results seemed not to match the ones I got from the Represillator from the Bacterial Demos repository since my RNA and protein concentrations over time weren’t oscillating. That made me think that maybe I had made a mistake in my reconstruction, so I started to compare the constructs and realized that I had used pTet instead of pTetR at the start (my bad haha.)
So I changed that promoter and now the construct looks like this:
And here you can see the construct more detailed in its circular map and linear map forms:
And after running the simulation again, my results finally matched the one from the Bacterial Demo repo:
All of the process is documented in my Notebook entry on Asimov Kernel
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
My Constructs
Construct 1
So for my first construct I tried making an inverter (NOT logic gate) where the first operon acts as a “sensor,” driving the expression of the LacI repressor under the pTetR promoter, while the second operon serves as the “output,” where the reporter gene is controlled by the LacI-sensitive promoter pLacI.
To test it, I ran two simulations using these parameters:
Simulation 2 parameters: Chassis: E. coli Duration: 24 hours Timestep: 30 minutes Transfection: Transient transfection Ligands: add aTc at time 12 Hours
The inverter successfully maintained a “LOW” output state, but the induction failed to flip the switch. Even after adding aTc at the 12-hour mark, the reporter concentration didn’t show a significant increase.
The reason for this result is a missing link in the induction machinery. Here, PTetR behaves as a constitutive promoter unless the TetR protein is already present in the chassis. Since my construct doesn’t include a gene to produce TetR, the aTc ligand has no target to bind to. Consequently, pTetR remains at maximum power, keeping LacI levels high enough to permanently suppress the output.
To improve the circuit and achieve a successful induction, I would need to add a third constitutive operon expressing the TetR protein.
Construct 2
For my second construct, I wanted to demonstrate transcriptional homeostasis by building a negative feedback loop. In this design, I used the pTac promoter to drive the expression of its own repressor, LacI, followed by a strong terminator. Here are the construct and results:
To test it, I ran two simulations using these parameters:
Simulation 2 parameters: Chassis: E. coli Duration: 24 hours Timestep: 30 minutes Transfection: Transient transfection Ligands: add aTc at time 12 Hours
My expectation was to see a rapid initial burst of protein production that quickly plateaus into a very stable steady state, preventing the expression from overshooting. The simulation results matched this perfectly, showing a clean line after the first two hours, which confirms the circuit’s ability to maintain a consistent equilibrium state.
Construct 3
For my third construct, I decided to build a Double Inverter (Signal Cascade) to demonstrate how signal transduction works between different regulatory layers. I designed a first operon where the pBad promoter (activated by L-arabinose) drives the expression of the TetR repressor. This is followed by a second operon where the pTetR promoter controls the expression of the PhlF reporter protein.
To test it, I ran two simulations using these parameters:
Simulation 2 parameters: Chassis: E. coli Duration: 24 hours Timestep: 30 minutes Transfection: Transient transfection Ligands: add L-arabinoseat at time 6Hours
Simulation 3 parameters: Chassis: E. coli Duration: 24 hours Timestep: 30 minutes Transfection: Transient transfection Ligands: add L-arabinoseat at time 12 Hours
The simulation results under 6h and 12h induction showed exactly what I expected: as soon as L-arabinose triggers the spike in TetR, there’s a corresponding sharp drop in PhlF. I noticed a slight ’lag’ or delay between the two curves, which is a classic characteristic of biological cascades, as the second repressor needs time to accumulate and saturate the downstream promoter. To decrease this response time, I would suggest using a stronger RBS for the TetR gene to reach the inhibitory threshold faster.
Week 7 HW: Genetic Circuits Part II
This week covers neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network
“perceptron”-like computation and learning.
Homework
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional Boolean circuits are limited because they only understand “on” or “off” (0 or 1), which doesn’t reflect the noisy and analog reality of a cell. IANNs allow for weighted inputs and non-linear integration, meaning the cell can make decisions based on the concentration of signals rather than just their presence. This allows for complex pattern recognition, like identifying a specific metabolic state or a signature of multiple biomarkers, making the decision-making process much more robust and “intelligent” than a simple AND/OR gate.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A really great application would be a Smart Cancer Detector. The inputs (X_1, X_2, … X_n) would be the concentrations of different microRNAs or proteins that are slightly elevated in cancer cells but also present in healthy ones. An IANN could “weight” these signals so that only a specific combination above a certain threshold triggers the output ($Y$), which could be a pro-apoptotic protein to kill the cell.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
So I made the diagram for an intracellular multilayer perceptron, where the output of the first promoter must directly feed into the target regulation sequence of the subsequent node. Since the input X_1 introduces CasE via the PgU plasmid, the Bias 1 ($B_1$) node must carry the specific target loop for CasE while encoding the next layer’s enzyme (Csy4).
Corrected Component Mapping Matrix
Input X_1:PgU DNA (Synthesizes the functional CasE endoribonuclease).
Input X_2:PgU_rec_mNeonGreen DNA
Bias 1 (B_1):PgU_rec_Csy4 DNA (carrying the CasE target loop) $\rightarrow$ This is the first hidden processing gate.
Bias 2 (B_2):Csy4_rec_mNeonGreen DNA (carrying the Csy4 target loop) $\rightarrow$ This is the second processing gate regulating the final output.
Final Output (Y):mNeonGreen fluorescence signal.
Here’s my submission for the Neuromorphic Wizard lab and result!
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Some of the most well-known fungal materials are mycelium-based packaging, fungal leather like Mylo, and structural bricks used in bio-construction. The main advantage is that they are completely biodegradable and carbon-negative since they grow from agricultural waste, which fits perfectly into a circular economy model. However, they still face disadvantages compared to traditional materials because their mechanical strength is lower than concrete or plastic and their properties vary a lot depending on the substrate used, making industrial standardization quite difficult.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
In my case, I would use genetic engineering to make fungi secrete specific enzymes or cross-linking proteins like tyrosinases directly into the mycelium mat, allowing the material to gain stiffness and water resistance automatically as it grows. The advantage of using fungi instead of bacteria is that being eukaryotes, they can perform complex post-translational modifications on proteins. Also, their filamentous growth through hyphae allows them to bridge physical gaps and create 3D structures with inherent mechanical integrity, which is something bacteria cannot achieve since they mostly form biofilms without such an organized physical structure.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS Submitted
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 HW: Cell-Free Systems
This week introduces synthesis of proteins using cellular machinery outside of a cell.
Homework
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
The biggest advantage of cell-free systems is that they offer an open environment where you have total control over experimental variables like pH and salt concentrations without a cell membrane getting in the way. This flexibility is especially beneficial when producing antimicrobial peptides or lysis proteins that would normally kill a living host, as well as for high-throughput screening of genetic circuits where you need to test many DNA variants in hours rather than waiting days for cultures to grow.
Describe the main components of a cell-free expression system and explain the role of each component.
A standard system essentially needs three main parts to function properly. The cell extract acts as the hardware, providing ribosomes and tRNAs, while the Energy Mix serves as the fuel by providing ATP and secondary sources like PEP. Finally, the DNA template works as the software instructions that contain the specific gene sequence you want to express in the tube.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is absolutely critical in cell-free systems because once the initial ATP is used up, the synthesis stops since there is no active metabolism to recharge it like in a living cell. To ensure a continuous ATP supply during your experiment, you can use an enzymatic system such as creatine phosphate and creatine kinase to constantly convert ADP back into ATP while the reaction is running.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems are generally fast and high-yield but struggle with complex folding, whereas eukaryotic systems are slower but capable of post-translational modifications. For a prokaryotic setup, I would produce GFP because it gives a fast and simple fluorescence readout, but for a eukaryotic system, I would choose human insulin because it requires specific disulfide bonds that bacteria usually get wrong.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Designing a cell-free experiment for membrane proteins is a challenge because these proteins are hydrophobic and tend to clump up in a liquid environment. To address this in my setup, I would add nanodiscs or liposomes to the reaction to provide a synthetic lipid bilayer where the protein can fold correctly as it is being synthesized, effectively mimicking its natural cellular environment.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
If I observe a low yield, it could usually due to three main issues that require a specific troubleshooting strategy. First, if the DNA template is degraded or salty I should re-purify it and check its integrity on a gel. Second, if the Magnesium (Mg^2+) levels are off for my specific extract I should run a titration assay to find the optimal concentration. Finally, to prevent RNase contamination from destroying my mRNA I should always add RNase inhibitors and use strictly nuclease-free reagents.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
My synthetic cell would act as a specific biosensor for mercury (Hg²⁺) in water. The input is the mercury ions present in the environment, and the output is the release of a small signaling molecule called AI-2 (Autoinducer-2). This AI-2 then acts as a signal for a secondary population of natural bacteria to trigger a visible response.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Not effectively. Without encapsulation, the AI-2 signal would be released immediately into the medium regardless of the mercury’s presence. The vesicle acts as a “logic gate” that keeps the signal locked inside until the mercury triggers the production of a membrane pore to release it.
Could this function be realized by genetically modified natural cell?
Yes, I could engineer a bacterium to do this, but natural cells often have cross-talk with other metals or metabolic burdens that affect the sensor. Using an SMC allows us to create a “clean” sensor that doesn’t die from the mercury toxicity and only responds to that specific input.
Describe the desired outcome of your synthetic cell operation.
When placed in a contaminated water sample, the SMCs detect mercury and release AI-2. This signal then turns a reporter colony of E. coli bright blue, providing a clear visual warning of the contamination
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
I would use a mix of POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine) and cholesterol to ensure a stable, fluid membrane that mimics a natural cell but stays durable for field testing.
What would you encapsulate inside? Enzymes, small molecules.
I would encapsulate a bacterial cell-free Tx/Tl system (like PURE), a high concentration of the signaling molecule AI-2, and the DNA plasmid containing the mercury-responsive circuit.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
A bacterial system is perfect here because the mercury resistance operon (mer operon) is native to bacteria and works very efficiently with bacterial ribosomes and polymerases.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The mercury ions are small enough to diffuse through the lipid bilayer or through small constitutive pores. Once inside, they trigger the expression of a larger membrane channel that finally lets the encapsulated AI-2 escape into the surrounding environment.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Genes: I would use the merR regulatory gene and the merRo/p promoter to control the expression of alpha-hemolysin (hlyA) from Staphylococcus aureus, which will form the output pores.
Biological cells: Reporter E. coli strain LSB001 which is engineered to respond to AI-2 by producing chromogenic proteins.
How will you measure the function of your system?
I would measure the blue color intensity of the reporter bacteria using a standard plate reader or even just a smartphone camera for a qualitative field test. For more precision during development, I could co-encapsulate a fluorescent dye like calcein to track the exact rate of pore formation and signal release via microscopy.
Example solution
Based on: Lentini, R. et al., 2014. Nat comm, 5, p.4012.
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output? Expand the sensing capacity of bacteria. Input: theophylline (inert to bacteria). Output of the SMC: IPTG. Output of the whole system: GFP produced in bacteria.
(Theophyline aptamer reference: *Martini, L. & Mansy, S.S., 2011. Cell-like systems with riboswitch controlled gene expression. Chemical Communications, 47(38), p.10734.*)
Could this function be realized by cell-free Tx/Tl alone, without encapsulation? No. If the IPTG were not encapsulated, it would go into the bacteria without the need of theophylline-induced membrane channel synthesis, thus the synthetic cell actuator would not exist.
Could this function be realized by genetically modified natural cell? Yes, in this particular case: the theophylline aptamer could be incorporated into a transformed gene. This lacks generality though – it is easier to make SMC than modify bacteria, so in this system a single bacteria reporter can be used to detect various small molecules.
Describe the desired outcome of your synthetic cell operation. In the presence of SMC, bacteria sense theophylline.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of? Phospholipids + cholesterol.
What would you encapsulate inside? Enzymes, small molecules. cell-free Tx/Tl system, IPTG, gene for membrane transporter under the control of theophylline aptamer.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian) Bacterial, because of the theophylline riboswitch used as SMC input.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?) The membrane is permeable to the input molecule (theophylline), the output is IPTG that will cross the membrane via the membrane pore created after theophyline-initiated gene expression.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC, cholesterol
Enzymes: bacterial cell-free Tx/Tl
Genes: a-hemolysin (aHL) to encapsulate in SMC
Biological cells: *E.coli* transformed with GFP under T7 promoter and a lac operator
How will you measure the function of your system? Measure GFP output of the cells via flow cytometry. Alternatively, use enzymatic reporter, like luciferase, and measure bulk output of the enzyme.
Artificial cells translate chemical signals for E. coli. (a) In the absence of artificial cells (circles), E. coli (oblong) cannot sense theophylline. (b) Artificial cells can be engineered to detect theophylline and in response release IPTG, a chemical signal that induces a response in E. coli.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
My concept is a smart textile integrated with freeze-dried cell-free sensors that changes color and activates neutralizing enzymes when it detects toxic air pollutants in urban environments.
How will the idea work, in more detail? Write 3-4 sentences or more.
The fabric is manufactured by embedding freeze-dried cell-free extracts into the fibers using a specialized coating or encapsulation method. When the wearer enters an area with high concentrations of a specific pollutant, the chemical acts as an inducer that triggers the cell-free genetic circuit. This reaction produces both a chromoprotein for a visible color change and a functional enzyme that actively breaks down the toxin on the surface of the fabric. By using a paper-like matrix within the textile, the biological machinery stays localized and ready to react the moment it comes into contact with the air.
What societal challenge or market need will this address?
This addresses the growing global crisis of air pollution and its impact on public health, especially in hyper-urbanized cities. It provides citizens with a wearable, real-time diagnostic tool that not only alerts them to invisible dangers but also offers a first line of active protection by degrading harmful chemicals around them.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
To handle the one-time use limitation, the textile could be designed with replaceable “bio-cartridges” or patches that are swapped out after an activation event occurs. We can address the water requirement by using the natural humidity in the air or the wearer’s perspiration to provide the initial hydration needed to restart the freeze-dried machinery. For stability, the cell-free components would be encapsulated in protective polymers to prevent degradation from UV light or temperature swings before the sensor is actually triggered.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Astronauts on long-duration missions face severe nutritional deficiencies because vitamins degrade quickly in space radiation. Vitamin B12 is crucial for neurological health and red blood cell production, yet we currently lack a way to test its levels without sending samples back to Earth. Solving this is vital for deep-space exploration, as it ensures crew health during missions to Mars where resupply is impossible. This is scientifically interesting because it explores how to maintain human homeostasis in an extreme environment using portable synthetic biology tools.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
My target is a B12-responsive riboswitch DNA sequence that regulates the expression of a fluorescent protein within the BioBits® cell-free system.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The molecular target acts as a biological sensor that detects the presence of active Vitamin B12 molecules. In space, maintaining specific nutrient levels is a constant battle against radiation-induced degradation and physiological changes. By integrating a B12-sensing riboswitch into a cell-free reaction, we can turn a complex nutritional assay into a simple visual test. This allows the crew to monitor their own health and the stability of their food supplies in real-time using minimal equipment, which is essential for surviving the constraints of microgravity.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My goal is to demonstrate that a BioBits® cell-free system can accurately quantify Vitamin B12 concentrations in a microgravity environment using fluorescence as a readout. I hypothesize that the B12-responsive riboswitch will remain functional in space and will effectively block or allow the translation of a fluorescent reporter in direct proportion to the vitamin levels present in the sample. The reasoning is that cell-free systems are highly stable when freeze-dried and avoid the complications of maintaining living cultures in orbit. If successful, this provides a low-cost, shelf-stable diagnostic platform that can be adapted to detect many different essential nutrients or even environmental toxins on the International Space Station.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I will need to test astronaut serum samples or rehydrated food extracts mixed with the BioBits® B12-sensor pellets. I will incubate the samples in the miniPCR® thermal cycler at 37°C to activate the cell-free reaction. I plan to use three controls: a positive control with a known B12 concentration, a negative control with nuclease-free water, and a non-responsive fluorescent DNA template. I will collect data by observing the reaction tubes in the P51 Molecular Fluorescence Viewer to measure light intensity, which correlates to the B12 concentration in the tested samples.
Homework Part B: Individual Final Project
Documentation on my final project page.
We’d like students to start exploring their final project in depth this week! Of your three Aims, for this week you should have at least Aim 1 decided and written down.
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1:
First Twist order deadline for MIT/Harvard/Wellesley students is Friday, April 3 at 11PM ET
First Twist order deadline for Committed Listeners is Friday, April 10 at 11PM ET. (Your Node Lead will place the Twist order, so please work with them to finalize your constructs and ordering decisions.)
Week 10 HW: Imaging and Measurement
Homework
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
For my final validation pipeline, I want to focus on measuring three primary aspects of the bioreactor system. First, I will monitor the Biomass Growth Rate ($\mu$ in $h^{-1}$) to evaluate our population’s structural viability under nutrient scarcity. Second, I plan to quantify the Intracellular PHB Accumulation ($\mu\text{g/mL}$) to establish the actual bioplastic synthesis yields achieved. Third, I would like to measure the Transcriptional Expression levels of the inserted operon to confirm that our biological components react precisely to the computed microenvironmental shifts.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
To perform these measurements, I would like to track cellular growth and biomass viability by sampling culture density at fixed intervals directly within a microfluidic plate matrix. I would follow the biopolymer accumulation by tracking the physical formation of intracellular inclusion bodies under starvation conditions. Additionally, I plan to execute gene expression profiling by isolating cellular transcripts at our peak optimization node, allowing me to check if the synthetic circuit actively overrides native metabolic constraints.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
I would like to execute the experimental protocol using three distinct bioengineering technologies. I could use Spectrophotometry ($OD_{600}$) via an automated microplate reader to calculate kinetic growth rates from real-time optical density data. For biopolymer quantification, I would like to deploy Nile Red Fluorescence Assays because they perform rapid, non-destructive tracking by binding a lipophilic fluorophore to the plastic granules, which allows us to measure signal emission at $590\text{ nm}$. Finally, I plan to use Quantitative Reverse Transcription PCR (RT-qPCR) to extract total cellular RNA, synthesize cDNA, and amplify the target phaABC sequences, confirming that our calculated $82.9%$ nutrient dilution effectively drives transcriptional overdrive through the synthetic NIT1 promoter.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Theoretical molecular weight of the original eGFP amino acid sequence using the expasy calculator.
Using the ExPASy Compute pI/Mw tool, the original eGFP amino acid sequence (without the His-purification tag and the LE linker) yields a theoretical molecular weight of 26941.48 Da.
Theoretical molecular weight of the modified eGFP amino acid sequence using the expasy calculator.
On the other hand, the modified eGFP amino acid sequence yields a theoretical molecular weight of 28006.60 Da.
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
I selected this pair of adjacent peaks from the intact LC-MS data to verify the experimental molecular weight calculation:
$$ \frac{m}{z_n} = 848.9162 $$ (left peak, high charge)
Determine z for each adjacent pair of peaks (n, n+1) using:
$ {\large z} = {\Large \frac{\frac{m}{z_{n+1}}} {\frac{m}{z_n} - \frac{m}{z_{n+1}}}} $
Using the adjacent pair formula:
$$ z = \frac{848.9162}{875.4421 - 848.9162} = \frac{848.9162}{26.5259} $$
$$ z = 32.003 \rightarrow \mathbf{z = +32} $$
This places the charge state of the first peak at $+32$ and the second peak at $+31$.
Determine the MW of the protein using the relationship between $\frac{m}{z_n}$, $MW$, and $z$
Using the relationship $$ MW = z \cdot (\frac{m}{z}) - (z \cdot m_{\text{proton}}) $$, where the mass of a proton ($H^+$) is $\approx 1.0078\text{ Da}$:
$$ MW_{\text{experiment}} = 32 \cdot 875.4421 - (32 \cdot 1.0078) = 28014.15 - 32.25 = \mathbf{27981.90\text{ Da}} $$
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with $ \frac{m}{z} $ values.
In this case, using the formula stated above, I’ve got this result:
$$ \text{Accuracy} = \frac{|27981.90 - 28006.60|}{28006.60} = \frac{24.7}{28006.60} \approx \mathbf{0.0008819\text{ (or 0.088%)}} $$
In ppm, the accuracy of the measurement is $$ \text{Accuracy} = \mathbf{0.0008819 \times 1,000,000} = 881.9 \approx 882 $$
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, I can’t observe the individual isotopic charge states in the zoom-in window. Although I see some minor peaks, they probably represent instrumental noise and overlapping profiles rather than clean, resolved isotopes, because the expected isotopic splitting distance at this high mass and charge ($z \approx 19$) is far too small ($\approx 0.053\text{ m/z}$) to be baseline resolved. I think it could be observed if the image had higher quality since I can’t really distinguish the small numbers at the top of the peaks.
Homework: Waters Part II — Secondary/Tertiary structure
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
Native proteins retain their compact, three-dimensional folded structures, stabilizing non-covalent interactions and burying hydrophobic residues. When a protein denatures, it unfolds into an extended, flexible, and disordered polymer chain, greatly increasing its solvent-accessible surface area.
In electrospray ionization mass spectrometry (ESI-MS), this structural change directly alters the Charge State Distribution (CSD):
Top Spectrum (Green, Denatured): Unfolding exposes a massive number of basic amino acid residues (like Lys, Arg, and His) that were previously buried in the core. This results in extensive protonation, yielding a broad distribution of peaks at a higher charge state (higher $z$), which shifts the entire envelope toward lower $m/z$ values (between 700 and 1300 $m/z$).
Bottom Spectrum (Red, Native): The tightly folded conformation restricts solvent and acid accessibility, leaving only a few surface residues available for protonation. Consequently, the protein carries fewer charges (lower $z$), shifting the signal to a tight, high-intensity peak envelope at higher $m/z$ values (predominantly around 2545 and 2799 $m/z$).
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 $ \frac{m}{z} $? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800 $ \frac{m}{z} $ on a mass spectrometer with 30,000 resolution.
Yes, the charge state can be clearly discerned from the high-resolution inset window because the compact, single-conformer nature of native eGFP allows the instrument to baseline-resolve individual isotopic peaks.
Taking two consecutive isotopic peaks from the zoom-in inset:
$(\frac{m}{z})_1 = 2545.3140$
$(\frac{m}{z})_2 = 2545.4058$
1. Difference in $\frac{m}{z}$ values ($\Delta \frac{m}{z}$):
Using the natural monoisotopic mass difference of a neutron:
$$\Delta m = {}{13}\text{C} - {}{12}\text{C} \approx 1.00335\text{ Da}$$
Based on this value and the fundamental mass-to-charge relationship:
$$\Delta \frac{m}{z} = \frac{\Delta m}{z}$$
Now, we can solve for z using the experimental data:
$$z = \frac{1.00335}{\Delta \frac{m}{z}} = \frac{1.00335}{0.0918} = 10.93 \rightarrow \mathbf{z = +11}$$
The charge state of the native protein peak cluster is $+11$. We can tell because the fine isotopic splitting distance of $\approx 0.092\text{ m/z}$ corresponds mathematically to a multi-protonated species carrying exactly 11 charges.
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
Here’s the amino acid sequence of eGFP with Lysines (K) and Arginines (R) circled!
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Figure 4. Example conditions for predicting the number of tryptic peptides from the eGFP standard. Please replicate all parameters shown above.
I followed the steps and got the following results:
Tryptic digestion results of eGFP using the expasy PeptideMass tool!
The PeptideMass Tool found that 19 peptides will be generated from eGFP tryptic digestion, and it covered 90.7% of the total amino acid sequence.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
First, to determine the 10% relative abundance threshold, I looked at the highest peak in the chromatogram (the peak at 4.87 minutes), which reaches the maximum intensity of $1.2 \times 10^7$ counts (100%).
Calculating 10% of this maximum intensity value gives our cutoff line:
$$I_{\text{cutoff}} = 0.10 \times (1.2 \times 10^7)$$
Any peak rising above this intensity line (just below the $2 \times 10^6$ mark on the TIC axis) is included. Looking at the Total Ion Chromatogram (TIC) between 0.5 and 6.0 minutes, I can clearly identify 17 distinct peaks that meet this criteria:
Between 0.5 - 1.0 min: 0.61, 0.79
Between 1.0 - 2.0 min: 1.43, 1.80, 1.85, 1.93
Between 2.0 - 3.0 min: 2.17, 2.26, 2.54, 2.78
Between 3.0 - 4.0 min: 3.27, 3.53, 3.59, 3.70
Between 4.0 - 5.0 min: 4.48, 4.64, 4.87
Just to mention, the 3.27 peak is also considered since it is above the threshold but just by a little. Besides that one, there are also 2 other peaks with no number that could also be considered since they are above the line too.
With those considerations, there are 17 identified numbered peaks + 2 unnumbered peaks = 19 total peaks.
TIC of the eGFP with the 10% threshold, numbered peaks are highlighted in orange and small peaks are highlighted in red.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
However, the 17 observed chromatographic peaks do not perfectly match the total number of predicted tryptic peptides from PetideMass, which is 19. Generally, a typical digest yields fewer visible peaks than theoretical fragments because of some of the following reasons:
Some small hydrophilic peptides elute in the void volume (before 0.5 min) and are lost.
Multiple distinct peptides can co-elute at the exact same retention time (overlapping peaks).
Incomplete or missed tryptic cleavages can create larger, alternative peptide fragments.
Identify the mass-to-charge ($\frac{m}{z}$) of the peptide shown in Figure 5b. What is the charge ($z$) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ($\small{[M\!\!+\!\!H]^+}$) based on its $\frac{m}{z}$ and $z$.
Figure 5b. Mass spectrum figure to show $\frac{m}{z}$ for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at $\frac{m}{z}$ 525.76, to discern the isotope peaks.
Figure 5c. Fragmentation spectrum of the peptide eluting at retention time 2.78 minutes in Figure 5a (above).
5.1 Mass-to-charge ratio ($\frac{m}{z}$)
The most abundant precursor peak eluting at 2.78 minutes has an $\frac{m}{z}$ of $525.76$ (specifically $525.76712$).
5.2 Charge state ($z$) determination
In the zoomed-in mass spectrum inset (Figure 5b), the spacing between the individual isotope peaks is consistently $0.5\text{ m/z}$.
Since $\Delta \frac{m}{z} = \frac{1}{z}$:**
$$z = \frac{1}{0.5} = \mathbf{2}$$
The peptide is in a doubly charged state ($z = +2$).
5.3 Calculation of the singly charged form ($[M+H]^+$)
Using the single charged mass relationship $[M+H]^+ = (z \times \frac{m}{z}) - (z - 1)$ to find the mass of the monoisotopic protonated form:
$$[M+H]^+ = (2 \times 525.76) - (2 - 1)$$
$$[M+H]^+ = 1051.52 - 1 = \mathbf{1050.52\text{ Da}}$$
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
(Recall that $ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $ )
By matching the experimental $[M+H]^+$ mass of 1050.52 Da against the predicted tryptic masses of eGFP using the PeptideMass tool, we find a perfect sequence match with the peptide fragment: FEGDTLVNR (Theoretical $MW_{\text{theory}} = 1050.5214\text{ Da}$).
Mass Accuracy Calculation (in ppm):
$$\text{Accuracy} = \frac{|1050.52 - 1050.5214|}{1050.5214} \times 1,000,000$$
$$\text{Accuracy} = \frac{0.0014}{1050.5214} \times 1,000,000 \approx \mathbf{1.33\text{ ppm}}$$
An error of 1.33 ppm is well within the high-resolution mass accuracy threshold ($<5\text{ ppm}$) of the Waters Xevo G3 QTof system, confidently confirming the identity of the peptide.
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Figure 6. Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data.
The percentage of the confirmed sequence by peptide mapping is 88% according to Figure 6.
Bonus Peptide Map Questions
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Yes, the peptide sequence can be definitively determined and verified by matching the fragment ions from the MS/MS spectrum in Figure 5c with the theoretical monoisotopic masses generated by the online Fragment Ion Calculator tool for the sequence FEGDTLVNR. The calculator results confirm a perfect structural match, resolving a clear N-terminal b-ion series containing $b_2$ (FE) at $277.11833\text{ m/z}$ and $b_3$ (FEG) at $334.13979\text{ m/z}$, alongside an intense C-terminal y-ion series anchored by the terminal Arginine.
Peptide sequence fragmentation pattern using the systemsbiology tool.
Specifically, the highly abundant experimental fragments track directly with the tool’s predictions for $y_1$ (R) at $175.11900\text{ m/z}$, $y_2$ (NR) at $289.16192\text{ m/z}$, and $y_3$ (VNR) at $388.23034\text{ m/z}$. Furthermore, the tool calculates the theoretical monoisotopic mass for the doubly charged precursor $(M+2H)^{2+}$ at $525.76441\text{ m/z}$, which aligns flawlessly with our dominant experimental signal at $525.76\text{ m/z}$ and solidifies this peptide identity.
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Yes, the peptide map data perfectly indicates that the protein sample is the eGFP standard because the results provide a definitive structural fingerprint. First, the automated peptide mapping achieved an exceptionally high 88% amino acid coverage, which easily exceeds the standard threshold required for confident protein identification. Furthermore, the experimental mass for the prominent tryptic peptide at 2.78 minutes matched the theoretical sequence with an incredibly tight error margin of only 1.33 ppm, a level of precision that virtually rules out false positives. Finally, the structural fragment markers verified in the MS/MS spectrum uniquely trace back to the primary sequence of the eGFP construct, confirming that both the intact mass and internal chemical structure align with the standard.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS).
CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer
8FU Didecamer
8FU 3-Decamer
8FU 4-Decamer
Polypeptide Subunit Name
Subunit Mass
7FU
340 kDa
8FU
400 kDa
Table 1: KLH Subunit Masses
Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
To determine the oligomeric states using CDMS, we first need to calculate the mass of each decamer. We also need to remember that a decamer is 10 identical subunits, so didecamer is 20 identical subunits, and 3 decamer is 30 and so on.
Making the calculations:
7FU Decamer: Mass = 10 x 340 kDa = 3400 kDa <> 3.4 MDa (Megadaltons)
8FU Didecamer: Mass = 20 x 400 kDa = 8000 kDa <> 8 MDa (Megadaltons)
8FU 3-Decamer: Mass = 30 x 400 kDa = 3400 kDa <> 12 MDa (Megadaltons)
8FU 4-Decamer: Mass = 40 x 400 kDa = 3400 kDa <> 16 MDa (Megadaltons)
Once calculated, I need to identify them in the graph. Here’s the result of that:
Oligomeric identificacion using the CDMS peaks graph.
In this case most peaks align decently with the theoretical calculations.
7FU Decamer: 3.4 observed peak as calculated before.
8FU Didecamer: 8.33 observed peak, which is near the 8MDa calculation made before.
8FU 3-Decamer: 12.67 observed peak, which is also near the 12MDa calculation made before
8FU 4-Decamer: 16 to 17 observed peaks, which should be near the 16 MDa mass calculation but are lower than the other peaks.
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28006.60
27981.90
882
Week 11 HW: Building Genomes
Cloud laboratories are making science accessible, affordable, and reproducible. Our aim this semester is to showcase how they can enable human creativity at scale, and how they provide a platform for collaboration and community.
How To Grow (Almost) Anything is about synthetic biology, bioengineering, robotics, automation, art, and AI. But it is also about friendship, shared purpose, and the freedom to build beyond what we know and to be inspired by what can be. To that end, the goal with this cloud lab unit and homework assignment is to inspire collaboration and creativity while designing a scientifically rigorous cell-free fluorescent protein optimization experiment together.
Tip
As you plan for final projects, you may want to refer to the provided non-exhaustive list of common Nebula protocols and their parameters in the “Reading & Resources” section below.
Homework
Info
Note that this homework is due a week later than it ordinarily would due to its release a week later than normal.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
So I contributed 29 pixels to the collective Artwork! Placing 39th on the dashboard (Not bad eh).
Essentially I wanted to leave a yellow heart since on the first day of going live there were three hearts that needed some color. So I first added yellow pixels mostly to try and make the heart more heart-shaped.
Then after a day or two I came back and noticed that the overall drawing completely changed, which was very fun to see. Adapting to that change, I filled another yellow heart, and it stayed like that for some time. Besides the yellow hearted adventure I added some cyan and red pixels to make some figures symmetrical.
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
I contributed with yellow hearts on the bottom right plate and on the upper left plate. However, these hearts were later changed since the overall scheme of the collective artwork changed too! I also contributed with some cyan and red pixels on the bottom left plate to “even out” some corners and make the drawings more symmetrical.
what you liked about the project, and
What I really liked about the collective artwork project is how community can change a blank slate (in this case an agar plate) and transform it into something memorable and beautiful in just some days! I remember very well how people chatted and discussed it over the classes and on the forum. I also loved the interface so much (Shoutout to Ronan!!!!), it was really well implemented (wish I could learn some of those interface creation abilities for more projects). I also really loved how much love and appreciation went into each pixel since it represented a person in a place in our world.
what about this collaborative art experiment could be made better for next year.
I think the only thing that could be better for next year is to maybe reduce the time limit just a little bit more :D (although there was a secret technique to get unlimited time!). And also it would be nice to expand the artwork every year (that way we can have more space for initial exploration). It would be really impressive to see a collective 4x4 plate artwork fully completed, too.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Provides the essential molecular machinery, including ribosomes, tRNAs, and initiation factors, required to execute the translation of target proteins. The BL21 (DE3) Star Lysate specifically includes T7 RNA Polymerase to drive highly efficient, coupled transcription from plasmids containing a T7 promoter.
Salts/Buffer
Potassium Glutamate
HEPES-KOH pH 7.5
Magnesium Glutamate
Potassium phosphate monobasic
Potassium phosphate dibasic
Maintains the optimal physiological pH and osmotic balance required to preserve protein stability and enzymatic activity throughout the long incubation. Potassium Glutamate and Magnesium Glutamate supply indispensable cofactors that stabilize mRNA structures, ribosomal subunits, and tRNA-aminoacyl interactions, while HEPES-KOH pH 7.5, Potassium phosphate monobasic, and Potassium phosphate dibasic establish a robust buffering system to prevent pH drifts.
Energy / Nucleotide System
Ribose
Glucose
AMP
CMP
GMP
UMP
Guanine
Functions as a sustainable recycling system that slowly generates ATP and GTP locally from simple precursors, preventing rapid phosphate accumulation while continuously fueling transcription and translation. Within this system, Ribose and Glucose serve as primary carbohydrate energy sources, while AMP, CMP, GMP, UMP, and Guanine act as nucleotide precursors that endogenous enzymes progressively phosphorylate into active substrate building blocks.
Translation Mix (Amino Acids)
17 Amino Acid Mix
Tyrosine
Cysteine
Supplies the fundamental substrate building blocks necessary for the ribosome to polymerize and synthesize the primary polypeptide chain of the target protein. The 17 Amino Acid Mix provides the majority of the standard genetic code requirements, while Tyrosine and Cysteine are added individually to bypass the solubility limits typical of highly concentrated master stocks, ensuring complete dissolution.
Additives
Nicotinamide
Acts as a stabilizing metabolic precursor that sustains the functional lifetime of essential nicotinamide adenine dinucleotide (NAD+/NADH) cofactors within the lysate. Specifically, Nicotinamide prevents the premature degradation of these electron carriers, supporting the homeostatic core metabolic pathways that generate chemical energy for the cell-free system.
Backfill
Nuclease Free Water
Brings the overall master mix to its exact final required reaction volume without introducing external biological contamination. Nuclease Free Water ensures that no trace DNA or RNA degrading enzymes jeopardize the stability of the genetic template or the newly synthesized transcripts.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
So, the 1-hour PEP-NTP system relies on a high concentration of readily available nucleoside triphosphates (NTPs) paired with phosphoenolpyruvate (PEP) as a rapid, direct phosphate donor for immediate transcription and translation. In contrast, the 20-hour system is designed for long-term sustainability, utilizing cheap precursors like ribose, glucose, and nucleoside monophosphates (NMPs) that endogenous enzymes slowly convert into energy. This metabolic pacing keeps chemical energy generation active over nearly a day and avoids the toxic accumulation of inorganic phosphate that prematurely halts the PEP-based reaction.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
This one is interesting! From what I’ve read, transcription can still proceed effectively because the E. coli lysate contains active endogenous salvage pathway enzymes, such as purine nucleoside phosphorylase and phosphoribosyltransferases. These enzymes can salvage free Guanine by combining it with ribose-1-phosphate or PRPP to generate GMP directly within the reaction. Once GMP is produced, native adenylate and nucleoside diphosphate kinases sequentially phosphorylate it into GDP and functional GTP, successfully fueling T7 transcription.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Superfolder GFP possesses an exceptionally robust folding kinetics network that prevents aggregation during rapid transcription-translation coupling, making it highly efficient in cell-free systems. However, its rapid expression can outpace the chemical maturation of its chromophore, which strictly requires molecular oxygen ($O_2$) to become fluorescent.
mRFP1: Monomeric RFP1 features a notoriously slow chromophore maturation time of over an hour, which delays real-time fluorescence readouts during the early phases of cell-free incubation. Additionally, it exhibits relatively low photostability and brightness compared to newer generation red fluorescent proteins, limiting its long-term detection sensitivity.
mKO2: Monomeric Kusabira Orange 2 is characterized by a high molar extinction coefficient and excellent brightness, but its unique azoline-containing chromophore exhibits significant acid sensitivity. As a cell-free reaction progresses and metabolic waste products accumulate, any decrease in pH can drastically quench mKO2 fluorescence.
mTurquoise2: This enhanced cyan fluorescent protein has a uniquely high quantum yield and fast maturation due to a stabilized tryptophan side chain within its beta-barrel core. A key functional constraint in cell-free platforms is its narrow excitation/emission gap, which requires precise optical filtering to avoid overlap with background autofluorescence from the lysate.
mScarlet_I: mScarlet_I is an engineered red fluorescent protein with a highly rigidified chromophore that yields record-breaking brightness and quantum efficiency. Its main operational challenge in extended reactions is its high susceptibility to photobleaching under continuous excitation, requiring carefully pulsed plate reader protocols over long timelines.
Electra2: Electra2 is a specialized, rapidly maturing yellow-green fluorescent protein specifically engineered for high-throughput visibility. Its primary biophysical limitation is its high sensitivity to chloride ion concentrations and ionic strength changes, meaning slight variations in the cell-free salt balance can destabilize its tertiary structure.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
High-Illumination and Metabolic Oscillator Hypotheses
To achieve high-illumination pixels and maximize absolute fluorescence over a 36-hour incubation, I hypothesize that supplementing high concentrations of HEPES-KOH pH 7.5 paired with a tightly metered dose of Magnesium glutamate will drive translation velocity while delaying system shutdown. The primary bottleneck for sustained cell-free brightness is the progressive acidification of the reaction combined with the accumulation of free inorganic phosphate from energy depletion, which typically kills translation within hours. By expanding the buffering capacity with HEPES-KOH, we actively neutralize the acid waste, preserving the unquenched state of mScarlet_I and sfGFP while magnesium glutamate supports ribosomal elongation stability to maximize the total number of synthesized protein copies before the system reaches exhaustion.
Alternatively, to explore the feasibility of an intermittent light effect or a metabolic blinking pixel, I hypothesize that a transient, self-limiting loop can be established by intentionally overloading the L-tyrosine substrate concentration against a minimal HEPES-KOH baseline using sfGFP. In this metabolic context, the cell-free machinery will initially synthesize highly fluorescent sfGFP, causing a rapid spike in light emission. Concurrently, the active MelA tyrosinase enzyme will consume the abundant L-tyrosine to produce dark melanin pigment while the unbuffered system undergoes natural metabolic acidification. As the reaction darkens and the pH drops, the optical interference of the melanin combined with the acid-induced quenching of the green reporter will systematically extinguish the visible light, creating a distinct, self-terminating pulse of fluorescence that mimics a delayed metabolic countdown.
These flowcharts illustrate the operational logic and downstream biological consequences of both experimental designs:
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Important
High-intensity orange/yellow display that counteracts the default acid sensitivity of the reporter.
Q3-I4
mKO2
Blinking / Intermittent Pixel 1
HEPES-KOH pH 7.5 Tyrosine pH 12
57.500 mM 8.438 mM
Yellow pixel engineered to shut down prematurely via high-velocity substrate-driven pigmentation under zero-water saturation.
Q3-H1
mKO2
Blinking / Intermittent Pixel 2
HEPES-KOH pH 7.5 Tyrosine pH 12
57.500 mM 8.438 mM
Replicate yellow blinking node to validate the kinetic timing of the metabolic countdown loop using interface limits.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!).
The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
This section will be finished once we get the results back from the global Cell-Free experiment!
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Assignees for the following sections
MIT/Harvard students
Optional
Committed Listeners
Optional
Ginkgo Nebula Cloud Laboratory Rendering, 2025
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Tip
Note from Ronan: If you are interested in helping me build out future HTGAA cloud lab software, please fill out this form!
Skipping this one for now.
Week 12 HW: Bioproduction
This week focuses on designing, synthesizing, and editing whole genomes, from minimal cells to refactored microbes and synthetic chromosomes.
Homework
Important
Be sure you’ve seen the updated week 11 homework which is due at the start of the April 28 lecture.
Tip
Continue making progress this week on your Individual Final Project and on DNA orders (due Friday midnight ET).
This week I’ve worked on the individual final project.
Week 13 HW: AI, SynBio, and Scaling Health Innovation (ARPA-H)
No Lab Assignment this week.
Final Project Lab time available
If your final project requires lab work, you can schedule a block of lab time this week.
Continued working on the final Individual project.
Week 14 HW: Bio Design & Bio Fabrication
We wrap up the term looking towards a future of Bio-Design and Bio-Fabrication.
Homework: Finish your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
Worked on final project and finished the slides on time!
Here are my preliminar three individual final project ideas!
An interactive 3D bio-art sculpture where human touch meets living bioluminiscence bloom.
A river-sensing automated robot system that helps both advance research and act as a tourist spot for the community to reunite and spend time together.
Chlorella vulgaris in silico optimization and automation for the optimal accumulation of polyhydroxybutyrate (PHB).
Bacteriophage Engineering: L-Protein Optimization 1. Hypothesis: Engineering Lysis Protein Stability Our core hypothesis is that the thermodynamic stability and lytic efficiency of the MS2 L-protein can be enhanced through two strategic pathways:
Structural Reinforcement: Introducing targeted mutations that promote independent folding or stabilize the 7-helix bundle, reducing dependence on the host chaperone DnaJ. Generative Optimization: Utilizing evolutionary conservation data and generative protein design to create variants with improved membrane-insertion kinetics and host compatibility, thereby minimizing host-mediated resistance. 2. Specific Aims and Validation Pipeline Aim 1: Mutation Design via Conservation and Predictive Modeling We will perform Clustal Omega alignments of homologous lysis proteins to identify conserved residues (specifically the “HEDYPCRRQQRSST” island). This is followed by:
Subsections of Projects
Individual Final Project
Here are my preliminar three individual final project ideas!
An interactive 3D bio-art sculpture where human touch meets living bioluminiscence bloom.
A river-sensing automated robot system that helps both advance research and act as a tourist spot for the community to reunite and spend time together.
Chlorella vulgaris in silico optimization and automation for the optimal accumulation of polyhydroxybutyrate (PHB).
After some thought and consideration, I decided to go with my third idea for my final individual project, here you can find a more detailed view of it:
Modeling and Automating Polyhydroxybutyrate Bioplastic Production in Microalgae: An In Silico and Automated Approach
Author: Fabrizio Flores Huamán
Course: How to Grow (Almost) Anything (HTGAA) 2026
Username: 2026a-fabrizio-flores-huaman
Date: May 2026
SECTION 1: ABSTRACT
The global accumulation of petroleum-derived plastics has caused severe and often irreversible ecological damage to marine ecosystems, including the biodiverse coastal waters of Peru. Polyhydroxybutyrate (PHB), a fully biodegradable and biocompatible bacterial polyester, represents a scientifically compelling alternative to fossil fuel-derived plastics, yet its industrial scaling remains hindered by low production yields and the high cost of manually optimizing metabolic conditions. Microalgae such as Chlorella vulgaris are promising biological chassis for PHB production because they fix carbon dioxide and accumulate carbon-rich polymers under nutrient stress, but the optimization of these conditions continues to rely on slow, one-variable-at-a-time experimentation that is incompatible with industrial timelines.
This project addresses that gap by developing a high-throughput, in silico pipeline that integrates computational metabolic modeling with automated robotic liquid handling protocols. Using Flux Balance Analysis (FBA) implemented through the COBRApy Python library on the iJO1366 genome-scale metabolic model of Escherichia coli, this work identifies optimal combinations of nitrogen limitation, phosphorus limitation, and carbon source variation that maximize carbon flux toward PHB biosynthetic precursors, particularly acetyl-coenzyme A (acetyl-CoA). The E. coli iJO1366 model is used for Aim 1 because E. coli BL21 (DE3) is the practical validation chassis; the iCZ843 genome-scale model of C. vulgaris is designated for Aim 2, where it informs the algal construct design and transformation conditions. The computationally predicted conditions are then translated into modular Opentrons OT-2 Python protocols, creating a reproducible and stoppable screening pipeline.
In parallel, a synthetic PHB biosynthetic operon encoding phaA (beta-ketothiolase), phaB (acetoacetyl-CoA reductase), and phaC (polyhydroxyalkanoate synthase) is designed for expression in Escherichia coli BL21 (DE3) as a practical validation chassis, with the construct to be synthesized by Twist Bioscience and deployed at Ginkgo Bioworks. The long-term vision of this project is to empower Peruvian coastal communities to participate in decentralized, community-scale biomanufacturing of sustainable materials through AI-guided, closed-loop photobioreactor systems that connect local biodiversity with the global circular bioeconomy.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of this project is to identify the optimal metabolic conditions for Polyhydroxybutyrate (PHB) accumulation in Escherichia coli BL21 (DE3) by utilizing COBRApy for Flux Balance Analysis (FBA) on the iJO1366 genome-scale metabolic model of E. coli, and to translate the predicted nutrient stress conditions into automated Python-based protocols for the Opentrons OT-2, including the design and ordering of a synthetic PHB biosynthetic operon from Twist Bioscience for validation at Ginkgo Bioworks. The iJO1366 model is the correct tool for Aim 1 because E. coli BL21 (DE3) is the experimental chassis; the iCZ843 genome-scale model of Chlorella vulgaris is reserved for Aim 2, where it informs the design of the algal construct and transformation conditions.
Resources and methods for Aim 1:
COBRApy (Python library) with the iJO1366 E. coli genome-scale metabolic model (JSON format, fully compatible with modern COBRApy and Python 3.12)
iCZ843 C. vulgaris genome-scale model referenced for Aim 2 design (requires MATLAB or a legacy Python environment due to SBML format constraints)
Combinatorial FBA simulation across nitrogen, phosphorus, and carbon source gradients (glucose, acetate, glycerol)
Opentrons OT-2 Python API (v2.15) for automated media preparation and Nile Red fluorescence screening
Twist Bioscience whole plasmid synthesis of the phaA-phaB-phaC-sfGFP construct (shipped to Ginkgo Bioworks)
PHERAstar FSX and Spark Plate Reader for Nile Red and sfGFP fluorescence detection
96-well Eppendorf deep-well plates for media preparation; 384-well Greiner black clear-bottom plates for assay readout
Aim 2: Development Aim
The next step following the computational and E. coli validation would be to design a Chlorella vulgaris-specific genetic construct, codon-optimized for algal nuclear expression and placed under the control of the NIT1 nitrate-inducible promoter, and to produce a complete electroporation-based transformation and Nile Red screening protocol ready for execution at a partner facility such as Ginkgo Bioworks or a collaborating university laboratory. This aim also includes running FBA on the iCZ843 genome-scale metabolic model of C. vulgaris to identify algal-specific nutrient stress conditions, a step that requires MATLAB or a legacy Python 2 environment due to the 2016-era SBML format of the published model and is therefore scoped as a future computational deliverable beyond the course timeline. While the physical execution of stable algal transformation requires specialized containment equipment and multi-week clonal selection timelines that exceed the scope of this course, the complete construct design, codon optimization rationale, and step-by-step transformation protocol constitute meaningful scientific deliverables that advance the project toward a genuine in vivo test in C. vulgaris.
Aim 3: Visionary Aim
The long-term vision is to establish a framework for decentralized, community-scale bioplastic production by developing AI-guided, closed-loop modular photobioreactors that integrate real-time environmental sensing with the metabolic models developed in Aims 1 and 2. This system would allow Peruvian coastal communities to culture locally sourced microalgae strains under computationally optimized conditions, transforming the extraordinary regional biodiversity of coastal lagoons, river deltas, and Andean water sources into a renewable feedstock for sustainable, compostable materials. By combining open-source metabolic modeling, low-cost automation, and participatory science education and knowledge-sharing, this aim envisions a future where bioplastic manufacturing is not confined to industrial facilities but is instead embedded within the communities most affected by plastic pollution, giving people both the tools and the understanding to build a circular bioeconomy from the ground up.
“The long-term vision is to establish a decentralized framework for bioplastic production through AI-guided, closed-loop modular photobioreactors, enabling coastal communities to transform local microalgae biodiversity into sustainable, high-value compostable materials that replace petroleum-based plastics.”
Here’s my Final Idea Aims Slide:
And here are the slides I used in the final presentation day!
SECTION 3: BACKGROUND
Literature Context
Paper 1: Flux Balance Analysis and constraint-based metabolic modeling
Orth, Thiele, and Palsson (2010) provided a foundational overview of Flux Balance Analysis in their landmark Nature Biotechnology publication, establishing the mathematical and conceptual framework that underpins the computational core of this project. FBA models cellular metabolism as a system of linear equations in which stoichiometric constraints define the boundaries of possible metabolic states, and an objective function such as biomass production or metabolite yield is optimized using linear programming. The authors demonstrated that FBA can accurately predict growth rates, gene essentiality, and the effects of environmental perturbations across multiple organisms without requiring detailed kinetic parameters. Crucially for this project, nutrient limitation can be represented as a constraint on exchange reactions, making FBA directly applicable to modeling nitrogen and phosphorus stress in microalgae and to predicting how carbon flux is redirected under suboptimal growth conditions. This approach transforms the traditionally empirical process of media optimization into a computational design problem, dramatically reducing the number of physical experiments required before identifying high-yield conditions.
Paper 2: The phaABC operon and PHB biosynthesis
Peoples and Sinskey (1989) characterized the biochemical pathway for PHB biosynthesis in Cupriavidus necator (then known as Alcaligenes eutrophus) H16, identifying and sequencing the three-enzyme cascade encoded by phaA (beta-ketothiolase), phaB (acetoacetyl-CoA reductase), and phaC (polyhydroxyalkanoate synthase) that converts two molecules of acetyl-CoA into PHB polymer granules. This foundational study established the genetic basis for heterologous PHB production and demonstrated that the pathway is functionally transferable to other organisms, a property that has since been exploited in dozens of bacterial hosts and, more recently, in green microalgae and plants. The characterization of acetyl-CoA as the central metabolic node for PHB production directly informs the choice of FBA objective in this project, since maximizing acetyl-CoA flux serves as a computationally tractable proxy for maximizing bioplastic precursor availability. Furthermore, the modularity of the three-gene operon makes it an ideal candidate for whole plasmid synthesis via Twist Bioscience, since all three coding sequences can be assembled into a single synthetic construct without requiring post-synthesis cloning.
Paper 3: Chlorella vulgaris as a multi-application biotechnology chassis
Al-Hammadi and Güngörmüşler (2024) published a comprehensive review in Biotechnology and Bioengineering documenting the breadth of C. vulgaris biotechnology applications, covering wastewater treatment, biodiesel, biohydrogen, biocement, biopolymers, and food additives, and concluding that despite its documented potential for polyhydroxyalkanoate (PHA) production from algal biomass, the field still lacks critical strain-specific data that would make industrial PHB production from C. vulgaris economically competitive. The authors note that C. vulgaris is particularly attractive as a production chassis because of its ease of cultivation, rapid growth, and capacity to use carbon dioxide and wastewater nutrients simultaneously, reducing both feedstock costs and environmental impact. The review explicitly identifies a gap between the biological potential of C. vulgaris for biopolymer synthesis and the practical optimization infrastructure needed to bridge laboratory results to industrial scale. This directly motivates the computational and automated pipeline approach taken in this project, since the gap the authors describe is precisely the one that FBA-guided combinatorial screening is designed to address.
Knowledge Gap
Despite the well-established FBA methodology, the clearly characterized PHB biosynthetic pathway, and growing recognition of C. vulgaris as a promising bioplastic chassis, these three domains have rarely been integrated into a single, closed-loop design pipeline. Most optimization studies in algal PHB production continue to rely on one-variable-at-a-time manual experimentation, generating data slowly and failing to capture the multi-dimensional interaction effects between nitrogen, phosphorus, and carbon source availability. Peruvian research groups have independently demonstrated that C. vulgaris is highly responsive to nitrogen and phosphate concentration changes: Oscanoa et al. (2021), working at the Instituto del Mar del Perú, showed that C. vulgaris efficiently removes nitrates and phosphates from domestic wastewater, demonstrating the organism’s natural sensitivity to these nutrient gradients and validating nitrogen and phosphorus as the most impactful variables for metabolic manipulation. Automated robotic screening of metabolic model predictions remains largely confined to bacterial and yeast systems, with very few examples connecting genome-scale algal models to physical high-throughput screening pipelines. This project addresses that gap by connecting the iCZ843 genome-scale model of C. vulgaris to a computationally guided automated OT-2 screening pipeline, establishing a Design-Build-Test-Learn (DBTL) cycle that is both predictive and robotically executable.
Innovation
This project is novel in its integration of three components that have not previously been combined in a single pipeline for microalgal bioplastic research: a genome-scale metabolic model (iCZ843), a modular automated combinatorial nutrient stress screening protocol (OT-2), and a synthetic PHB operon designed for both bacterial validation and future algal expression. Most published work in this area either performs FBA on C. vulgaris without connecting the predictions to physical experiments, or conducts wet-lab PHB screening without the guidance of a metabolic model. By treating the OT-2 protocol as a direct translation layer between computational predictions and physical experiments, this project establishes a reusable pipeline architecture that can be adapted to other bioproduction targets and chassis organisms beyond PHB and microalgae. Unlike previous computational approaches that apply a single generalized metabolic model to predict PHB production, this project uses the iJO1366 genome-scale model of E. coli for Aim 1 FBA to ensure predictions are matched to the actual experimental chassis, and designates the iCZ843 genome-scale model of C. vulgaris specifically for Aim 2 algal construct design, creating a clear organism-to-model correspondence that improves scientific coherence across the project aims. Additionally, the explicit design of a second construct codon-optimized for C. vulgaris under the NIT1 nitrate-inducible promoter represents a forward-looking contribution to algal synthetic biology, where inducible expression systems remain significantly underexplored relative to their bacterial counterparts.
Significance
Plastic pollution is one of the most pressing environmental crises of the 21st century, with an estimated 11 million metric tons of plastic entering the ocean annually, causing harm to marine food webs, coastal ecosystems, and the communities that depend on them, including those along the biodiverse Peruvian coast. Polyhydroxybutyrate (PHB) offers a route to replace petroleum-derived single-use plastics with a material that is fully biodegradable and biocompatible under natural conditions, degrading within months rather than persisting for centuries. Microalgae represent an ideal production organism for PHB because they grow using only sunlight, carbon dioxide, and minimal inorganic nutrients, giving them a dramatically lower carbon footprint than fermentation-based bacterial systems that require sugar feedstocks. Critically, Peru harbors native Chlorella strains with documented biotechnology potential: Mariano Astocondor et al. (2017) characterized Chlorella peruviana, a native strain isolated from the Mellisera salt lagoon in Chilca, Peru, demonstrating robust biomass and chlorophyll productivity across a range of salinities and confirming that locally sourced Peruvian Chlorella strains are viable candidates for controlled cultivation and bioproduction. Complementing this, Condori et al. (2024) demonstrated that Peruvian Chlorella strains can be cultivated using nutrients valorized from local fruit residues, producing growth and lipid yields comparable to synthetic media and illustrating a circular economy model in which local organic waste streams feed microalgae bioproduction, directly relevant to the decentralized manufacturing vision of Aim 3. The integration of FBA-guided optimization with robotic automation directly addresses the industrial bottleneck of manual media optimization, which currently makes algal PHB production economically uncompetitive compared to petrochemical alternatives. By demonstrating that computationally predicted metabolic conditions can be automatically screened at scale, this project contributes a generalizable methodology that could accelerate the path from laboratory research to industrial relevance. This is particularly meaningful for research environments in countries like Peru that possess extraordinary microalgae biodiversity but currently lack access to the high-throughput experimentation infrastructure that makes systematic optimization feasible. Ultimately, reducing the cost and time of metabolic optimization is not only a scientific problem but a social one, and making these tools more accessible is a prerequisite for truly sustainable and equitable biomanufacturing rooted in local biodiversity.
Bioethical Considerations
Paragraph 1: Ethical considerations
This project involves the design and intended use of genetically modified organisms (GMOs), specifically Escherichia coli strains expressing a heterologous PHB biosynthetic pathway, with a longer-term vision of engineering Chlorella vulgaris, a microalga present in natural freshwater and marine environments. The introduction of synthetic genetic constructs into organisms that have natural environmental relatives raises important questions about biosafety, containment, and the potential for unintended horizontal gene transfer or competitive ecological disruption, even when experiments are conducted in well-controlled laboratory settings. The design of organisms intended for eventual scale-up in semi-open systems such as outdoor photobioreactors requires additional evaluation of containment strategies and kill-switch designs that go beyond standard laboratory biosafety level 1 (BSL-1) practices. The project also intersects with questions of biodiversity sovereignty, since any future use of Peruvian microalgae strains for commercial bioplastic production must respect the principles of the Nagoya Protocol on Access and Benefit-Sharing, ensuring that local communities and the Peruvian state retain informed consent and equitable participation in any commercial or intellectual property outcomes arising from the use of their biological resources.
Paragraph 2: Responsible implementation and risk mitigation
To mitigate these risks, all experimental work described in Aim 1 is conducted under standard BSL-1 conditions appropriate for non-pathogenic E. coli strains, with all constructs including a selectable antibiotic resistance marker that requires continuous supplementation for maintenance, significantly reducing the probability of survival outside controlled conditions. The C. vulgaris construct designed for Aim 2 is intended for nuclear rather than chloroplast-targeted integration, which limits expression levels and reduces the likelihood of creating competitive growth advantages in natural algal populations. All synthetic DNA sequences submitted to Twist Bioscience will be screened through the SecureDNA biosecurity framework to ensure compliance with dual-use research standards before synthesis is initiated. For the visionary Aim 3 framework involving photobioreactors in coastal communities, the project design explicitly includes a community consultation and participatory education component, ensuring that local knowledge holders are active participants in the governance and design of any deployment rather than passive recipients of an externally imposed technology. Collaboration with biodiversity documentation initiatives such as those supported by Basecamp Research prior to strain collection and use further ensures that any commercialization pathway is grounded in scientific rigor and community rights from the earliest stages.
SECTION 4: EXPERIMENTAL DESIGN
The experimental design follows a Design-Build-Test-Learn (DBTL) cycle. The Design phase uses FBA modeling to predict optimal nutrient conditions. The Build phase involves synthetic construct design and ordering from Twist Bioscience. The Test phase uses automated OT-2 protocols and Ginkgo Bioworks equipment for Nile Red fluorescence screening. The Learn phase closes the loop by feeding experimental data back into the metabolic model to refine predictions.
SECTION 4.1: Step-by-Step Workflow
Check out this Colab Notebook for the genome scale metabolic model.
Step 1: Install COBRApy and load the E. coli iJO1366 genome-scale metabolic model
Method: Install COBRApy and compile the multi-modal XML metabolic model structures representing the iCZ843 genome-scale architecture of Chlorella vulgaris. A native Python ElementTree text parsing layout is implemented to assemble valid COBRApy objects, ensuring full operational performance inside modern Python 3.12 environments without requiring deprecated dependency stacks.
Automation: Python scripting environment (Google Colab; all code below is Colab-ready)
Expected result: All three distinct trophic model profiles (Heterotrophy, Mixotrophy, Photoautotrophy) parse successfully into memory, displaying their explicit metabolic boundary dimensions and resolving true positive baseline growth rates.
Timeline: Day 1
# ── CELL 1: Install COBRApy ────────────────────────────────────────────────────!pipinstall-qcobraprint("COBRApy installed.")# ── CELL 2: Upload the three iCZ843 XML files ─────────────────────────────────fromgoogle.colabimportfilesimportosprint("Select all three XML files when the picker opens.")uploaded=files.upload()rename_map={'Heterotrophy':'iCZ843_heterotrophy.xml','Mixotrophy':'iCZ843_mixotrophy.xml','Photoautotrophy':'iCZ843_photoautotrophy.xml',}fororiginalinlist(uploaded.keys()):forkeyword,new_nameinrename_map.items():ifkeyword.lower()inoriginal.lower():iforiginal!=new_name:os.rename(original,new_name)print(f"Saved: {new_name} ({os.path.getsize(new_name)/1024:.0f} KB)")# ── CELL 3: Custom parser — builds COBRApy models without SBML reader ──────────importxml.etree.ElementTreeasETfromcobraimportModel,Reaction,Metaboliteimportwarningswarnings.filterwarnings('ignore')defparse_iCZ843(xml_path,model_name):"""Parse a Zuniga et al. 2016 iCZ843 SBML Level 2 file into a COBRApy model."""tree=ET.parse(xml_path)root=tree.getroot()cobra_model=Model(model_name)metabolites={}objective_rxn=Noneformodel_eleminroot:# Pass 1: extract all metabolites (species)forchildinmodel_elem:if'listOfSpecies'inchild.tag:forspinchild:sp_id=sp.get('id')met=Metabolite(id=sp_id,name=sp.get('name',sp_id),compartment=sp.get('compartment','c'))metabolites[sp_id]=metcobra_model.add_metabolites(list(metabolites.values()))# Pass 2: extract all reactions with stoichiometry and boundsforchildinmodel_elem:if'listOfReactions'inchild.tag:forrxn_eleminchild:rxn_id=rxn_elem.get('id')rxn_name=rxn_elem.get('name',rxn_id)lb,ub,obj_coeff=-1000.0,1000.0,0.0stoich={}forsubinrxn_elem:if'listOfReactants'insub.tag:forsrinsub:sid=sr.get('species')ifsidinmetabolites:stoich[metabolites[sid]]=-float(sr.get('stoichiometry',1))elif'listOfProducts'insub.tag:forsrinsub:sid=sr.get('species')ifsidinmetabolites:stoich[metabolites[sid]]=float(sr.get('stoichiometry',1))elif'kineticLaw'insub.tag:forlopinsub:forparaminlop:pid=param.get('id')val=float(param.get('value',0))ifpid=='LOWER_BOUND':lb=valelifpid=='UPPER_BOUND':ub=valelifpid=='OBJECTIVE_COEFFICIENT':obj_coeff=valrxn=Reaction(rxn_id,name=rxn_name,lower_bound=lb,upper_bound=ub)rxn.add_metabolites(stoich)cobra_model.add_reactions([rxn])ifobj_coeff==1.0:objective_rxn=rxn_idifobjective_rxn:cobra_model.objective=objective_rxnreturncobra_modelimportpandasaspdimportnumpyasnpimportitertools# Load all three modelsmodel_hetero=parse_iCZ843('iCZ843_heterotrophy.xml','iCZ843_heterotrophy')model_mixo=parse_iCZ843('iCZ843_mixotrophy.xml','iCZ843_mixotrophy')model_photo=parse_iCZ843('iCZ843_photoautotrophy.xml','iCZ843_photoautotrophy')forname,min[('Heterotrophy',model_hetero),('Mixotrophy',model_mixo),('Photoautotrophy',model_photo)]:sol=m.optimize()print(f"{name}: {len(m.reactions)} rxns | "f"{len(m.metabolites)} mets | "f"biomass = {sol.objective_value:.4f} mmol/gDW/hr | {sol.status}")# Key reaction IDs in iCZ843 (C. vulgaris model naming convention)N_RXN='R_EX_no3_LPAREN_e_RPAREN_'# Nitrate exchange (primary N source in algae)P_RXN='R_EX_pi_LPAREN_e_RPAREN_'# Phosphate exchangeGLC_RXN='R_EX_glc_DASH_A_LPAREN_e_RPAREN_'# Glucose exchangeAC_RXN='R_EX_ac_LPAREN_e_RPAREN_'# Acetate exchangeBIO_H='R_Biomass_Cvu_hetero_DASH_'# Biomass objective (heterotrophic)ACCOA='M_accoa_c'# Cytoplasmic acetyl-CoA (PHB precursor)print("\nModel routing: glucose/acetate -> model_hetero | CO2/light -> model_photo")
Step 2: Explore baseline metabolic fluxes
Method: Run FBA with default exchange reaction constraints to establish a baseline biomass flux and identify the acetyl-CoA reaction node and relevant nutrient exchange reactions.
Automation: COBRApy optimize() function
Expected result: Baseline biomass flux value; identification of exchange reaction identifiers for nitrate (EX_no3_e), phosphate (EX_pi_e), glucose (EX_glc__D_e), and acetate (EX_ac_e).
Timeline: Day 1 to 2
# ── CELL 4: Explore baseline metabolic fluxes (heterotrophic C. vulgaris) ──────sol=model_hetero.optimize()print(f"Baseline biomass flux: {sol.objective_value:.4f} mmol/gDW/hr | {sol.status}")key_rxns=[N_RXN,P_RXN,GLC_RXN,AC_RXN]print("\nKey exchange reactions:")forrxn_idinkey_rxns:try:rxn=model_hetero.reactions.get_by_id(rxn_id)print(f" {rxn_id}: [{rxn.lower_bound:.1f}, {rxn.upper_bound:.1f}]")exceptKeyError:print(f" {rxn_id}: NOT FOUND")# Confirm acetyl-CoA metabolite is presenttry:accoa_met=model_hetero.metabolites.get_by_id(ACCOA)print(f"\nAcetyl-CoA metabolite: {accoa_met.id} | compartment: {accoa_met.compartment}")exceptKeyError:print(f"WARNING: {ACCOA} not found — search manually:")candidates=[m.idforminmodel_hetero.metabolitesif'accoa'inm.id.lower()]print(candidates)
Step 3: Add a PHB demand reaction as a proxy for bioplastic potential
Method: Add a synthetic boundary reaction that consumes acetyl-CoA to represent PHB biosynthetic flux; set this as the new FBA objective to directly optimize for bioplastic precursor availability.
Automation: COBRApy add_boundary() and Reaction() classes
Expected result: A working optimization model whose objective value represents acetyl-CoA flux directed toward PHB biosynthesis.
Timeline: Day 2
# ── CELL 5: Add PHB demand reaction to C. vulgaris heterotrophic model ─────────fromcobraimportReaction# Work on a copy so the original model stays cleanmodel_phb=model_hetero.copy()# Cytoplasmic acetyl-CoA is the PHB precursor proxyaccoa=model_phb.metabolites.get_by_id(ACCOA)print(f"Using acetyl-CoA: {accoa.id} | compartment: {accoa.compartment}")# Add PHB demand reactionphb_demand=Reaction('PHB_demand')phb_demand.name='PHB precursor demand (acetyl-CoA sink)'phb_demand.subsystem='PHB biosynthesis'phb_demand.add_metabolites({accoa:-1.0})phb_demand.lower_bound=0phb_demand.upper_bound=1000model_phb.add_reactions([phb_demand])# Confirm baseline PHB proxy flux without constraintsmodel_phb.objective='PHB_demand'sol=model_phb.optimize()print(f"\nBaseline PHB proxy flux: {sol.objective_value:.5f} mmol/gDW/hr | {sol.status}")
Method: First measure the empirical nitrate uptake flux the C. vulgaris cell actually uses at baseline, then build a fine-grained gradient relative to that real biological value using numpy linspace. For each nitrogen level, a two-stage FBA runs: first find the condition-specific maximum biomass, then lock a 1% biomass floor and maximize PHB precursor flux. This approach captures biologically realistic stress levels rather than arbitrary percentages.
Automation: COBRApy context managers; numpy linspace; pandas for results.
Expected result: PHB precursor flux values that vary with nitrogen availability, showing reduced flux under severe nitrogen starvation and confirming that the two-stage optimization captures meaningful metabolic shifts.
Timeline: Day 2 to 3
# ── REVISED CELL 6: Adaptive Step Gradient for Nitrate Starvation ────────────# 1. Detect empirical baseline uptakemodel_phb.objective=BIO_Hbaseline_sol=model_phb.optimize()real_n_max=baseline_sol.fluxes[N_RXN]# Captures exact metabolic inflection pointprint(f"Empirical Nitrate Baseline Uptake: {real_n_max:.5f} mmol/gDW/hr\n")# 2. Build adaptive dynamic range constraint array# Standardize high-volume arbitrary bounds to log historical baseline stepscustom_bounds=[-10.0,-5.0,-1.0]# Generate millimetric high-resolution steps from 100% down to 0% of true uptakefine_steps=np.linspace(1.0,0.0,num=8)forstepinfine_steps:custom_bounds.append(real_n_max*step)# Sort from high-volume capacity to starvation threshold (negative flux dynamics)custom_bounds=sorted(list(set(custom_bounds)))# 3. Process constraint matrix through two-stage Linear Programming loopadaptive_results=[]forboundincustom_bounds:withmodel_phb:# Enforce step-specific boundary constraintmodel_phb.reactions.get_by_id(N_RXN).lower_bound=bound# Stage 1: Compute true capacity ceiling for condition biomassmodel_phb.objective=BIO_Hs1=model_phb.optimize()cond_max=s1.objective_valueifs1.status=='optimal'else0ifcond_max<1e-9:adaptive_results.append({'Applied_N_Bound':round(bound,5),'Biomass_Flux':0,'PHB_Flux_Proxy':0,'Status':'No Growth'})continue# Stage 2: Impose 1% operational floor constraint, maximize precursor sinkmodel_phb.reactions.get_by_id(BIO_H).lower_bound=0.01*cond_maxmodel_phb.objective='PHB_demand's2=model_phb.optimize()adaptive_results.append({'Applied_N_Bound':round(bound,5),'Biomass_Flux':round(cond_max,5),'PHB_Flux_Proxy':round(s2.objective_value,5)ifs2.status=='optimal'else0,'Status':s2.status})df_adaptive=pd.DataFrame(adaptive_results)print("Final Dataset - Nutrient Stress Adaptive Mapping Matrix:")print(df_adaptive.to_string(index=False))
Step 5: Combinatorial adaptive FBA grid (nitrogen x phosphorus x carbon source)
Method: Run the full combinatorial grid using empirically-calibrated nutrient baselines per carbon source. For each carbon source (glucose and acetate), the actual N and P uptake fluxes the cell uses during optimal growth are measured first; stress fractions then scale relative to those real biological values. This is more accurate than scaling from model lower bound constants, which may not reflect actual in silico uptake. The two-stage optimization applies throughout.
Expected result: A ranked table showing that carbon source type is the primary FBA determinant of acetyl-CoA flux in C. vulgaris, with glucose significantly outperforming acetate. This finding motivates the NIT1 promoter design in Aim 2, since nutrient limitation alone is insufficient to drive PHB accumulation without active transcriptional control.
Timeline: Day 3 to 4
# ── CELL 7: Combinatorial adaptive FBA grid (iCZ843 C. vulgaris) ──────────────# Measures empirical N and P baseline uptake per carbon source, then applies# stress fractions relative to those real biological values.withmodel_phb:model_phb.reactions.get_by_id(GLC_RXN).lower_bound=default_glcmodel_phb.reactions.get_by_id(AC_RXN).lower_bound=0model_phb.objective=BIO_Hsol_glc=model_phb.optimize()real_n_glc=sol_glc.fluxes[N_RXN]ifsol_glc.status=='optimal'elsedefault_n_lbreal_p_glc=sol_glc.fluxes[P_RXN]ifsol_glc.status=='optimal'elsedefault_p_lbwithmodel_phb:model_phb.reactions.get_by_id(AC_RXN).lower_bound=default_acmodel_phb.reactions.get_by_id(GLC_RXN).lower_bound=0model_phb.objective=BIO_Hsol_ac=model_phb.optimize()real_n_ac=sol_ac.fluxes[N_RXN]ifsol_ac.status=='optimal'elsedefault_n_lbreal_p_ac=sol_ac.fluxes[P_RXN]ifsol_ac.status=='optimal'elsedefault_p_lbprint(f"Glucose baseline: N = {real_n_glc:.5f} | P = {real_p_glc:.5f}")print(f"Acetate baseline: N = {real_n_ac:.5f} | P = {real_p_ac:.5f}")n_fracs=[0.1,0.25,0.5,0.75,1.0]p_fracs=[0.1,0.25,0.5,0.75,1.0]carbon_modes={'glucose':(GLC_RXN,default_glc,AC_RXN,0,real_n_glc,real_p_glc),'acetate':(AC_RXN,default_ac,GLC_RXN,0,real_n_ac,real_p_ac),}results_3d=[]forn_f,p_f,(c_name,(open_rxn,open_val,close_rxn,close_val,target_n,target_p))initertools.product(n_fracs,p_fracs,carbon_modes.items()):withmodel_phb:iftarget_n==0ortarget_p==0:continuemodel_phb.reactions.get_by_id(N_RXN).lower_bound=target_n*n_fmodel_phb.reactions.get_by_id(P_RXN).lower_bound=target_p*p_fmodel_phb.reactions.get_by_id(open_rxn).lower_bound=open_valmodel_phb.reactions.get_by_id(close_rxn).lower_bound=close_valmodel_phb.objective=BIO_Hs1=model_phb.optimize()cond_max=s1.objective_valueifs1.status=='optimal'else0ifcond_max<1e-9:results_3d.append({'N':n_f,'P':p_f,'Carbon':c_name,'PHB_flux':0,'status':'no_growth'})continuemodel_phb.reactions.get_by_id(BIO_H).lower_bound=0.01*cond_maxmodel_phb.objective='PHB_demand's2=model_phb.optimize()results_3d.append({'N':n_f,'P':p_f,'Carbon':c_name,'PHB_flux':round(s2.objective_value,5)ifs2.status=='optimal'else0,'status':s2.status})df_3d=pd.DataFrame(results_3d).sort_values('PHB_flux',ascending=False)print("\nTop 15 conditions by predicted PHB precursor flux:")print(df_3d.head(15).to_string(index=False))print(f"\nGlucose max PHB = {df_3d[df_3d.Carbon=='glucose']['PHB_flux'].max():.5f}")print(f"Acetate max PHB = {df_3d[df_3d.Carbon=='acetate']['PHB_flux'].max():.5f}")
Step 6: Export top conditions and generate FBA landscape visualizations
Method: Export the top conditions as a CSV for OT-2 protocol parameterization, then generate heatmaps and line plots showing the PHB precursor flux landscape across the nitrogen-phosphorus-carbon combinatorial space.
Automation: pandas to_csv; matplotlib and seaborn for visualization; files.download for Colab export.
Expected result: A CSV file ready for OT-2 parameterization and two publication-quality figures showing the FBA optimization landscape.
Step 7: Design the PHB biosynthetic construct in Benchling
Method: Design a synthetic operon containing phaA, phaB, and phaC genes from Cupriavidus necator H16, codon-optimized for E. coli BL21 (DE3) expression; add an sfGFP transcriptional reporter under the BBa_J23119 constitutive promoter as a proxy for construct activity; use the T7 promoter for operon expression and the pET-28a(+) backbone with kanamycin resistance and ColE1 high-copy origin for selection and propagation. The insert (4,725 bp) was designed in Benchling and submitted to Twist Bioscience as a clonal gene synthesis order for insertion into pET-28a(+), producing a complete 10,017 bp expression plasmid.
Automation: Benchling sequence editor for annotation; IDT Codon Optimization Tool or Benchling codon optimizer for sequence design.
Expected result: A fully annotated plasmid sequence in GenBank format confirmed by Twist Bioscience tools, ready for synthesis and delivery to Ginkgo Bioworks.
Timeline: Day 4 to 5
The E. coli expression insert (phaABC_Ecoli_Construct_2, 4,725 bp) GenBank file is shown below. This insert was submitted to Twist Bioscience for clonal synthesis into the pET-28a(+) backbone, producing the complete 10,017 bp expression plasmid (phaABC_Ecoli_Final_Con):
The complete expression plasmid (phaABC_Ecoli_Final_Con, 10,017 bp circular) with the pET-28a(+) backbone is the Twist Bioscience synthesis target. Key backbone features are summarized below:
LOCUS phaABC_Ecoli_Final_Con 10017 bp ds-DNA circular 12-MAY-2026
DEFINITION phaABC_Ecoli_Construct_2 in pET-28a(+). Complete expression plasmid
for PHB biosynthesis in E. coli BL21(DE3). Insert (1-4725) contains
T7-driven phaABC operon and sfGFP reporter. Backbone (4726-10017)
provides kanamycin resistance, ColE1 high-copy origin, lacI repressor,
and T7/lac regulatory elements. Created using Twist Bioscience tools.
FEATURES Location/Qualifiers
misc_feature 1..4725
/label="insert"
/note="phaABC operon + sfGFP reporter; see
phaABC_Ecoli_Construct_2.gb for full annotation"
misc_feature 4732..4749
/label="6x_His_tag"
/note="C-terminal His-tag in backbone for optional
protein purification"
misc_feature 4760..4888
/label="T7"
/note="T7 promoter in backbone (upstream read-through)"
misc_feature 5447..6262
/label="kan"
/note="Kanamycin resistance; select at 50 ug/mL;
NOTE: pET-28a uses kanamycin, NOT ampicillin"
misc_feature 6384..6972
/label="colE1_high_copy"
/note="ColE1 high-copy origin; ~15-20 copies per cell"
misc_feature 7399..7590
/label="ROP"
/note="ROP protein; regulates plasmid copy number"
misc_feature 8365..8386
/label="CAP_binding"
misc_feature 8399..9490
/label="lacI"
/note="Lac repressor; represses T7lac promoter
until IPTG induction"
misc_feature 9482..9559
/label="lacIQ_promoter"
misc_feature 9868..9886
/label="T7"
/note="T7lac promoter driving insert expression"
misc_feature 9886..9913
/label="lacO"
/note="Lac operator; IPTG-inducible expression"
misc_feature 9926..9948
/label="RBS_3"
/note="Ribosome binding site upstream of insert"
misc_feature 9967..9983
/label="6x_His_tag"
/note="N-terminal His-tag"
//
The Chlorella vulgaris expression construct (pPHB_Chlorella, 5,614 bp linear) GenBank annotation for Aim 2 is provided below. This construct was designed in Benchling with phaA, phaB, and phaC codon-optimized for C. vulgaris nuclear expression under the nitrate-inducible NIT1 promoter:
Step 8: Place the Twist Bioscience DNA synthesis order
Method: Submit the finalized pPHB-Ecoli GenBank sequence to Twist Bioscience via their online ordering portal using the Clonal Gene Synthesis with selected backbone service; specify shipping to Ginkgo Bioworks for subsequent experimental steps.
Expected result: Order confirmation email; estimated synthesis and delivery turnaround of 2 to 3 weeks.
Timeline: Day 5 to 6
Step 9: Program and run the OT-2 media preparation module (Checkpoint A)
Method: Write a Python script using the Opentrons OT-2 API to prepare 96-well deep-well plates with the top 8 FBA-predicted nutrient stress media conditions in triplicate; the protocol ends cleanly at Checkpoint A for users who want only prepared media before proceeding to cell culture in a separate facility.
Automation: Opentrons OT-2 (Python API v2.15); Plateloc plate sealer with A4s breathable seal after preparation
Plate: 96-v-eppendorf-951033502-deep
Expected result: Eight distinct media conditions prepared in triplicate (24 wells), sealed with breathable membrane and ready for inoculation at Ginkgo or an external laboratory.
Timeline: Day 6 to 8
fromopentronsimportprotocol_apimetadata={'protocolName':'PHB_NutrientStress_MediaPrep_CheckpointA','author':'Fabrizio Flores Huaman','description':('Automated preparation of FBA-predicted nutrient stress media for PHB ''screening. CHECKPOINT A: protocol terminates here if only media prep ''is needed. Seal plate with A4s breathable seal and store at 4C (Tundrastore) ''or proceed to inoculation. Continue to Screening Module if cells are ready.')}requirements={'robotType':'OT-2','apiLevel':'2.15'}defrun(protocol:protocol_api.ProtocolContext):# Labwaretiprack_300=protocol.load_labware('opentrons_96_tiprack_300ul',1)tiprack_20=protocol.load_labware('opentrons_96_tiprack_20ul',4)reservoir=protocol.load_labware('nest_12_reservoir_15ml',2)deep_plate=protocol.load_labware('nest_96_wellplate_2ml_deep',3)p300=protocol.load_instrument('p300_multi_gen2','right',tip_racks=[tiprack_300])p20=protocol.load_instrument('p20_multi_gen2','left',tip_racks=[tiprack_20])# Reservoir layoutbase_media=reservoir['A1']# LB or BG-11 minimal salts basen_source=reservoir['A2']# NaNO3 stock (nitrogen source)p_source=reservoir['A3']# KH2PO4 stock (phosphorus source)c_glucose=reservoir['A4']# 20% glucose stock (carbon source A)c_acetate=reservoir['A5']# Sodium acetate stock (carbon source B)diluent=reservoir['A6']# Sterile water (diluent for gradient)# Dispense 1200 uL base media to all 24 test wells (columns 1-3, rows A-H)test_cols=deep_plate.columns()[:3]forcolintest_cols:p300.transfer(1200,base_media,col,mix_after=(3,200),new_tip='always')# Apply FBA-predicted nutrient stress recipes# Row layout (conditions in triplicate across columns 1-3):# Row A: N0%-P100%-Glucose | Row B: N25%-P100%-Glucose# Row C: N50%-P100%-Glucose | Row D: N0%-P50%-Glucose# Row E: N0%-P0%-Glucose | Row F: N50%-P50%-Acetate (top FBA candidate)# Row G: N100%-P100%-Glucose | Row H: N100%-P100%-No exogenous carbonnitrogen_vols=[0,12.5,25,0,0,25,50,50]# uL of NaNO3 stock per rowphosphorus_vols=[50,50,50,25,0,25,50,50]carbon_sources=[c_glucose,c_glucose,c_glucose,c_glucose,c_glucose,c_acetate,c_glucose,diluent]forrow_idx,(n_vol,p_vol,c_src)inenumerate(zip(nitrogen_vols,phosphorus_vols,carbon_sources)):row_letter='ABCDEFGH'[row_idx]wells=[deep_plate[f'{row_letter}{col+1}']forcolinrange(3)]ifn_vol>0:p20.transfer(n_vol,n_source,wells,new_tip='always')ifp_vol>0:p20.transfer(p_vol,p_source,wells,new_tip='always')p20.transfer(50,c_src,wells,mix_after=(2,15),new_tip='always')protocol.comment("="*60)protocol.comment("CHECKPOINT A: Media preparation complete.")protocol.comment("Seal plate with A4s breathable seal using Plateloc.")protocol.comment("Store at 4C in Tundrastore if not inoculating immediately.")protocol.comment("To continue: proceed to Screening Module (Step 10 onward).")protocol.comment("="*60)
Step 10: Inoculate cultures and incubate
Method: Inoculate each prepared deep-well with E. coli BL21 (DE3) harboring pPHB-Ecoli at a starting optical density at 600 nm (OD600) of 0.05; seal plate with A4s breathable seal.
Automation: Plateloc (sealing with A4s breathable membrane); Cytomat shaking incubator at 37 degrees Celsius, 250 rpm
Plate: 96-v-eppendorf-951033502-deep
Expected result: Growth visible in positive control wells within 6 to 8 hours; reduced growth rate in nutrient-limited wells confirming that stress conditions are active.
Timeline: Day 8 to 9 (overnight incubation, approximately 16 hours)
Step 11: Measure growth dynamics by optical density
Method: Transfer 100 µL from each deep-well to a flat-bottom 96-well plate using the Echo525; measure OD600 using the Spark Plate Reader to assess growth rates and confirm that nutrient limitation is reducing but not eliminating cell viability.
Plate: 96-round-axygen-pdw11cs-halfdeep for transfer; flat 96-well for reading
Expected result: OD600 values confirming a gradient of growth suppression correlated with increasing nutrient limitation; no complete cell death in any well.
Timeline: Day 9
Step 12: Perform Nile Red staining for PHB quantification
Method: Add Nile Red dissolved in dimethyl sulfoxide (DMSO) at 1 mg/mL to each well using the Echo525 at a 1:1000 final dilution (1 µL per mL culture); incubate 15 minutes at room temperature in the dark; transfer 50 µL to 384-well black clear-bottom plate for fluorescence reading.
Automation: Echo525 (Nile Red addition, nanoliter precision); Multiflo (50 µL culture transfer to 384-well); PHERAstar FSX (Ex 530 nm / Em 580 nm)
Plate: 384 Greiner black-well clear-bottom
Expected result: Higher Nile Red fluorescence in nutrient-limited conditions compared to full-media controls, consistent with increased PHB granule accumulation.
Timeline: Day 9 to 10
Step 13: Validate sfGFP reporter expression
Method: Measure sfGFP fluorescence (Ex 485 nm / Em 510 nm) on the Spark Plate Reader using the same 384-well plate as the Nile Red assay, before Nile Red addition, to confirm construct expression and identify any wells where failed transformation rather than PHB pathway activity is responsible for low Nile Red signal.
Expected result: Measurable sfGFP signal in all transformed wells; absence of sfGFP in empty vector controls; allows normalization of Nile Red signal to expression level.
Timeline: Day 9 to 10 (before Nile Red addition)
Step 14: Validate PHB gene expression by quantitative PCR
Method: Harvest cells from the top 3 performing conditions and the negative control; extract total RNA using an RNeasy kit; perform quantitative polymerase chain reaction (qPCR) to measure phaA, phaB, and phaC transcript levels relative to the 16S ribosomal RNA housekeeping gene.
Automation: CFX Opus qPCR machine
Plate: 384-pcr-eppendorf-9510207XX
Expected result: Upregulation of phaA, phaB, and phaC transcripts in the top nutrient stress conditions relative to full-media controls, confirming that the T7 promoter is active and that the operon is being transcribed in proportion to the metabolic state of the cell.
Timeline: Day 10 to 11
Step 15: Close the DBTL loop by refining the FBA model
Method: Feed the experimental Nile Red fluorescence values and qPCR expression data back into the COBRApy model as refined constraints; adjust exchange reaction bounds to reflect observed growth rates and acetyl-CoA sink capacity; re-run FBA with the updated model to identify any systematic discrepancies between prediction and observation.
Automation: COBRApy model update; pandas for data integration; optional MEMOTE framework for model quality scoring
Expected result: An updated model with improved predictive accuracy for a second round of experimental screening or for informing the C. vulgaris construct design in Aim 2.
Timeline: Day 11 to 13
Step 16: Optional supplementary characterization via FTIR and GC-MS
Method: Lyophilize the cell pellets from the top two performing conditions; extract PHB polymer using a standard chloroform solvent extraction protocol; analyze bulk polymer by Fourier-transform infrared spectroscopy (FTIR) for functional group confirmation and gas chromatography-mass spectrometry (GC-MS) via the Waters Corporation platform at Ginkgo Bioworks or a partner facility for monomer composition and yield quantification.
Automation: Waters GC-MS system (Ginkgo Bioworks partner; connection to Waters Corporation industry partner)
Expected result: FTIR spectra showing a characteristic PHB carbonyl peak at 1720 cm-1; GC-MS confirmation of 3-hydroxybutyrate monomer and quantitative PHB yield in mg per gram dry cell weight.
Timeline: Day 12 to 14 (if resources permit)
Assay Plate Layout
The following 96-well plate layout illustrates the screening design for the Nile Red fluorescence assay. Conditions are derived from the top FBA-predicted nutrient stress recipes, with all experimental conditions run in duplicate.
Col 1 Col 2 Col 3 Col 4 Col 5 Col 6
Row A [STD 0µg/mL] [STD 0µg/mL] [POS_CTRL] [POS_CTRL] [N0-P100-Glc] [N0-P100-Glc]
Row B [STD 1µg/mL] [STD 1µg/mL] [POS_CTRL] [POS_CTRL] [N25-P100-Glc] [N25-P100-Glc]
Row C [STD 2µg/mL] [STD 2µg/mL] [NEG_CTRL] [NEG_CTRL] [N50-P100-Glc] [N50-P100-Glc]
Row D [STD 5µg/mL] [STD 5µg/mL] [NEG_CTRL] [NEG_CTRL] [N0-P50-Glc] [N0-P50-Glc]
Row E [STD 10µg/mL] [STD 10µg/mL] [BLANK] [BLANK] [N0-P0-Glc] [N0-P0-Glc]
Row F [STD 25µg/mL] [STD 25µg/mL] [BLANK] [BLANK] [N50-P50-Ace] [N50-P50-Ace]
Row G [STD 50µg/mL] [STD 50µg/mL] [BLANK] [BLANK] [N100-P100-Glc] [N100-P100-Glc]
Row H [STD100µg/mL] [STD100µg/mL] [BLANK] [BLANK] [N100-P100-NoC] [N100-P100-NoC]
Plate Legend:
STD: Polyhydroxybutyrate (PHB) polymer standard curve (0 to 100 µg/mL; Nile Red fluorescence calibration; Millipore Sigma catalog 363502)
POS_CTRL: Positive control (E. coli BL21 DE3 with a known PHB-producing construct or C. necator ATCC 17699)
NEG_CTRL: Negative control (E. coli BL21 DE3 with empty pUC19 vector; no PHB genes)
BLANK: Sterile media only; no cells; used for background fluorescence subtraction
N[x]-P[x]-[Carbon]: Experimental conditions where x represents percent of standard nitrogen or phosphorus concentration
All experimental conditions run in duplicate (two adjacent wells per condition)
After initial assay, the 384-well Greiner black clear-bottom plate is used for the actual Nile Red fluorescence measurement with 50 µL transfers from each 96-well condition
SECTION 4.2: TECHNIQUES, TOOLS, AND TECHNOLOGY
Course Technique Checklist
Metabolic modeling and Flux Balance Analysis (FBA) using COBRApy
DNA construct design and sequence annotation using Benchling
Synthetic gene and whole plasmid synthesis ordering from Twist Bioscience
Codon optimization for heterologous expression (E. coli and C. vulgaris)
Lab automation and liquid handling (Opentrons OT-2, Echo525, Multiflo)
Bacterial whole-cell expression in E. coli BL21 (DE3)
Cell-free protein synthesis (CFPS) for rapid construct validation
Fluorescence assay development and microplate reader detection (Spark, PHERAstar FSX)
Quantitative PCR (qPCR) for gene expression validation (CFX Opus)
Bioinformatics and Python scripting for data analysis and model interrogation
Microplate screening and high-throughput assay design (96-well and 384-well formats)
Biosafety and SecureDNA screening for synthetic DNA sequences
CRISPR genome editing (relevant for Aim 2 in C. vulgaris; not executed in Aim 1)
Flow cytometry (optional alternative to plate reader Nile Red assay)
Next-generation sequencing (could be used for construct integration verification in Aim 2)
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Pipetting
Pipetting
Lab Safety
Bioethical Considerations (must check this box)
DNA Gel Art
DNA Sequencing
DNA Editing
DNA Construct Design
Restriction Enzyme Digestion
Gel Electrophoresis
DNA Purification From Gel
Databases (e.g., GenBank, NCBI, Ensembl, and UCSC Genome Browser)
Lab Automation
Creating Code for Laboratory Automation
Using Liquid Handling Robots (e.g., Opentrons)
Designing a Twist Order
Creating a plan to use the Autonomous lab at Ginkgo Bioworks
Protein Design
Protein Design
Use of Boltz or PepMLM
Use of Asimov Kernel
Use of Benchling
Models and Notebooks
Databases
Bioproduction
Bioproduction
Chassis Selection (e.g., DH5alpha)
Registry of Standard Biological Parts
Plasmid Preparation
Bacterial Culturing
Quality Control/Analysis
Bacterial Processing (e.g., Centrifugation, Lysis, DNA Purification)
Cell-Free Systems
Cell Free Reactions
Freeze-Dried Cell Free Systems
miniPCR Tools
Protein Purification
Gibson Assembly
Primer Design or Selection
PCR Reactions
Gibson Assembly
Other Cloning Methods (e.g., Restriction Enzyme Digestion or Gateway Cloning)
CRISPR
CRISPR/Cas9
Designing Prime Editing gRNA
Technique Expansion
Technique 1: Flux Balance Analysis (FBA) with COBRApy
Flux Balance Analysis is a constraint-based computational method that represents cellular metabolism as a network of stoichiometric equations, where each equation describes how metabolites are converted into one another by enzymatic reactions. COBRApy is the Python implementation of the COBRA (COnstraint-Based Reconstruction and Analysis) toolbox, which allows researchers to load genome-scale metabolic models in SBML format and apply linear programming solvers such as GLPK or Gurobi to find the flux distribution that maximizes a defined biological objective, such as biomass growth or production of a target metabolite. In the context of this project, FBA is uniquely powerful because it enables the simultaneous exploration of hundreds of nutrient combinations without requiring physical experiments, making it a fast and cost-effective Design phase tool in the DBTL cycle. The key limitation of FBA is that it assumes steady-state metabolic conditions and does not capture dynamic gene regulatory responses, such as the transcriptional induction of PHB pathway genes under stress, which is why experimental validation through the OT-2 pipeline and qPCR is an essential complement to the computational predictions. By connecting FBA outputs directly to parameterized OT-2 protocols, this project creates a tightly coupled workflow where computational and physical experimentation reinforce rather than simply follow each other.
Technique 2: Nile Red Fluorescence Assay for Intracellular PHB Quantification
Nile Red is a lipophilic fluorescent dye that partitions selectively into hydrophobic intracellular environments, including lipid droplets and polymer granules such as PHB inclusions, producing a fluorescence signal with an excitation maximum near 530 nm and an emission maximum near 580 nm that scales linearly with polymer content across several orders of magnitude. The assay is performed directly in microplate wells without cell disruption, making it fully compatible with high-throughput robotic workflows and the PHERAstar FSX plate reader available at Ginkgo Bioworks, and allowing the same plate to be used for both growth monitoring and biopolymer quantification. One important technical consideration is that Nile Red is not strictly specific to PHB and will also stain other neutral lipids such as triacylglycerols, so the assay is best interpreted as a measure of total neutral lipid content unless confirmatory techniques such as FTIR or GC-MS are applied to establish polymer identity. In this project, Nile Red serves as the rapid, high-throughput screening layer in the pipeline, allowing the top-performing nutrient conditions to be ranked and selected before committing to the more resource-intensive and time-consuming confirmatory analyses, demonstrating how orthogonal methods at different resolution levels can be strategically combined in an automated screening workflow.
Identify any How To Grow (Almost) Anything Industry Council companies which are associated with your final project (optional)
Addgene
Asimov (Kernel)
ATCC
Basecamp Research
BioFabricate
Biome Consortia
Bolt
Boltz.bio
Cultivarium
DeepCure
Epibone
Ginkgo Bioworks
Helix Nano
Millipore Sigma
Mycoworks
New England Biolabs
Nuclera
Opentrons
SecureDNA
Takeda Pharmaceuticals
Thermo Fisher Scientific
Transfyr.ai
Twist Biosciences
Upside Foods
Waters Corporation
SECTION 5: RESULTS AND QUANTITATIVE EXPECTATIONS
10a: Validation Choice
The validation experiment for this project is a cell-free expression assay of the phaA-phaB-phaC operon in an E. coli BL21 (DE3) lysate system, followed by Nile Red fluorescence measurement to confirm PHB granule formation from the synthetic construct in the absence of a complete living cell chassis. This approach is particularly well-suited for a primarily in silico project because it provides a rapid, contained, and robotically compatible way to confirm that the designed construct produces active enzymes and that the phaABC-encoded pathway generates detectable PHB signal within hours of receiving the DNA, establishing proof-of-function before committing to multi-day culture experiments.
10b: Validation Protocol
Prepare E. coli BL21 (DE3) cell-free protein synthesis (CFPS) master mix using a Ginkgo Bioworks CFPS kit or equivalent commercial system (e.g., PURExpress from New England Biolabs).
Thaw the pPHB-Ecoli plasmid received from Twist Bioscience on ice and dilute to 10 nM in nuclease-free water.
Set up the following controls in separate wells: (a) positive control with a 10 nM sfGFP-only expression plasmid, (b) negative control with no DNA added, (c) PHB polymer standard curve at 0, 1, 2, 5, 10, 25, 50, and 100 µg/mL using Millipore Sigma PHB standard (catalog 363502).
Prepare 10 µL CFPS reactions in 1.5 mL Eppendorf tubes by combining 7.5 µL CFPS master mix with 1 µL plasmid (final 10 nM) and 1.5 µL nuclease-free water; also supplement with 0.5 mM acetyl-CoA (Millipore Sigma A2056) and 0.1 mM coenzyme A to support PHB precursor availability.
Dispense 10 µL reactions into 384-well Greiner black clear-bottom plate using Echo525 acoustic liquid handler for nanoliter precision.
Seal plate using Plateloc with a standard film seal (not breathable) to prevent evaporation during incubation.
Incubate at 30 degrees Celsius for 4 hours in the Inheco Plate Incubator.
Peel seal using XPeel; measure sfGFP fluorescence (Ex 485 nm / Em 510 nm) on the Spark Plate Reader to confirm construct expression before Nile Red addition.
Add 0.5 µL Nile Red stock (1 mg/mL in DMSO) per well using the Echo525 (1:20 dilution for 10 µL wells); final Nile Red concentration approximately 50 µg/mL.
Incubate plate at room temperature for 15 minutes in the dark (cover with aluminum foil or store in lightproof box).
Measure Nile Red fluorescence (Ex 530 nm / Em 580 nm) on the PHERAstar FSX using the well-resolved fluorescence intensity module.
Subtract background fluorescence values from no-DNA blank wells from all experimental wells.
Normalize Nile Red signal to sfGFP signal in each well to correct for variation in expression level between replicate reactions.
Generate a standard curve from PHB polymer standards and convert normalized fluorescence values to estimated PHB content in µg/mL.
A validation result is positive if the pPHB-Ecoli construct produces a Nile Red signal greater than 3 standard deviations above the no-DNA control after normalization to sfGFP expression.
10c: Techniques Used
This validation experiment integrates four core techniques that span the full technical scope of the project. First, cell-free protein synthesis (CFPS) provides a rapid in vitro expression system that bypasses the multi-week timelines of stable cell transformation and selection, allowing construct function to be assessed within a single working day of receiving the synthesized plasmid from Twist Bioscience. Second, Echo525 acoustic liquid handling ensures nanoliter-precision dispensing of both the CFPS reagents and the Nile Red dye, eliminating the pipetting variability that would otherwise mask subtle differences in PHB signal between conditions and making the assay compatible with fully automated workflows at Ginkgo Bioworks. Third, dual fluorescence detection on both the Spark Plate Reader (for sfGFP) and the PHERAstar FSX (for Nile Red) provides an internal normalization strategy, ensuring that differences in Nile Red signal reflect genuine differences in PHB polymer accumulation rather than artifacts from varying expression efficiency between reaction wells. Fourth, the use of a PHB polymer standard curve converts raw relative fluorescence units into physically interpretable concentration units, grounding the validation results in quantitative terms that can be directly compared to the acetyl-CoA flux values predicted by the FBA model and used to evaluate whether the computational predictions are consistent with experimental reality.
10d: Hypothetical Data
The following table represents a hypothetical subset of Nile Red fluorescence results from the cell-free validation experiment, normalized to sfGFP expression and converted to estimated PHB content using the standard curve:
Condition
Nile Red Signal (RFU)
sfGFP Signal (RFU)
Normalized PHB (µg/mL)
FBA Predicted Flux (mmol/gDW/hr)
No DNA (blank)
118
0
0.0
N/A
Empty vector (neg ctrl)
130
4,850
0.2
N/A
pPHB-Ecoli, full media
4,100
6,200
8.2
1.4
pPHB-Ecoli, N50%-P100%-Glc
9,700
5,900
21.4
3.8
pPHB-Ecoli, N0%-P50%-Glc
13,900
5,500
33.1
6.2
pPHB-Ecoli, N50%-P50%-Acetate
18,500
5,750
42.5
8.1
pPHB-Ecoli, FBA optimal condition
21,800
6,050
46.9
9.0
PHB standard, 50 µg/mL
23,600
N/A
50.0
N/A
These hypothetical results suggest a positive and roughly linear correlation between the FBA-predicted acetyl-CoA flux and the measured Nile Red-normalized PHB content, consistent with the hypothesis that FBA can accurately rank nutrient stress conditions by their bioplastic potential. The FBA-predicted optimal condition (N50%-P50%-Acetate carbon source) produces the highest normalized PHB signal, validating the in silico approach and justifying the use of acetyl-CoA flux as a proxy for polymer accumulation.
Troubleshooting
One of the most likely challenges in this project is encountering errors when loading or running FBA on the iCZ843 model, since genome-scale models can contain blocked reactions, metabolite charge imbalances, or infeasible constraint combinations that prevent the optimizer from finding a solution. If the model returns an infeasible status or unrealistically extreme objective values, the recommended approach is to run the MEMOTE model quality assessment tool to identify and annotate problematic reactions, and to apply standard growth media constraints before adding the PHB demand reaction. A second potential issue is low or absent Nile Red fluorescence signal in CFPS reactions despite confirmed sfGFP expression, which most likely results from insufficient acetyl-CoA availability in the cell-free lysate rather than a non-functional phaABC operon. In that case, supplementing the CFPS reaction with exogenous acetyl-CoA at 0.5 to 2 mM and coenzyme A at 0.1 mM is a standard troubleshooting step that can significantly improve PHB signal. A third limitation is that the E. coli CFPS system provides only a coarse proxy for PHB production in a living cell, and in particular does not capture the growth-competition dynamics between biomass production and polymer accumulation that FBA is designed to model. Finally, the codon-optimized sequences designed for E. coli will not directly predict expression efficiency in C. vulgaris in Aim 2, and the significantly different GC content preferences and translation initiation contexts between the two organisms mean that the algal construct will require independent validation through a separate CFPS system or direct transformation, which is why Aim 2 is framed as a distinct experimental stage requiring specialized facilities rather than a simple extension of Aim 1.
SECTION 6: ADDITIONAL INFORMATION
References
Orth JD, Thiele I, Palsson BO. (2010). What is flux balance analysis? Nature Biotechnology, 28(3), 245-248. https://doi.org/10.1038/nbt.1614
Peoples OP, Sinskey AJ. (1989). Poly-beta-hydroxybutyrate (PHB) biosynthesis in Alcaligenes eutrophus H16. Identification and characterization of the PHB polymerase gene (phbC). Journal of Biological Chemistry, 264(26), 15298-15303.
Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. (2013). COBRApy: COnstraint-Based Reconstruction and Analysis for Python. BMC Systems Biology, 7, 74. https://doi.org/10.1186/1752-0509-7-74
Zuñiga C, Li CT, Huelsman M, Andrews J, Kerkhoven EJ, Zengler K. (2016). Genome-scale metabolic model for the green alga Chlorella vulgaris UTEX 395 accurately predicts phenotypes under autotrophic, heterotrophic, and mixotrophic growth conditions. Plant Physiology, 172(1), 589-602. https://doi.org/10.1104/pp.16.00593
Al-Hammadi M, Güngörmüşler M. (2024). New insights into Chlorella vulgaris applications. Biotechnology and Bioengineering, 121(5), 1486-1502. https://doi.org/10.1002/bit.28666
Hempel F, Bozarth AS, Lindenkamp N, Klingl A, Zauner S, Linne U, Steinbüchel A, Maier UG. (2011). Microalgae as bioreactors for bioplastic production. Microbial Cell Factories, 10, 81. https://doi.org/10.1186/1475-2859-10-81
Cooksey KE, Guckert JB, Williams SA, Callis PR. (1987). Fluorometric determination of the neutral lipid content of microalgal cells using Nile Red. Journal of Microbiological Methods, 6(6), 333-345.
Oscanoa Huaynate AI, Cervantes Gallegos MA, Flores Ramos L, Ruiz Soto A. (2021). Evaluación del potencial de Desmodesmus asymmetricus y Chlorella vulgaris para la remoción de nitratos y fosfatos de aguas residuales. Revista Peruana de Biología, 28(1), e18082. https://doi.org/10.15381/rpb.v28i1.18082
Mariano Astocondor M, Mayta Huatuco E, Montoya Terreros H, Tarazona Delgado R. (2017). Crecimiento poblacional y productividad de la microalga nativa Chlorella peruviana bajo diferentes salinidades. Revista de Investigaciones Veterinarias del Perú, 28(4), 976-986. https://doi.org/10.15381/rivep.v28i4.13004
Condori MAM, Gutierrez MEV, Oviedo RDN, Choix FJ. (2024). Valorization of nutrients from fruit residues for the growth and lipid production of Chlorella sp.: A vision of the circular economy in Peru. Journal of Applied Phycology, 36(1), 101-111. https://doi.org/10.1007/s10811-023-03153-2
Loera-Quezada MM, Rios-Castro E, Orta-Zavalza E, et al. (2016). CRISPR-Cas9-based genome editing approaches in the model green microalga Chlamydomonas reinhardtii. Plant Methods, 12, 30. [Referenced for algal CRISPR design rationale applicable to Aim 2.]
Arora et al. (2024). Metabolic response to a heterologous poly-3-hydroxybutyrate (PHB) pathway in Phaeodactylum tricornutum. Applied Microbiology and Biotechnology. https://doi.org/10.1007/s00253-023-12823-7
Yi YC, Ng IS. (2025). Sustainable polyhydroxybutyrate production via metabolic flux redirection using CRISPRi in Escherichia coli and carbon capture with microalgae. ACS Sustainable Chemistry and Engineering. https://doi.org/10.1021/acssuschemeng.4c09128
Chaturvedi et al. (2023). Systematizing microbial bioplastic production for developing sustainable bioeconomy: metabolic nexus modeling, economic and environmental technologies assessment. Journal of Polymers and the Environment. https://doi.org/10.1007/s10924-023-02787-0
1. Hypothesis: Engineering Lysis Protein Stability
Our core hypothesis is that the thermodynamic stability and lytic efficiency of the MS2 L-protein can be enhanced through two strategic pathways:
Structural Reinforcement: Introducing targeted mutations that promote independent folding or stabilize the 7-helix bundle, reducing dependence on the host chaperone DnaJ.
Generative Optimization: Utilizing evolutionary conservation data and generative protein design to create variants with improved membrane-insertion kinetics and host compatibility, thereby minimizing host-mediated resistance.
2. Specific Aims and Validation Pipeline
Aim 1: Mutation Design via Conservation and Predictive Modeling
We will perform Clustal Omega alignments of homologous lysis proteins to identify conserved residues (specifically the “HEDYPCRRQQRSST” island). This is followed by:
In silico Mutational Scanning: Using ESM-2 embeddings and LLR scores to nominate stabilizing mutations.
Folding Assessment: Validation of fold accuracy via ESMFold and AlphaFold-Multimer to ensure independent folding propensity and multimeric pore symmetry.
Aim 2: Generative Design for Chaperone Independence
We propose using generative models (like ProteinMPNN or RFdiffusion) to optimize the soluble N-terminal domain. The goal is to redesign the interface to either:
Enhance co-folding with DnaJ under controlled structural constraints.
Enable “folding rescue” by alternative or orthogonal chaperones (e.g., DnaK or GroEL) to bypass host adaptation.
Aim 3: Evolutionary Analysis and Host Factor Integration
Using pBLAST to survey orthologs, we will reconstruct evolutionary trajectories of stability. Candidates will be screened against E. coli host factors to minimize proteotoxicity while maximizing the “aggressive” lytic potential identified through Genomic Language Models (GLMs).
3. Computational Tools and Workflow
Our design-build-test-learn (DBTL) framework utilizes the following stack:
Sequence & Conservation: Clustal Omega and pBLAST for “evolutionary grammar” analysis.
Mutational Analysis: ESM-2 (LLR scores) for high-speed structural feedback on the 7-helix bundle.
Generative Design: ProteinMPNN for sequence backbone optimization.
Structural Validation: AlphaFold 3 and AlphaFold-Multimer to ensure biophysical plausibility of the 8-chain pore assembly.
Contextual Gap: A lack of specific data regarding the host bacteria’s in vivo environment may lead to unexpected results despite positive simulations.
Functional Trade-offs: Mutations that improve structural stability might inadvertently perturb the membrane-interaction properties or the native lytic activity, leading to a loss of function.
Misfolding Risk: Compact lysis proteins are highly sensitive; even high-confidence predicted folds (pLDDT > 80) may aggregate or fail to insert into the membrane in a real biological system.
5. Expected Outcomes
If successful, this framework will yield L-protein variants with:
Increased Stability: Robust functionality under diverse environmental conditions.
Reduced Host Dependency: Decreased reliance on native DnaJ interactions, making the phage less vulnerable to host-dependent failure modes.
Optimized Lysis: Retention of a mature fold compatible with aggressive lytic activity, establishing a generalizable template for synthetic antimicrobial modules.
The study explains that the MS2 phage L protein is a 75 amino acid polypeptide that kills bacteria through a unique mechanism. Unlike other proteins like E or A2, which block cell wall synthesis, the L protein does not affect peptidoglycan production. Using a smart screening system with a lacZ reporter to filter out false positives, the researchers discovered that L depends entirely on the host chaperone DnaJ to function. Interestingly, a specific mutation in DnaJ called P330Q completely blocks lysis at 30°C. Through pulldown assays, they confirmed that DnaJ physically binds to the N-terminal “head” of the protein, which is full of basic charges and is actually dispensable for the killing process, serving instead as a control unit.
The final model proposes that this N-terminal domain of L acts as a biological brake that auto-inhibits the protein. The DnaJ chaperone acts like a key that unlocks this brake, allowing the hydrophobic tail of the protein to reach its actual target inside the cell. This was proven with Lodj mutants, which are versions of the L protein lacking the head. These mutants do not need DnaJ and kill the bacteria 20 minutes faster than the wild type. This system mirrors what happens with the E protein and its chaperone SlyD, suggesting that phages evolved these charged domains as a strategy to control lysis timing and ensure the virus has enough time to replicate before destroying the host.
The MS2 lysis protein (L) is a 75-amino acid polypeptide that triggers bacterial cell death without disrupting net peptidoglycan synthesis. Research reveals a conserved LS (Leu-Ser) dipeptide motif at residues Leu48-Ser49, which serves as the essential core for protein-protein interactions. While the N-terminal half of the protein is dispensable for lytic activity, the C-terminal domain is critical; specifically, the S49C mutation in the LS motif causes an absolute lysis defect. This motif is highly conserved across diverse phages, indicating it is a universal structural requirement for the lytic function in amurins.
The study suggests that the L protein interacts with a host membrane target through the LS motif and surrounding essential domains. The N-terminus functions as a regulatory domain that naturally inhibits this interaction, while the host chaperone DnaJ binds to the N-terminus to displace it from its inhibitory position. Interestingly, deleting the basic N-terminal domain allows the protein to bypass the need for DnaJ entirely. This confirms that the N-terminus acts as a regulatory gatekeeper, and DnaJ is the key that unlocks the protein’s ability to engage its cellular target.
The MS2 bacteriophage lysis protein (MS2L) facilitates host cell escape by punching holes in the bacterial wall through a dual-domain mechanism. It consists of a soluble HEAD domain and a transmembrane TAIL domain that anchors into membranes, behaving similarly to soap or micelles. A key finding is that the TAIL domain drives oligomerization, causing 10 or more proteins to clump into large complexes. CryoEM data confirms these clusters gather at specific spots to trigger a sequential rupture: first the outer membrane breaks, followed by the peptidoglycan layer, and finally the inner membrane, causing the cell contents to leak out.
The researchers identified the HEAD domain as a biological brake that regulates the timing of lysis. While the full MS2L protein is difficult to insert into membranes, removing the HEAD allows for relatively easy insertion, suggesting it functions as a timer to prevent premature cell death. Additionally, the helper protein DnaJ binds to MS2L but does not influence its membrane entry or oligomerization. From an engineering perspective, removing the HEAD domain could bypass this brake to achieve a “quicker kill,” a strategic goal for optimizing lytic toxicity in synthetic biology.
This paper explains how phages have evolved from a biological curiosity into a sophisticated therapeutic tool by focusing on their life cycles and resistance mechanisms. The review highlights that success in therapy depends on more than just injecting phages; it requires a deep understanding of pharmacokinetics and the patient’s immune response, as the body might neutralize the viruses before they reach the infection site. A key advancement mentioned is the use of genetic engineering to create “designer” phages that do more than just kill bacteria, such as degrading biofilms or working alongside traditional antibiotics to restore drug sensitivity. The future of the field points toward precision medicine where specific phages are selected or edited for each patient to overcome the regulatory and technical barriers that previously limited mass clinical use.
This text explores the historical evolution and the modern resurgence of phage therapy in response to the global antibiotic resistance crisis. It begins by reminding us that phages were used long before penicillin but were largely forgotten in the West due to a lack of standardized protocols and the convenience of broad-spectrum antibiotics. Currently, we are in a stage of “compassionate use” where phages are successfully applied in desperate cases of multi-drug resistant infections, which is driving new controlled clinical trials. The study concludes that the biggest challenge today is not just biological but also logistical and legal, as a global infrastructure is needed to collect and characterize phage libraries that can be quickly deployed against emerging pathogens. This marks a shift from general treatments to a completely personalized paradigm.
This research utilizes the Evo 1 and Evo 2 DNA foundation models to design functional biological systems at the whole-genome scale. Using the phiX174 lytic phage as a chassis, the AI successfully generated 16 viable phages with substantial evolutionary novelty. Some variants were highly distant from common natural sequences, proving that genomic language models (GLMs) can expand the known biological space. This is critical for phage therapy, as these AI-designed variants demonstrated a superior ability to overcome bacterial resistance in E. coli strains where natural phages failed.
The computational method employed taxonomic prompting (e.g., Riboviria) to guide the generative process toward specific viral realms. Novelty was rigorously validated using nucleotide BLAST against core databases to confirm the emergence of original sequences. This strategy offers a robust framework for creating diverse phage cocktails, a key requirement for modern antimicrobial treatments. By leveraging taxonomic labels and pretraining, the study establishes a “design-build-test” workflow for engineering complex, multi-gene systems beyond the limits of natural evolution.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
We will focus on increasing the structural stability of the L protein to ensure it remains functional under different environmental conditions.
We will also attempt to increase the toxicity of the lysis protein by optimizing its target regions to enhance bacterial cell wall disruption.
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
We will use ESMFold to perform in silico mutational scanning and identify target regions in the L protein.
We propose using Genomic Language Models (GLMs) to design and optimize sequences with higher lytic potential.
Finally, we will use AlphaFold-Multimer to validate the folding and stability of the engineered protein complexes.
Why do you think those tools might help solve your chosen sub-problem?
ESMFold allows for high-speed structural feedback, making it easier to test how mutations affect the 7-helix bundle.
GLMs are essential for capturing the “evolutionary grammar” of toxicity, helping to design proteins that are more aggressive than natural variants.
AlphaFold ensures that our computational designs are biophysically plausible and stable before any potential wet-lab implementation.
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Contextual Gap: There is a lack of specific data regarding the host bacteria’s environment, which might lead to unexpected results in vivo.
Misfolding Risk: The engineered protein might still misfold or aggregate in a real biological system despite having positive simulation results in the pipeline.
Include a schematic of your pipeline.
Here’s a short written schematic of our pipeline:
[Sequence Input] → [ESM-2 Mutational Scan] → [GLM Toxicity Optimization] → [AlphaFold Validation] → [Final Design]
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a
MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
L-Protein Engineering - Option 1: Mutagenesis
Step 1: Information Gathering
Here are the L-protein and Dnaj sequences
Lysis Protein Sequence (UniProtKB ID: P03609)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
Soluble N-terminal domain: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYV
TM domain: LIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Additionally, here’s a screenshot of the BLAST results for L-protein:
Lastly, these results were aligned using Clustal Omega, revealing a highly conserved “island” (HEDYPCRRQQRSST) at residues 24-38. These sites will be avoided during mutagenesis to preserve the critical interaction with DnaJ and overall biological function of the phage.
My approach is very straightforward: I combined computational LLR scores with experimental lab data using a copy of the HTGAA Colab. I filtered for mutations that showed “active lysis” (value 1) in the experimental spreadsheet and high positive LLR scores in the notebook.
Step 3: Filtering and Ranking
I used AlphaFold 3 to model the 8-chain assembly. This step was used to rank candidates that showed both positive computational scores and confirmed experimental activity, ensuring they don’t disrupt the pore’s symmetry.
Step 4: Final Mutated Sequences
These 5 mutations were selected because they are experimentally proven to maintain lysis (score 1) and show improved or stable computational scores.
Region
Mutation
LLR Score (ESM-2)
Experimental Lysis
Rationale
Soluble
S9Q
2.01
Active (1)
High computational confidence; replaces Serine with Glutamine to stabilize the N-terminal loop.
Soluble
C29R
2.39
Active (1)
One of the top scores; removing this Cysteine likely prevents incorrect disulfide bonding.
TM Domain
Y39L
2.24
Active (1)
High confidence score in the TM interface; optimizes hydrophobicity for membrane entry.
TM Domain
A45L
1.53
Active (1)
Consistent with experimental data; improves the hydrophobic core of the lytic pore.
TM Domain
N53L
1.86
Active (1)
Replaces a polar Asparagine with Leucine, significantly improving helix-helix packing in the multimer.
S9Q mutation 8-chain assembly:
C29R mutation 8-chain assembly:
Y39L mutation 8-chain assembly:
A45L mutation 8-chain assembly:
N53L mutation 8-chain assembly:
While AF3 structures were used to visualize the multimeric orientation, the ipTM scores remained low (~0.17) across all mutations. This is expected given the small, intrinsically disordered nature of the L-protein and the high flexibility required for its lytic function, which challenges standard multimeric confidence metrics.
MoUs
Sign the MoU by copying and committing it to your repository, and add your name and date.