Group Final Project
Bacteriophage Engineering: L-Protein Optimization
1. Hypothesis: Engineering Lysis Protein Stability
Our core hypothesis is that the thermodynamic stability and lytic efficiency of the MS2 L-protein can be enhanced through two strategic pathways:
- Structural Reinforcement: Introducing targeted mutations that promote independent folding or stabilize the 7-helix bundle, reducing dependence on the host chaperone DnaJ.
- Generative Optimization: Utilizing evolutionary conservation data and generative protein design to create variants with improved membrane-insertion kinetics and host compatibility, thereby minimizing host-mediated resistance.
2. Specific Aims and Validation Pipeline
Aim 1: Mutation Design via Conservation and Predictive Modeling
We will perform Clustal Omega alignments of homologous lysis proteins to identify conserved residues (specifically the “HEDYPCRRQQRSST” island). This is followed by:
- In silico Mutational Scanning: Using ESM-2 embeddings and LLR scores to nominate stabilizing mutations.
- Folding Assessment: Validation of fold accuracy via ESMFold and AlphaFold-Multimer to ensure independent folding propensity and multimeric pore symmetry.
Aim 2: Generative Design for Chaperone Independence
We propose using generative models (like ProteinMPNN or RFdiffusion) to optimize the soluble N-terminal domain. The goal is to redesign the interface to either:
- Enhance co-folding with DnaJ under controlled structural constraints.
- Enable “folding rescue” by alternative or orthogonal chaperones (e.g., DnaK or GroEL) to bypass host adaptation.
Aim 3: Evolutionary Analysis and Host Factor Integration
Using pBLAST to survey orthologs, we will reconstruct evolutionary trajectories of stability. Candidates will be screened against E. coli host factors to minimize proteotoxicity while maximizing the “aggressive” lytic potential identified through Genomic Language Models (GLMs).
3. Computational Tools and Workflow
Our design-build-test-learn (DBTL) framework utilizes the following stack:
- Sequence & Conservation: Clustal Omega and pBLAST for “evolutionary grammar” analysis.
- Mutational Analysis: ESM-2 (LLR scores) for high-speed structural feedback on the 7-helix bundle.
- Generative Design: ProteinMPNN for sequence backbone optimization.
- Structural Validation: AlphaFold 3 and AlphaFold-Multimer to ensure biophysical plausibility of the 8-chain pore assembly.
Schematic Pipeline:
[Sequence Input] → [ESM-2 Mutational Scan] → [GLM Toxicity Optimization] → [AlphaFold/AF3 Validation] → [Final Design Selection]
4. Potential Pitfalls
- Contextual Gap: A lack of specific data regarding the host bacteria’s in vivo environment may lead to unexpected results despite positive simulations.
- Functional Trade-offs: Mutations that improve structural stability might inadvertently perturb the membrane-interaction properties or the native lytic activity, leading to a loss of function.
- Misfolding Risk: Compact lysis proteins are highly sensitive; even high-confidence predicted folds (pLDDT > 80) may aggregate or fail to insert into the membrane in a real biological system.
5. Expected Outcomes
If successful, this framework will yield L-protein variants with:
- Increased Stability: Robust functionality under diverse environmental conditions.
- Reduced Host Dependency: Decreased reliance on native DnaJ interactions, making the phage less vulnerable to host-dependent failure modes.
- Optimized Lysis: Retention of a mature fold compatible with aggressive lytic activity, establishing a generalizable template for synthetic antimicrobial modules.
6. Group Brainstorming Details
- Collaborators: Sheila Ramani, Ganapathi Naayagam, Deep Dalvi and Fabrizio Flores.
- Selected Goals: * Stabilization: Increasing structural integrity.
- Toxicity Optimization: Enhancing bacterial cell wall disruption.
Part D. Group Brainstorm on Bacteriophage Engineering
- Find a group of ~3–4 students
I found a group of 4 students: Sheila Ramani, Ganapathi Naayagam, Deep Dalvi and I, Fabrizio Flores
- Read through the Phage Reading material listed under “Reading & Resources” below.
Here are the summaries of the phage reading materials:
Phage Reading
Identification MS2 lysis protein dependency on DnaJ By: @2026a-fabrizio-flores-huaman
The study explains that the MS2 phage L protein is a 75 amino acid polypeptide that kills bacteria through a unique mechanism. Unlike other proteins like E or A2, which block cell wall synthesis, the L protein does not affect peptidoglycan production. Using a smart screening system with a lacZ reporter to filter out false positives, the researchers discovered that L depends entirely on the host chaperone DnaJ to function. Interestingly, a specific mutation in DnaJ called P330Q completely blocks lysis at 30°C. Through pulldown assays, they confirmed that DnaJ physically binds to the N-terminal “head” of the protein, which is full of basic charges and is actually dispensable for the killing process, serving instead as a control unit.
The final model proposes that this N-terminal domain of L acts as a biological brake that auto-inhibits the protein. The DnaJ chaperone acts like a key that unlocks this brake, allowing the hydrophobic tail of the protein to reach its actual target inside the cell. This was proven with Lodj mutants, which are versions of the L protein lacking the head. These mutants do not need DnaJ and kill the bacteria 20 minutes faster than the wild type. This system mirrors what happens with the E protein and its chaperone SlyD, suggesting that phages evolved these charged domains as a strategy to control lysis timing and ensure the virus has enough time to replicate before destroying the host.
Mutational analysis of the MS2 lysis protein L By: @2026a-sheila-ramani
The MS2 lysis protein (L) is a 75-amino acid polypeptide that triggers bacterial cell death without disrupting net peptidoglycan synthesis. Research reveals a conserved LS (Leu-Ser) dipeptide motif at residues Leu48-Ser49, which serves as the essential core for protein-protein interactions. While the N-terminal half of the protein is dispensable for lytic activity, the C-terminal domain is critical; specifically, the S49C mutation in the LS motif causes an absolute lysis defect. This motif is highly conserved across diverse phages, indicating it is a universal structural requirement for the lytic function in amurins.
The study suggests that the L protein interacts with a host membrane target through the LS motif and surrounding essential domains. The N-terminus functions as a regulatory domain that naturally inhibits this interaction, while the host chaperone DnaJ binds to the N-terminus to displace it from its inhibitory position. Interestingly, deleting the basic N-terminal domain allows the protein to bypass the need for DnaJ entirely. This confirms that the N-terminus acts as a regulatory gatekeeper, and DnaJ is the key that unlocks the protein’s ability to engage its cellular target.
Characterization of the MS2 lysis protein properties By: @2026a-deep-dalvi
The MS2 bacteriophage lysis protein (MS2L) facilitates host cell escape by punching holes in the bacterial wall through a dual-domain mechanism. It consists of a soluble HEAD domain and a transmembrane TAIL domain that anchors into membranes, behaving similarly to soap or micelles. A key finding is that the TAIL domain drives oligomerization, causing 10 or more proteins to clump into large complexes. CryoEM data confirms these clusters gather at specific spots to trigger a sequential rupture: first the outer membrane breaks, followed by the peptidoglycan layer, and finally the inner membrane, causing the cell contents to leak out.
The researchers identified the HEAD domain as a biological brake that regulates the timing of lysis. While the full MS2L protein is difficult to insert into membranes, removing the HEAD allows for relatively easy insertion, suggesting it functions as a timer to prevent premature cell death. Additionally, the helper protein DnaJ binds to MS2L but does not influence its membrane entry or oligomerization. From an engineering perspective, removing the HEAD domain could bypass this brake to achieve a “quicker kill,” a strategic goal for optimizing lytic toxicity in synthetic biology.
Phage therapy: From biological mechanisms to future directions By: all
This paper explains how phages have evolved from a biological curiosity into a sophisticated therapeutic tool by focusing on their life cycles and resistance mechanisms. The review highlights that success in therapy depends on more than just injecting phages; it requires a deep understanding of pharmacokinetics and the patient’s immune response, as the body might neutralize the viruses before they reach the infection site. A key advancement mentioned is the use of genetic engineering to create “designer” phages that do more than just kill bacteria, such as degrading biofilms or working alongside traditional antibiotics to restore drug sensitivity. The future of the field points toward precision medicine where specific phages are selected or edited for each patient to overcome the regulatory and technical barriers that previously limited mass clinical use.
Phage Therapy: Past, Present and Future By: all
This text explores the historical evolution and the modern resurgence of phage therapy in response to the global antibiotic resistance crisis. It begins by reminding us that phages were used long before penicillin but were largely forgotten in the West due to a lack of standardized protocols and the convenience of broad-spectrum antibiotics. Currently, we are in a stage of “compassionate use” where phages are successfully applied in desperate cases of multi-drug resistant infections, which is driving new controlled clinical trials. The study concludes that the biggest challenge today is not just biological but also logistical and legal, as a global infrastructure is needed to collect and characterize phage libraries that can be quickly deployed against emerging pathogens. This marks a shift from general treatments to a completely personalized paradigm.
Generative design of novel bacteriophages with genome language models By: @2026a-ganapathi-naayagam
This research utilizes the Evo 1 and Evo 2 DNA foundation models to design functional biological systems at the whole-genome scale. Using the phiX174 lytic phage as a chassis, the AI successfully generated 16 viable phages with substantial evolutionary novelty. Some variants were highly distant from common natural sequences, proving that genomic language models (GLMs) can expand the known biological space. This is critical for phage therapy, as these AI-designed variants demonstrated a superior ability to overcome bacterial resistance in E. coli strains where natural phages failed.
The computational method employed taxonomic prompting (e.g., Riboviria) to guide the generative process toward specific viral realms. Novelty was rigorously validated using nucleotide BLAST against core databases to confirm the emergence of original sequences. This strategy offers a robust framework for creating diverse phage cocktails, a key requirement for modern antimicrobial treatments. By leveraging taxonomic labels and pretraining, the study establishes a “design-build-test” workflow for engineering complex, multi-gene systems beyond the limits of natural evolution.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
Brainstorm Session
- Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
- We will focus on increasing the structural stability of the L protein to ensure it remains functional under different environmental conditions.
- We will also attempt to increase the toxicity of the lysis protein by optimizing its target regions to enhance bacterial cell wall disruption.
- Write a 1-page proposal (bullet points or short paragraphs) describing:
- Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
- We will use ESMFold to perform in silico mutational scanning and identify target regions in the L protein.
- We propose using Genomic Language Models (GLMs) to design and optimize sequences with higher lytic potential.
- Finally, we will use AlphaFold-Multimer to validate the folding and stability of the engineered protein complexes.
- Why do you think those tools might help solve your chosen sub-problem?
- ESMFold allows for high-speed structural feedback, making it easier to test how mutations affect the 7-helix bundle.
- GLMs are essential for capturing the “evolutionary grammar” of toxicity, helping to design proteins that are more aggressive than natural variants.
- AlphaFold ensures that our computational designs are biophysically plausible and stable before any potential wet-lab implementation.
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
- Contextual Gap: There is a lack of specific data regarding the host bacteria’s environment, which might lead to unexpected results in vivo.
- Misfolding Risk: The engineered protein might still misfold or aggregate in a real biological system despite having positive simulation results in the pipeline.
- Include a schematic of your pipeline.
Here’s a short written schematic of our pipeline: [Sequence Input] → [ESM-2 Mutational Scan] → [GLM Toxicity Optimization] → [AlphaFold Validation] → [Final Design]
- This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
- Include your group’s short plan for engineering a bacteriophage
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
L-Protein Engineering - Option 1: Mutagenesis
Step 1: Information Gathering
Here are the L-protein and Dnaj sequences
Lysis Protein Sequence (UniProtKB ID: P03609)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DnaJ sequence (UniProtKB ID: P03609)
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ. Soluble N-terminal domain:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVTM domain:LIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
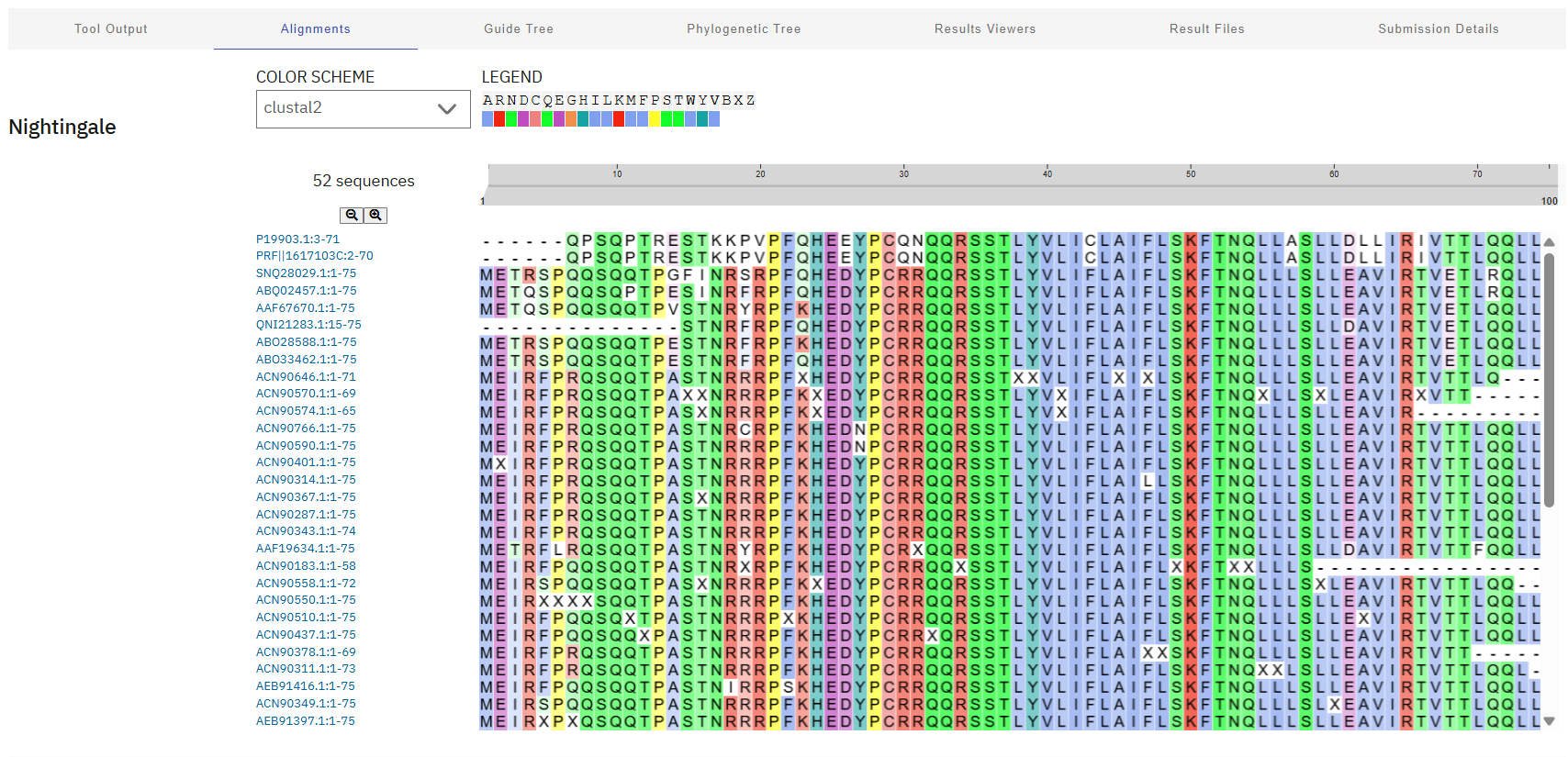
Additionally, here’s a screenshot of the BLAST results for L-protein:

Lastly, these results were aligned using Clustal Omega, revealing a highly conserved “island” (HEDYPCRRQQRSST) at residues 24-38. These sites will be avoided during mutagenesis to preserve the critical interaction with DnaJ and overall biological function of the phage.
Clustal Job ID: clustalo-I20260311-043120-0780-2033785-p2m
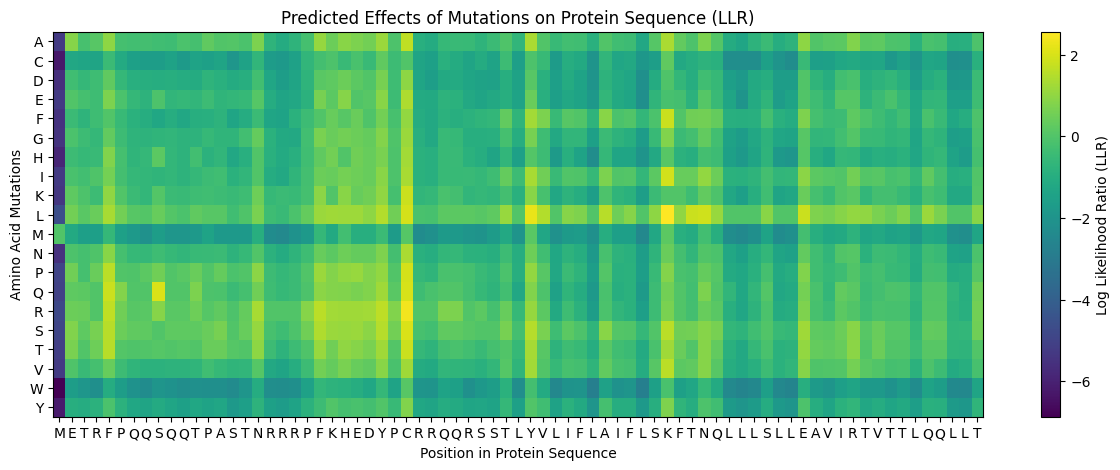
Step 2: Variant Selection Approach
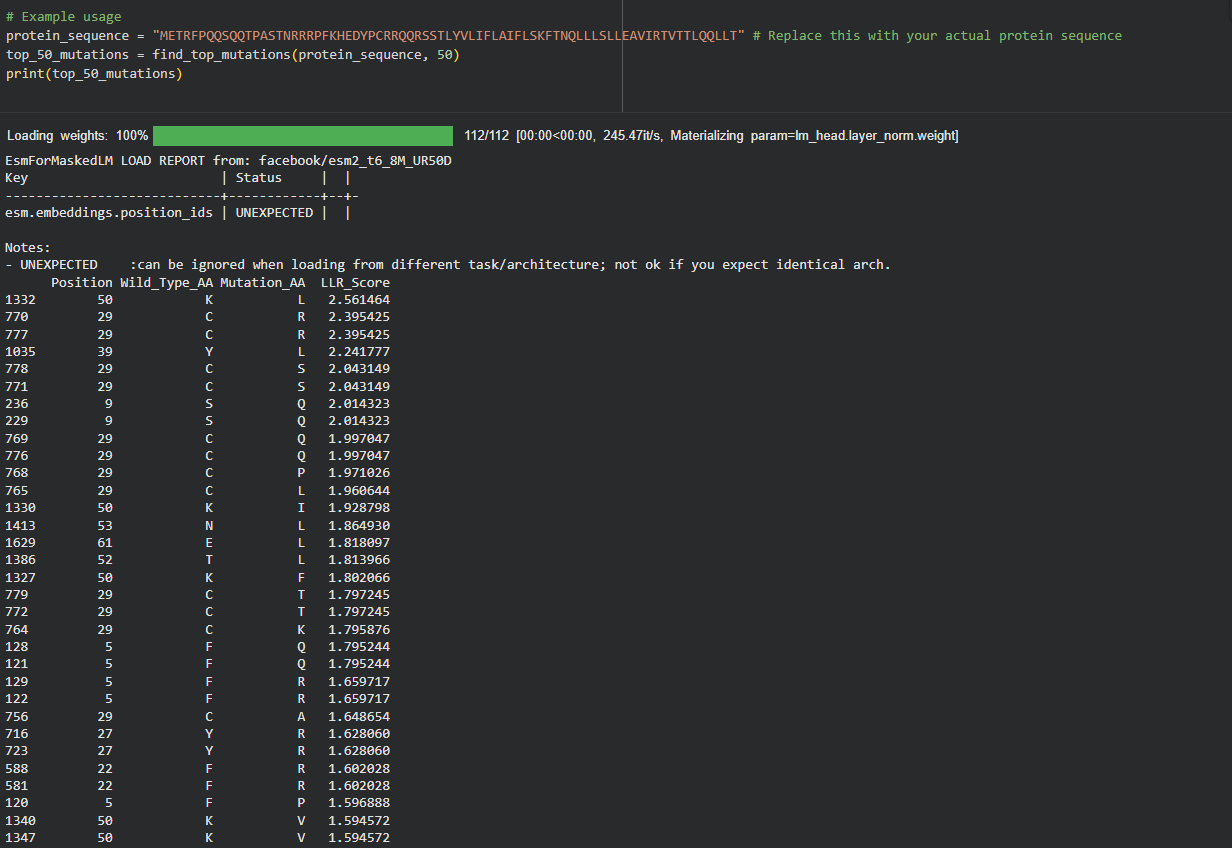
My approach is very straightforward: I combined computational LLR scores with experimental lab data using a copy of the HTGAA Colab. I filtered for mutations that showed “active lysis” (value 1) in the experimental spreadsheet and high positive LLR scores in the notebook.
Step 3: Filtering and Ranking
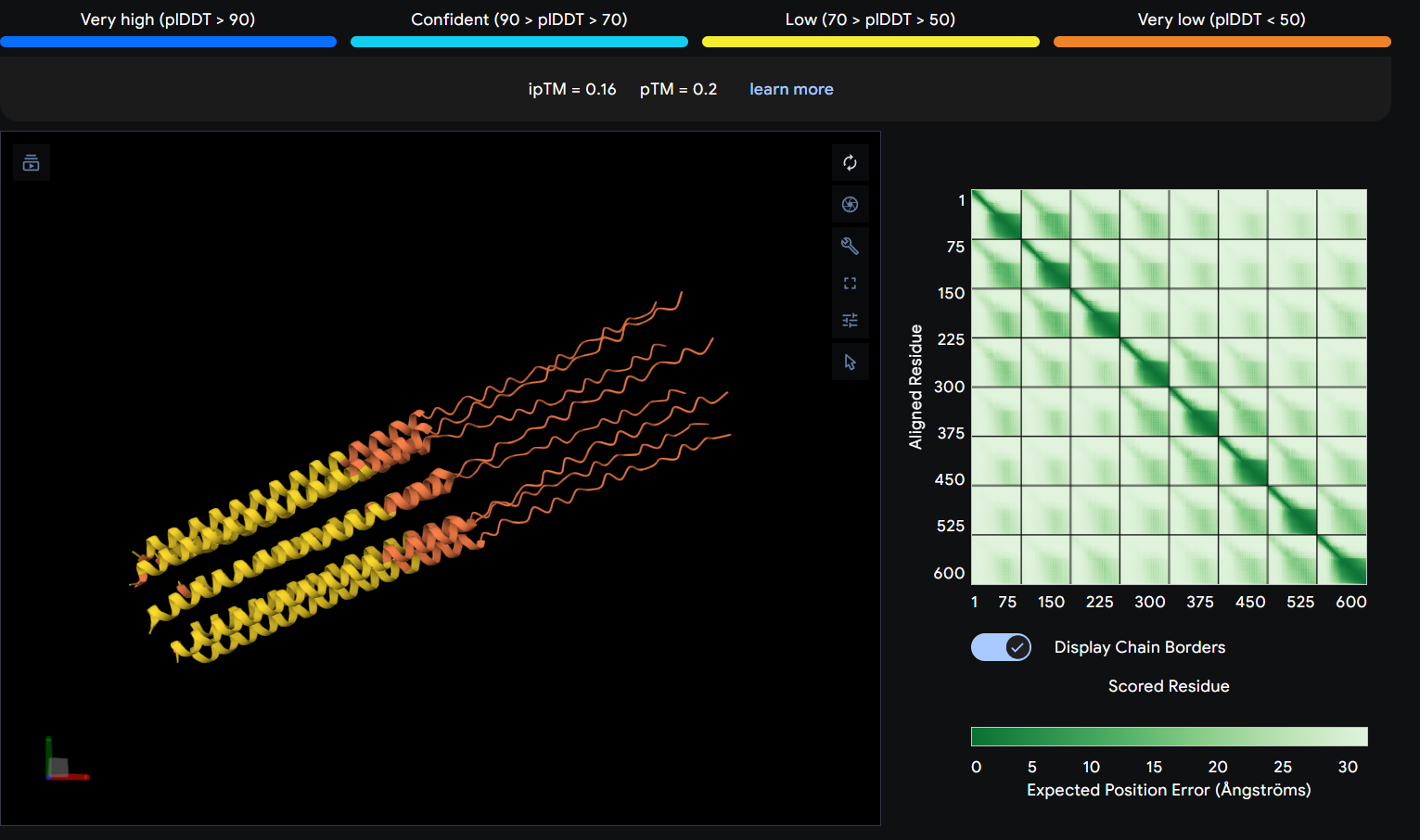
I used AlphaFold 3 to model the 8-chain assembly. This step was used to rank candidates that showed both positive computational scores and confirmed experimental activity, ensuring they don’t disrupt the pore’s symmetry.
Step 4: Final Mutated Sequences
These 5 mutations were selected because they are experimentally proven to maintain lysis (score 1) and show improved or stable computational scores.
| Region | Mutation | LLR Score (ESM-2) | Experimental Lysis | Rationale |
|---|---|---|---|---|
| Soluble | S9Q | 2.01 | Active (1) | High computational confidence; replaces Serine with Glutamine to stabilize the N-terminal loop. |
| Soluble | C29R | 2.39 | Active (1) | One of the top scores; removing this Cysteine likely prevents incorrect disulfide bonding. |
| TM Domain | Y39L | 2.24 | Active (1) | High confidence score in the TM interface; optimizes hydrophobicity for membrane entry. |
| TM Domain | A45L | 1.53 | Active (1) | Consistent with experimental data; improves the hydrophobic core of the lytic pore. |
| TM Domain | N53L | 1.86 | Active (1) | Replaces a polar Asparagine with Leucine, significantly improving helix-helix packing in the multimer. |
S9Q mutation 8-chain assembly:
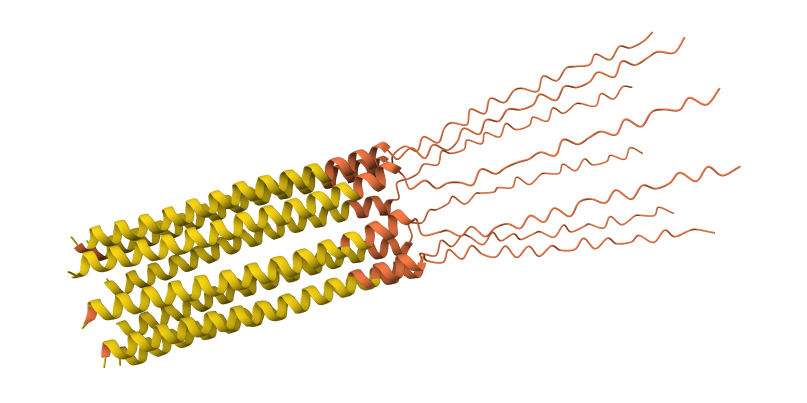
C29R mutation 8-chain assembly:
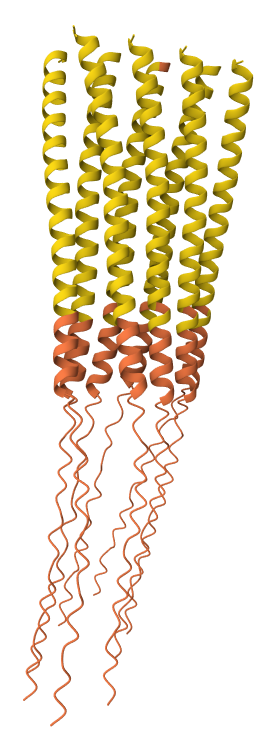
Y39L mutation 8-chain assembly:
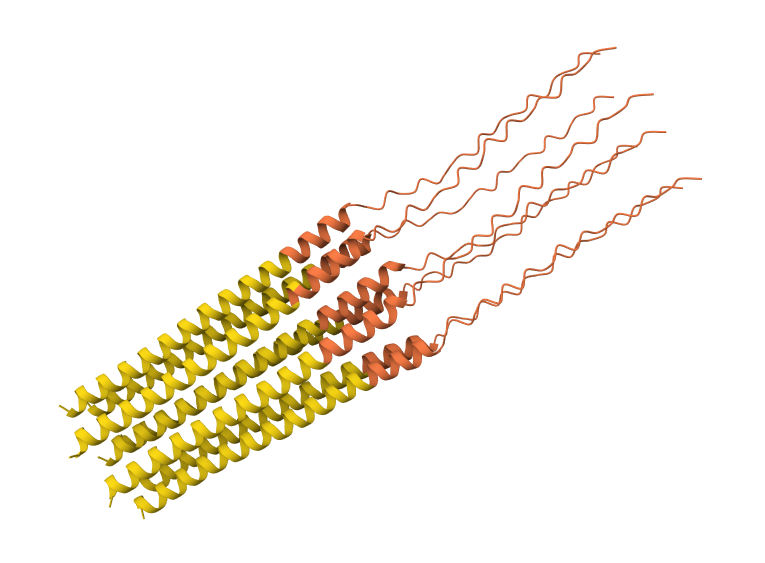
A45L mutation 8-chain assembly:

N53L mutation 8-chain assembly:
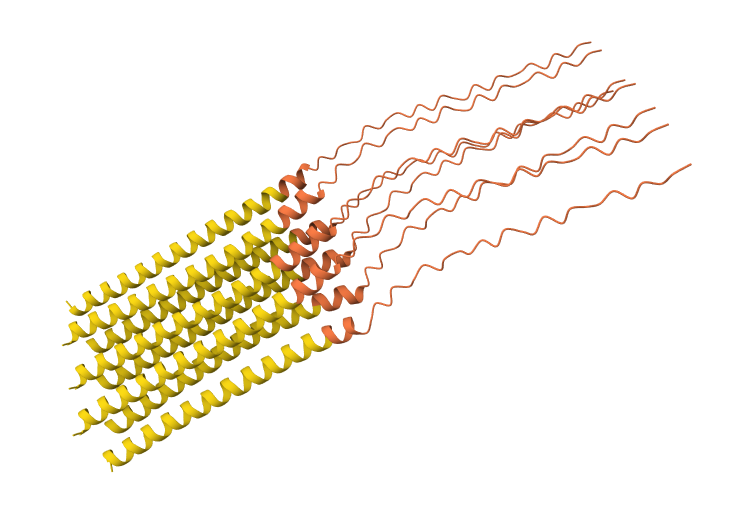
While AF3 structures were used to visualize the multimeric orientation, the ipTM scores remained low (~0.17) across all mutations. This is expected given the small, intrinsically disordered nature of the L-protein and the high flexibility required for its lytic function, which challenges standard multimeric confidence metrics.