Here are my preliminar three individual final project ideas!
An interactive 3D bio-art sculpture where human touch meets living bioluminiscence bloom.
A river-sensing automated robot system that helps both advance research and act as a tourist spot for the community to reunite and spend time together.
Chlorella vulgaris in silico optimization and automation for the optimal accumulation of polyhydroxybutyrate (PHB).
Bacteriophage Engineering: L-Protein Optimization 1. Hypothesis: Engineering Lysis Protein Stability Our core hypothesis is that the thermodynamic stability and lytic efficiency of the MS2 L-protein can be enhanced through two strategic pathways:
Structural Reinforcement: Introducing targeted mutations that promote independent folding or stabilize the 7-helix bundle, reducing dependence on the host chaperone DnaJ. Generative Optimization: Utilizing evolutionary conservation data and generative protein design to create variants with improved membrane-insertion kinetics and host compatibility, thereby minimizing host-mediated resistance. 2. Specific Aims and Validation Pipeline Aim 1: Mutation Design via Conservation and Predictive Modeling We will perform Clustal Omega alignments of homologous lysis proteins to identify conserved residues (specifically the “HEDYPCRRQQRSST” island). This is followed by:
Subsections of Projects
Individual Final Project
Here are my preliminar three individual final project ideas!
An interactive 3D bio-art sculpture where human touch meets living bioluminiscence bloom.
A river-sensing automated robot system that helps both advance research and act as a tourist spot for the community to reunite and spend time together.
Chlorella vulgaris in silico optimization and automation for the optimal accumulation of polyhydroxybutyrate (PHB).
After some thought and consideration, I decided to go with my third idea for my final individual project, here you can find a more detailed view of it:
Modeling and Automating Polyhydroxybutyrate Bioplastic Production in Microalgae: An In Silico and Automated Approach
Author: Fabrizio Flores Huamán
Course: How to Grow (Almost) Anything (HTGAA) 2026
Username: 2026a-fabrizio-flores-huaman
Date: May 2026
SECTION 1: ABSTRACT
The global accumulation of petroleum-derived plastics has caused severe and often irreversible ecological damage to marine ecosystems, including the biodiverse coastal waters of Peru. Polyhydroxybutyrate (PHB), a fully biodegradable and biocompatible bacterial polyester, represents a scientifically compelling alternative to fossil fuel-derived plastics, yet its industrial scaling remains hindered by low production yields and the high cost of manually optimizing metabolic conditions. Microalgae such as Chlorella vulgaris are promising biological chassis for PHB production because they fix carbon dioxide and accumulate carbon-rich polymers under nutrient stress, but the optimization of these conditions continues to rely on slow, one-variable-at-a-time experimentation that is incompatible with industrial timelines.
This project addresses that gap by developing a high-throughput, in silico pipeline that integrates computational metabolic modeling with automated robotic liquid handling protocols. Using Flux Balance Analysis (FBA) implemented through the COBRApy Python library on the iJO1366 genome-scale metabolic model of Escherichia coli, this work identifies optimal combinations of nitrogen limitation, phosphorus limitation, and carbon source variation that maximize carbon flux toward PHB biosynthetic precursors, particularly acetyl-coenzyme A (acetyl-CoA). The E. coli iJO1366 model is used for Aim 1 because E. coli BL21 (DE3) is the practical validation chassis; the iCZ843 genome-scale model of C. vulgaris is designated for Aim 2, where it informs the algal construct design and transformation conditions. The computationally predicted conditions are then translated into modular Opentrons OT-2 Python protocols, creating a reproducible and stoppable screening pipeline.
In parallel, a synthetic PHB biosynthetic operon encoding phaA (beta-ketothiolase), phaB (acetoacetyl-CoA reductase), and phaC (polyhydroxyalkanoate synthase) is designed for expression in Escherichia coli BL21 (DE3) as a practical validation chassis, with the construct to be synthesized by Twist Bioscience and deployed at Ginkgo Bioworks. The long-term vision of this project is to empower Peruvian coastal communities to participate in decentralized, community-scale biomanufacturing of sustainable materials through AI-guided, closed-loop photobioreactor systems that connect local biodiversity with the global circular bioeconomy.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of this project is to identify the optimal metabolic conditions for Polyhydroxybutyrate (PHB) accumulation in Escherichia coli BL21 (DE3) by utilizing COBRApy for Flux Balance Analysis (FBA) on the iJO1366 genome-scale metabolic model of E. coli, and to translate the predicted nutrient stress conditions into automated Python-based protocols for the Opentrons OT-2, including the design and ordering of a synthetic PHB biosynthetic operon from Twist Bioscience for validation at Ginkgo Bioworks. The iJO1366 model is the correct tool for Aim 1 because E. coli BL21 (DE3) is the experimental chassis; the iCZ843 genome-scale model of Chlorella vulgaris is reserved for Aim 2, where it informs the design of the algal construct and transformation conditions.
Resources and methods for Aim 1:
COBRApy (Python library) with the iJO1366 E. coli genome-scale metabolic model (JSON format, fully compatible with modern COBRApy and Python 3.12)
iCZ843 C. vulgaris genome-scale model referenced for Aim 2 design (requires MATLAB or a legacy Python environment due to SBML format constraints)
Combinatorial FBA simulation across nitrogen, phosphorus, and carbon source gradients (glucose, acetate, glycerol)
Opentrons OT-2 Python API (v2.15) for automated media preparation and Nile Red fluorescence screening
Twist Bioscience whole plasmid synthesis of the phaA-phaB-phaC-sfGFP construct (shipped to Ginkgo Bioworks)
PHERAstar FSX and Spark Plate Reader for Nile Red and sfGFP fluorescence detection
96-well Eppendorf deep-well plates for media preparation; 384-well Greiner black clear-bottom plates for assay readout
Aim 2: Development Aim
The next step following the computational and E. coli validation would be to design a Chlorella vulgaris-specific genetic construct, codon-optimized for algal nuclear expression and placed under the control of the NIT1 nitrate-inducible promoter, and to produce a complete electroporation-based transformation and Nile Red screening protocol ready for execution at a partner facility such as Ginkgo Bioworks or a collaborating university laboratory. This aim also includes running FBA on the iCZ843 genome-scale metabolic model of C. vulgaris to identify algal-specific nutrient stress conditions, a step that requires MATLAB or a legacy Python 2 environment due to the 2016-era SBML format of the published model and is therefore scoped as a future computational deliverable beyond the course timeline. While the physical execution of stable algal transformation requires specialized containment equipment and multi-week clonal selection timelines that exceed the scope of this course, the complete construct design, codon optimization rationale, and step-by-step transformation protocol constitute meaningful scientific deliverables that advance the project toward a genuine in vivo test in C. vulgaris.
Aim 3: Visionary Aim
The long-term vision is to establish a framework for decentralized, community-scale bioplastic production by developing AI-guided, closed-loop modular photobioreactors that integrate real-time environmental sensing with the metabolic models developed in Aims 1 and 2. This system would allow Peruvian coastal communities to culture locally sourced microalgae strains under computationally optimized conditions, transforming the extraordinary regional biodiversity of coastal lagoons, river deltas, and Andean water sources into a renewable feedstock for sustainable, compostable materials. By combining open-source metabolic modeling, low-cost automation, and participatory science education and knowledge-sharing, this aim envisions a future where bioplastic manufacturing is not confined to industrial facilities but is instead embedded within the communities most affected by plastic pollution, giving people both the tools and the understanding to build a circular bioeconomy from the ground up.
“The long-term vision is to establish a decentralized framework for bioplastic production through AI-guided, closed-loop modular photobioreactors, enabling coastal communities to transform local microalgae biodiversity into sustainable, high-value compostable materials that replace petroleum-based plastics.”
Here’s my Final Idea Aims Slide:
And here are the slides I used in the final presentation day!
SECTION 3: BACKGROUND
Literature Context
Paper 1: Flux Balance Analysis and constraint-based metabolic modeling
Orth, Thiele, and Palsson (2010) provided a foundational overview of Flux Balance Analysis in their landmark Nature Biotechnology publication, establishing the mathematical and conceptual framework that underpins the computational core of this project. FBA models cellular metabolism as a system of linear equations in which stoichiometric constraints define the boundaries of possible metabolic states, and an objective function such as biomass production or metabolite yield is optimized using linear programming. The authors demonstrated that FBA can accurately predict growth rates, gene essentiality, and the effects of environmental perturbations across multiple organisms without requiring detailed kinetic parameters. Crucially for this project, nutrient limitation can be represented as a constraint on exchange reactions, making FBA directly applicable to modeling nitrogen and phosphorus stress in microalgae and to predicting how carbon flux is redirected under suboptimal growth conditions. This approach transforms the traditionally empirical process of media optimization into a computational design problem, dramatically reducing the number of physical experiments required before identifying high-yield conditions.
Paper 2: The phaABC operon and PHB biosynthesis
Peoples and Sinskey (1989) characterized the biochemical pathway for PHB biosynthesis in Cupriavidus necator (then known as Alcaligenes eutrophus) H16, identifying and sequencing the three-enzyme cascade encoded by phaA (beta-ketothiolase), phaB (acetoacetyl-CoA reductase), and phaC (polyhydroxyalkanoate synthase) that converts two molecules of acetyl-CoA into PHB polymer granules. This foundational study established the genetic basis for heterologous PHB production and demonstrated that the pathway is functionally transferable to other organisms, a property that has since been exploited in dozens of bacterial hosts and, more recently, in green microalgae and plants. The characterization of acetyl-CoA as the central metabolic node for PHB production directly informs the choice of FBA objective in this project, since maximizing acetyl-CoA flux serves as a computationally tractable proxy for maximizing bioplastic precursor availability. Furthermore, the modularity of the three-gene operon makes it an ideal candidate for whole plasmid synthesis via Twist Bioscience, since all three coding sequences can be assembled into a single synthetic construct without requiring post-synthesis cloning.
Paper 3: Chlorella vulgaris as a multi-application biotechnology chassis
Al-Hammadi and Güngörmüşler (2024) published a comprehensive review in Biotechnology and Bioengineering documenting the breadth of C. vulgaris biotechnology applications, covering wastewater treatment, biodiesel, biohydrogen, biocement, biopolymers, and food additives, and concluding that despite its documented potential for polyhydroxyalkanoate (PHA) production from algal biomass, the field still lacks critical strain-specific data that would make industrial PHB production from C. vulgaris economically competitive. The authors note that C. vulgaris is particularly attractive as a production chassis because of its ease of cultivation, rapid growth, and capacity to use carbon dioxide and wastewater nutrients simultaneously, reducing both feedstock costs and environmental impact. The review explicitly identifies a gap between the biological potential of C. vulgaris for biopolymer synthesis and the practical optimization infrastructure needed to bridge laboratory results to industrial scale. This directly motivates the computational and automated pipeline approach taken in this project, since the gap the authors describe is precisely the one that FBA-guided combinatorial screening is designed to address.
Knowledge Gap
Despite the well-established FBA methodology, the clearly characterized PHB biosynthetic pathway, and growing recognition of C. vulgaris as a promising bioplastic chassis, these three domains have rarely been integrated into a single, closed-loop design pipeline. Most optimization studies in algal PHB production continue to rely on one-variable-at-a-time manual experimentation, generating data slowly and failing to capture the multi-dimensional interaction effects between nitrogen, phosphorus, and carbon source availability. Peruvian research groups have independently demonstrated that C. vulgaris is highly responsive to nitrogen and phosphate concentration changes: Oscanoa et al. (2021), working at the Instituto del Mar del Perú, showed that C. vulgaris efficiently removes nitrates and phosphates from domestic wastewater, demonstrating the organism’s natural sensitivity to these nutrient gradients and validating nitrogen and phosphorus as the most impactful variables for metabolic manipulation. Automated robotic screening of metabolic model predictions remains largely confined to bacterial and yeast systems, with very few examples connecting genome-scale algal models to physical high-throughput screening pipelines. This project addresses that gap by connecting the iCZ843 genome-scale model of C. vulgaris to a computationally guided automated OT-2 screening pipeline, establishing a Design-Build-Test-Learn (DBTL) cycle that is both predictive and robotically executable.
Innovation
This project is novel in its integration of three components that have not previously been combined in a single pipeline for microalgal bioplastic research: a genome-scale metabolic model (iCZ843), a modular automated combinatorial nutrient stress screening protocol (OT-2), and a synthetic PHB operon designed for both bacterial validation and future algal expression. Most published work in this area either performs FBA on C. vulgaris without connecting the predictions to physical experiments, or conducts wet-lab PHB screening without the guidance of a metabolic model. By treating the OT-2 protocol as a direct translation layer between computational predictions and physical experiments, this project establishes a reusable pipeline architecture that can be adapted to other bioproduction targets and chassis organisms beyond PHB and microalgae. Unlike previous computational approaches that apply a single generalized metabolic model to predict PHB production, this project uses the iJO1366 genome-scale model of E. coli for Aim 1 FBA to ensure predictions are matched to the actual experimental chassis, and designates the iCZ843 genome-scale model of C. vulgaris specifically for Aim 2 algal construct design, creating a clear organism-to-model correspondence that improves scientific coherence across the project aims. Additionally, the explicit design of a second construct codon-optimized for C. vulgaris under the NIT1 nitrate-inducible promoter represents a forward-looking contribution to algal synthetic biology, where inducible expression systems remain significantly underexplored relative to their bacterial counterparts.
Significance
Plastic pollution is one of the most pressing environmental crises of the 21st century, with an estimated 11 million metric tons of plastic entering the ocean annually, causing harm to marine food webs, coastal ecosystems, and the communities that depend on them, including those along the biodiverse Peruvian coast. Polyhydroxybutyrate (PHB) offers a route to replace petroleum-derived single-use plastics with a material that is fully biodegradable and biocompatible under natural conditions, degrading within months rather than persisting for centuries. Microalgae represent an ideal production organism for PHB because they grow using only sunlight, carbon dioxide, and minimal inorganic nutrients, giving them a dramatically lower carbon footprint than fermentation-based bacterial systems that require sugar feedstocks. Critically, Peru harbors native Chlorella strains with documented biotechnology potential: Mariano Astocondor et al. (2017) characterized Chlorella peruviana, a native strain isolated from the Mellisera salt lagoon in Chilca, Peru, demonstrating robust biomass and chlorophyll productivity across a range of salinities and confirming that locally sourced Peruvian Chlorella strains are viable candidates for controlled cultivation and bioproduction. Complementing this, Condori et al. (2024) demonstrated that Peruvian Chlorella strains can be cultivated using nutrients valorized from local fruit residues, producing growth and lipid yields comparable to synthetic media and illustrating a circular economy model in which local organic waste streams feed microalgae bioproduction, directly relevant to the decentralized manufacturing vision of Aim 3. The integration of FBA-guided optimization with robotic automation directly addresses the industrial bottleneck of manual media optimization, which currently makes algal PHB production economically uncompetitive compared to petrochemical alternatives. By demonstrating that computationally predicted metabolic conditions can be automatically screened at scale, this project contributes a generalizable methodology that could accelerate the path from laboratory research to industrial relevance. This is particularly meaningful for research environments in countries like Peru that possess extraordinary microalgae biodiversity but currently lack access to the high-throughput experimentation infrastructure that makes systematic optimization feasible. Ultimately, reducing the cost and time of metabolic optimization is not only a scientific problem but a social one, and making these tools more accessible is a prerequisite for truly sustainable and equitable biomanufacturing rooted in local biodiversity.
Bioethical Considerations
Paragraph 1: Ethical considerations
This project involves the design and intended use of genetically modified organisms (GMOs), specifically Escherichia coli strains expressing a heterologous PHB biosynthetic pathway, with a longer-term vision of engineering Chlorella vulgaris, a microalga present in natural freshwater and marine environments. The introduction of synthetic genetic constructs into organisms that have natural environmental relatives raises important questions about biosafety, containment, and the potential for unintended horizontal gene transfer or competitive ecological disruption, even when experiments are conducted in well-controlled laboratory settings. The design of organisms intended for eventual scale-up in semi-open systems such as outdoor photobioreactors requires additional evaluation of containment strategies and kill-switch designs that go beyond standard laboratory biosafety level 1 (BSL-1) practices. The project also intersects with questions of biodiversity sovereignty, since any future use of Peruvian microalgae strains for commercial bioplastic production must respect the principles of the Nagoya Protocol on Access and Benefit-Sharing, ensuring that local communities and the Peruvian state retain informed consent and equitable participation in any commercial or intellectual property outcomes arising from the use of their biological resources.
Paragraph 2: Responsible implementation and risk mitigation
To mitigate these risks, all experimental work described in Aim 1 is conducted under standard BSL-1 conditions appropriate for non-pathogenic E. coli strains, with all constructs including a selectable antibiotic resistance marker that requires continuous supplementation for maintenance, significantly reducing the probability of survival outside controlled conditions. The C. vulgaris construct designed for Aim 2 is intended for nuclear rather than chloroplast-targeted integration, which limits expression levels and reduces the likelihood of creating competitive growth advantages in natural algal populations. All synthetic DNA sequences submitted to Twist Bioscience will be screened through the SecureDNA biosecurity framework to ensure compliance with dual-use research standards before synthesis is initiated. For the visionary Aim 3 framework involving photobioreactors in coastal communities, the project design explicitly includes a community consultation and participatory education component, ensuring that local knowledge holders are active participants in the governance and design of any deployment rather than passive recipients of an externally imposed technology. Collaboration with biodiversity documentation initiatives such as those supported by Basecamp Research prior to strain collection and use further ensures that any commercialization pathway is grounded in scientific rigor and community rights from the earliest stages.
SECTION 4: EXPERIMENTAL DESIGN
The experimental design follows a Design-Build-Test-Learn (DBTL) cycle. The Design phase uses FBA modeling to predict optimal nutrient conditions. The Build phase involves synthetic construct design and ordering from Twist Bioscience. The Test phase uses automated OT-2 protocols and Ginkgo Bioworks equipment for Nile Red fluorescence screening. The Learn phase closes the loop by feeding experimental data back into the metabolic model to refine predictions.
SECTION 4.1: Step-by-Step Workflow
Check out this Colab Notebook for the genome scale metabolic model.
Step 1: Install COBRApy and load the E. coli iJO1366 genome-scale metabolic model
Method: Install COBRApy and compile the multi-modal XML metabolic model structures representing the iCZ843 genome-scale architecture of Chlorella vulgaris. A native Python ElementTree text parsing layout is implemented to assemble valid COBRApy objects, ensuring full operational performance inside modern Python 3.12 environments without requiring deprecated dependency stacks.
Automation: Python scripting environment (Google Colab; all code below is Colab-ready)
Expected result: All three distinct trophic model profiles (Heterotrophy, Mixotrophy, Photoautotrophy) parse successfully into memory, displaying their explicit metabolic boundary dimensions and resolving true positive baseline growth rates.
Timeline: Day 1
# ── CELL 1: Install COBRApy ────────────────────────────────────────────────────!pipinstall-qcobraprint("COBRApy installed.")# ── CELL 2: Upload the three iCZ843 XML files ─────────────────────────────────fromgoogle.colabimportfilesimportosprint("Select all three XML files when the picker opens.")uploaded=files.upload()rename_map={'Heterotrophy':'iCZ843_heterotrophy.xml','Mixotrophy':'iCZ843_mixotrophy.xml','Photoautotrophy':'iCZ843_photoautotrophy.xml',}fororiginalinlist(uploaded.keys()):forkeyword,new_nameinrename_map.items():ifkeyword.lower()inoriginal.lower():iforiginal!=new_name:os.rename(original,new_name)print(f"Saved: {new_name} ({os.path.getsize(new_name)/1024:.0f} KB)")# ── CELL 3: Custom parser — builds COBRApy models without SBML reader ──────────importxml.etree.ElementTreeasETfromcobraimportModel,Reaction,Metaboliteimportwarningswarnings.filterwarnings('ignore')defparse_iCZ843(xml_path,model_name):"""Parse a Zuniga et al. 2016 iCZ843 SBML Level 2 file into a COBRApy model."""tree=ET.parse(xml_path)root=tree.getroot()cobra_model=Model(model_name)metabolites={}objective_rxn=Noneformodel_eleminroot:# Pass 1: extract all metabolites (species)forchildinmodel_elem:if'listOfSpecies'inchild.tag:forspinchild:sp_id=sp.get('id')met=Metabolite(id=sp_id,name=sp.get('name',sp_id),compartment=sp.get('compartment','c'))metabolites[sp_id]=metcobra_model.add_metabolites(list(metabolites.values()))# Pass 2: extract all reactions with stoichiometry and boundsforchildinmodel_elem:if'listOfReactions'inchild.tag:forrxn_eleminchild:rxn_id=rxn_elem.get('id')rxn_name=rxn_elem.get('name',rxn_id)lb,ub,obj_coeff=-1000.0,1000.0,0.0stoich={}forsubinrxn_elem:if'listOfReactants'insub.tag:forsrinsub:sid=sr.get('species')ifsidinmetabolites:stoich[metabolites[sid]]=-float(sr.get('stoichiometry',1))elif'listOfProducts'insub.tag:forsrinsub:sid=sr.get('species')ifsidinmetabolites:stoich[metabolites[sid]]=float(sr.get('stoichiometry',1))elif'kineticLaw'insub.tag:forlopinsub:forparaminlop:pid=param.get('id')val=float(param.get('value',0))ifpid=='LOWER_BOUND':lb=valelifpid=='UPPER_BOUND':ub=valelifpid=='OBJECTIVE_COEFFICIENT':obj_coeff=valrxn=Reaction(rxn_id,name=rxn_name,lower_bound=lb,upper_bound=ub)rxn.add_metabolites(stoich)cobra_model.add_reactions([rxn])ifobj_coeff==1.0:objective_rxn=rxn_idifobjective_rxn:cobra_model.objective=objective_rxnreturncobra_modelimportpandasaspdimportnumpyasnpimportitertools# Load all three modelsmodel_hetero=parse_iCZ843('iCZ843_heterotrophy.xml','iCZ843_heterotrophy')model_mixo=parse_iCZ843('iCZ843_mixotrophy.xml','iCZ843_mixotrophy')model_photo=parse_iCZ843('iCZ843_photoautotrophy.xml','iCZ843_photoautotrophy')forname,min[('Heterotrophy',model_hetero),('Mixotrophy',model_mixo),('Photoautotrophy',model_photo)]:sol=m.optimize()print(f"{name}: {len(m.reactions)} rxns | "f"{len(m.metabolites)} mets | "f"biomass = {sol.objective_value:.4f} mmol/gDW/hr | {sol.status}")# Key reaction IDs in iCZ843 (C. vulgaris model naming convention)N_RXN='R_EX_no3_LPAREN_e_RPAREN_'# Nitrate exchange (primary N source in algae)P_RXN='R_EX_pi_LPAREN_e_RPAREN_'# Phosphate exchangeGLC_RXN='R_EX_glc_DASH_A_LPAREN_e_RPAREN_'# Glucose exchangeAC_RXN='R_EX_ac_LPAREN_e_RPAREN_'# Acetate exchangeBIO_H='R_Biomass_Cvu_hetero_DASH_'# Biomass objective (heterotrophic)ACCOA='M_accoa_c'# Cytoplasmic acetyl-CoA (PHB precursor)print("\nModel routing: glucose/acetate -> model_hetero | CO2/light -> model_photo")
Step 2: Explore baseline metabolic fluxes
Method: Run FBA with default exchange reaction constraints to establish a baseline biomass flux and identify the acetyl-CoA reaction node and relevant nutrient exchange reactions.
Automation: COBRApy optimize() function
Expected result: Baseline biomass flux value; identification of exchange reaction identifiers for nitrate (EX_no3_e), phosphate (EX_pi_e), glucose (EX_glc__D_e), and acetate (EX_ac_e).
Timeline: Day 1 to 2
# ── CELL 4: Explore baseline metabolic fluxes (heterotrophic C. vulgaris) ──────sol=model_hetero.optimize()print(f"Baseline biomass flux: {sol.objective_value:.4f} mmol/gDW/hr | {sol.status}")key_rxns=[N_RXN,P_RXN,GLC_RXN,AC_RXN]print("\nKey exchange reactions:")forrxn_idinkey_rxns:try:rxn=model_hetero.reactions.get_by_id(rxn_id)print(f" {rxn_id}: [{rxn.lower_bound:.1f}, {rxn.upper_bound:.1f}]")exceptKeyError:print(f" {rxn_id}: NOT FOUND")# Confirm acetyl-CoA metabolite is presenttry:accoa_met=model_hetero.metabolites.get_by_id(ACCOA)print(f"\nAcetyl-CoA metabolite: {accoa_met.id} | compartment: {accoa_met.compartment}")exceptKeyError:print(f"WARNING: {ACCOA} not found — search manually:")candidates=[m.idforminmodel_hetero.metabolitesif'accoa'inm.id.lower()]print(candidates)
Step 3: Add a PHB demand reaction as a proxy for bioplastic potential
Method: Add a synthetic boundary reaction that consumes acetyl-CoA to represent PHB biosynthetic flux; set this as the new FBA objective to directly optimize for bioplastic precursor availability.
Automation: COBRApy add_boundary() and Reaction() classes
Expected result: A working optimization model whose objective value represents acetyl-CoA flux directed toward PHB biosynthesis.
Timeline: Day 2
# ── CELL 5: Add PHB demand reaction to C. vulgaris heterotrophic model ─────────fromcobraimportReaction# Work on a copy so the original model stays cleanmodel_phb=model_hetero.copy()# Cytoplasmic acetyl-CoA is the PHB precursor proxyaccoa=model_phb.metabolites.get_by_id(ACCOA)print(f"Using acetyl-CoA: {accoa.id} | compartment: {accoa.compartment}")# Add PHB demand reactionphb_demand=Reaction('PHB_demand')phb_demand.name='PHB precursor demand (acetyl-CoA sink)'phb_demand.subsystem='PHB biosynthesis'phb_demand.add_metabolites({accoa:-1.0})phb_demand.lower_bound=0phb_demand.upper_bound=1000model_phb.add_reactions([phb_demand])# Confirm baseline PHB proxy flux without constraintsmodel_phb.objective='PHB_demand'sol=model_phb.optimize()print(f"\nBaseline PHB proxy flux: {sol.objective_value:.5f} mmol/gDW/hr | {sol.status}")
Method: First measure the empirical nitrate uptake flux the C. vulgaris cell actually uses at baseline, then build a fine-grained gradient relative to that real biological value using numpy linspace. For each nitrogen level, a two-stage FBA runs: first find the condition-specific maximum biomass, then lock a 1% biomass floor and maximize PHB precursor flux. This approach captures biologically realistic stress levels rather than arbitrary percentages.
Automation: COBRApy context managers; numpy linspace; pandas for results.
Expected result: PHB precursor flux values that vary with nitrogen availability, showing reduced flux under severe nitrogen starvation and confirming that the two-stage optimization captures meaningful metabolic shifts.
Timeline: Day 2 to 3
# ── REVISED CELL 6: Adaptive Step Gradient for Nitrate Starvation ────────────# 1. Detect empirical baseline uptakemodel_phb.objective=BIO_Hbaseline_sol=model_phb.optimize()real_n_max=baseline_sol.fluxes[N_RXN]# Captures exact metabolic inflection pointprint(f"Empirical Nitrate Baseline Uptake: {real_n_max:.5f} mmol/gDW/hr\n")# 2. Build adaptive dynamic range constraint array# Standardize high-volume arbitrary bounds to log historical baseline stepscustom_bounds=[-10.0,-5.0,-1.0]# Generate millimetric high-resolution steps from 100% down to 0% of true uptakefine_steps=np.linspace(1.0,0.0,num=8)forstepinfine_steps:custom_bounds.append(real_n_max*step)# Sort from high-volume capacity to starvation threshold (negative flux dynamics)custom_bounds=sorted(list(set(custom_bounds)))# 3. Process constraint matrix through two-stage Linear Programming loopadaptive_results=[]forboundincustom_bounds:withmodel_phb:# Enforce step-specific boundary constraintmodel_phb.reactions.get_by_id(N_RXN).lower_bound=bound# Stage 1: Compute true capacity ceiling for condition biomassmodel_phb.objective=BIO_Hs1=model_phb.optimize()cond_max=s1.objective_valueifs1.status=='optimal'else0ifcond_max<1e-9:adaptive_results.append({'Applied_N_Bound':round(bound,5),'Biomass_Flux':0,'PHB_Flux_Proxy':0,'Status':'No Growth'})continue# Stage 2: Impose 1% operational floor constraint, maximize precursor sinkmodel_phb.reactions.get_by_id(BIO_H).lower_bound=0.01*cond_maxmodel_phb.objective='PHB_demand's2=model_phb.optimize()adaptive_results.append({'Applied_N_Bound':round(bound,5),'Biomass_Flux':round(cond_max,5),'PHB_Flux_Proxy':round(s2.objective_value,5)ifs2.status=='optimal'else0,'Status':s2.status})df_adaptive=pd.DataFrame(adaptive_results)print("Final Dataset - Nutrient Stress Adaptive Mapping Matrix:")print(df_adaptive.to_string(index=False))
Step 5: Combinatorial adaptive FBA grid (nitrogen x phosphorus x carbon source)
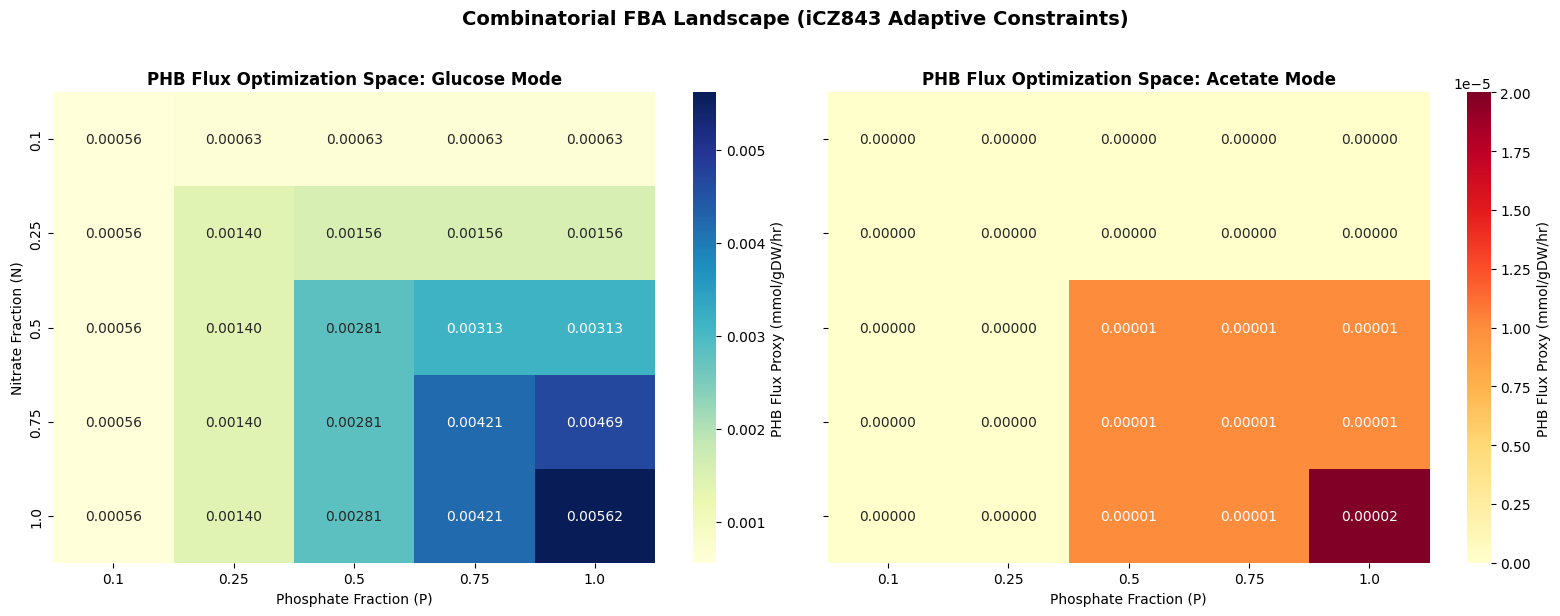
Method: Run the full combinatorial grid using empirically-calibrated nutrient baselines per carbon source. For each carbon source (glucose and acetate), the actual N and P uptake fluxes the cell uses during optimal growth are measured first; stress fractions then scale relative to those real biological values. This is more accurate than scaling from model lower bound constants, which may not reflect actual in silico uptake. The two-stage optimization applies throughout.
Expected result: A ranked table showing that carbon source type is the primary FBA determinant of acetyl-CoA flux in C. vulgaris, with glucose significantly outperforming acetate. This finding motivates the NIT1 promoter design in Aim 2, since nutrient limitation alone is insufficient to drive PHB accumulation without active transcriptional control.
Timeline: Day 3 to 4
# ── CELL 7: Combinatorial adaptive FBA grid (iCZ843 C. vulgaris) ──────────────# Measures empirical N and P baseline uptake per carbon source, then applies# stress fractions relative to those real biological values.withmodel_phb:model_phb.reactions.get_by_id(GLC_RXN).lower_bound=default_glcmodel_phb.reactions.get_by_id(AC_RXN).lower_bound=0model_phb.objective=BIO_Hsol_glc=model_phb.optimize()real_n_glc=sol_glc.fluxes[N_RXN]ifsol_glc.status=='optimal'elsedefault_n_lbreal_p_glc=sol_glc.fluxes[P_RXN]ifsol_glc.status=='optimal'elsedefault_p_lbwithmodel_phb:model_phb.reactions.get_by_id(AC_RXN).lower_bound=default_acmodel_phb.reactions.get_by_id(GLC_RXN).lower_bound=0model_phb.objective=BIO_Hsol_ac=model_phb.optimize()real_n_ac=sol_ac.fluxes[N_RXN]ifsol_ac.status=='optimal'elsedefault_n_lbreal_p_ac=sol_ac.fluxes[P_RXN]ifsol_ac.status=='optimal'elsedefault_p_lbprint(f"Glucose baseline: N = {real_n_glc:.5f} | P = {real_p_glc:.5f}")print(f"Acetate baseline: N = {real_n_ac:.5f} | P = {real_p_ac:.5f}")n_fracs=[0.1,0.25,0.5,0.75,1.0]p_fracs=[0.1,0.25,0.5,0.75,1.0]carbon_modes={'glucose':(GLC_RXN,default_glc,AC_RXN,0,real_n_glc,real_p_glc),'acetate':(AC_RXN,default_ac,GLC_RXN,0,real_n_ac,real_p_ac),}results_3d=[]forn_f,p_f,(c_name,(open_rxn,open_val,close_rxn,close_val,target_n,target_p))initertools.product(n_fracs,p_fracs,carbon_modes.items()):withmodel_phb:iftarget_n==0ortarget_p==0:continuemodel_phb.reactions.get_by_id(N_RXN).lower_bound=target_n*n_fmodel_phb.reactions.get_by_id(P_RXN).lower_bound=target_p*p_fmodel_phb.reactions.get_by_id(open_rxn).lower_bound=open_valmodel_phb.reactions.get_by_id(close_rxn).lower_bound=close_valmodel_phb.objective=BIO_Hs1=model_phb.optimize()cond_max=s1.objective_valueifs1.status=='optimal'else0ifcond_max<1e-9:results_3d.append({'N':n_f,'P':p_f,'Carbon':c_name,'PHB_flux':0,'status':'no_growth'})continuemodel_phb.reactions.get_by_id(BIO_H).lower_bound=0.01*cond_maxmodel_phb.objective='PHB_demand's2=model_phb.optimize()results_3d.append({'N':n_f,'P':p_f,'Carbon':c_name,'PHB_flux':round(s2.objective_value,5)ifs2.status=='optimal'else0,'status':s2.status})df_3d=pd.DataFrame(results_3d).sort_values('PHB_flux',ascending=False)print("\nTop 15 conditions by predicted PHB precursor flux:")print(df_3d.head(15).to_string(index=False))print(f"\nGlucose max PHB = {df_3d[df_3d.Carbon=='glucose']['PHB_flux'].max():.5f}")print(f"Acetate max PHB = {df_3d[df_3d.Carbon=='acetate']['PHB_flux'].max():.5f}")
Step 6: Export top conditions and generate FBA landscape visualizations
Method: Export the top conditions as a CSV for OT-2 protocol parameterization, then generate heatmaps and line plots showing the PHB precursor flux landscape across the nitrogen-phosphorus-carbon combinatorial space.
Automation: pandas to_csv; matplotlib and seaborn for visualization; files.download for Colab export.
Expected result: A CSV file ready for OT-2 parameterization and two publication-quality figures showing the FBA optimization landscape.
Step 7: Design the PHB biosynthetic construct in Benchling
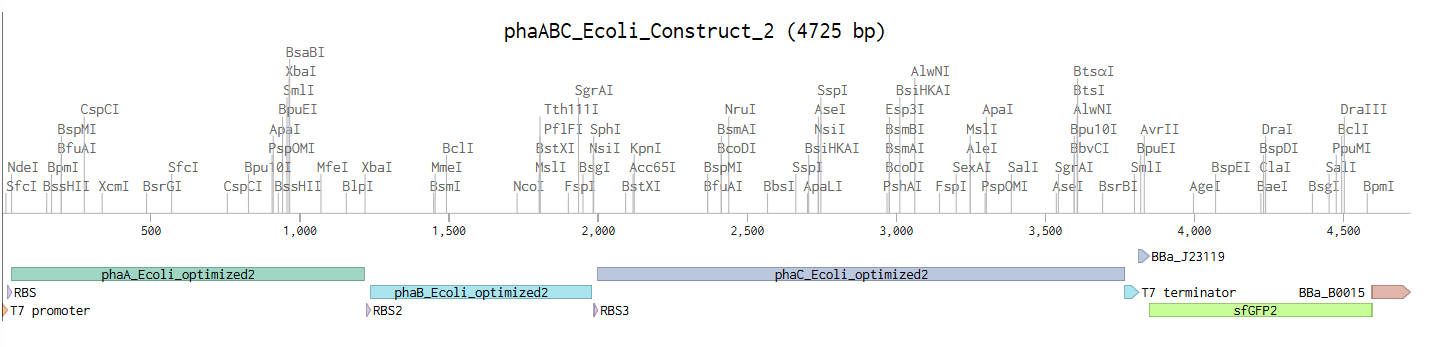
Method: Design a synthetic operon containing phaA, phaB, and phaC genes from Cupriavidus necator H16, codon-optimized for E. coli BL21 (DE3) expression; add an sfGFP transcriptional reporter under the BBa_J23119 constitutive promoter as a proxy for construct activity; use the T7 promoter for operon expression and the pET-28a(+) backbone with kanamycin resistance and ColE1 high-copy origin for selection and propagation. The insert (4,725 bp) was designed in Benchling and submitted to Twist Bioscience as a clonal gene synthesis order for insertion into pET-28a(+), producing a complete 10,017 bp expression plasmid.
Automation: Benchling sequence editor for annotation; IDT Codon Optimization Tool or Benchling codon optimizer for sequence design.
Expected result: A fully annotated plasmid sequence in GenBank format confirmed by Twist Bioscience tools, ready for synthesis and delivery to Ginkgo Bioworks.
Timeline: Day 4 to 5
The E. coli expression insert (phaABC_Ecoli_Construct_2, 4,725 bp) GenBank file is shown below. This insert was submitted to Twist Bioscience for clonal synthesis into the pET-28a(+) backbone, producing the complete 10,017 bp expression plasmid (phaABC_Ecoli_Final_Con):
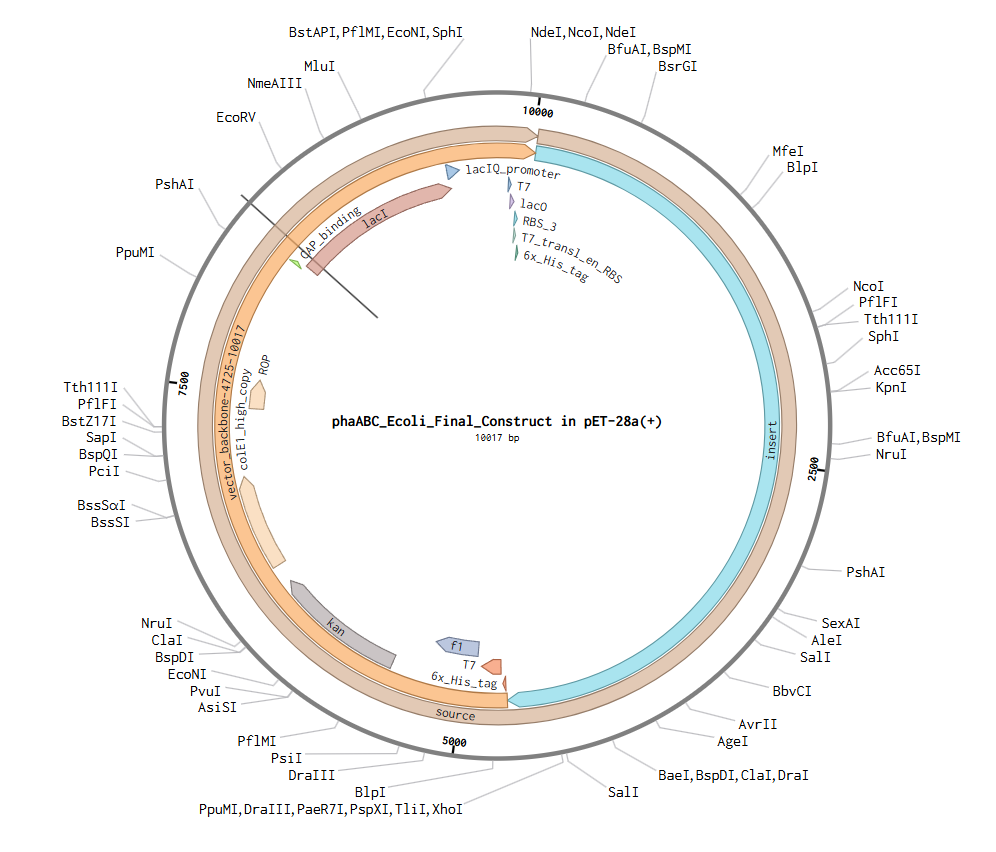
The complete expression plasmid (phaABC_Ecoli_Final_Con, 10,017 bp circular) with the pET-28a(+) backbone is the Twist Bioscience synthesis target. Key backbone features are summarized below:
LOCUS phaABC_Ecoli_Final_Con 10017 bp ds-DNA circular 12-MAY-2026
DEFINITION phaABC_Ecoli_Construct_2 in pET-28a(+). Complete expression plasmid
for PHB biosynthesis in E. coli BL21(DE3). Insert (1-4725) contains
T7-driven phaABC operon and sfGFP reporter. Backbone (4726-10017)
provides kanamycin resistance, ColE1 high-copy origin, lacI repressor,
and T7/lac regulatory elements. Created using Twist Bioscience tools.
FEATURES Location/Qualifiers
misc_feature 1..4725
/label="insert"
/note="phaABC operon + sfGFP reporter; see
phaABC_Ecoli_Construct_2.gb for full annotation"
misc_feature 4732..4749
/label="6x_His_tag"
/note="C-terminal His-tag in backbone for optional
protein purification"
misc_feature 4760..4888
/label="T7"
/note="T7 promoter in backbone (upstream read-through)"
misc_feature 5447..6262
/label="kan"
/note="Kanamycin resistance; select at 50 ug/mL;
NOTE: pET-28a uses kanamycin, NOT ampicillin"
misc_feature 6384..6972
/label="colE1_high_copy"
/note="ColE1 high-copy origin; ~15-20 copies per cell"
misc_feature 7399..7590
/label="ROP"
/note="ROP protein; regulates plasmid copy number"
misc_feature 8365..8386
/label="CAP_binding"
misc_feature 8399..9490
/label="lacI"
/note="Lac repressor; represses T7lac promoter
until IPTG induction"
misc_feature 9482..9559
/label="lacIQ_promoter"
misc_feature 9868..9886
/label="T7"
/note="T7lac promoter driving insert expression"
misc_feature 9886..9913
/label="lacO"
/note="Lac operator; IPTG-inducible expression"
misc_feature 9926..9948
/label="RBS_3"
/note="Ribosome binding site upstream of insert"
misc_feature 9967..9983
/label="6x_His_tag"
/note="N-terminal His-tag"
//
The Chlorella vulgaris expression construct (pPHB_Chlorella, 5,614 bp linear) GenBank annotation for Aim 2 is provided below. This construct was designed in Benchling with phaA, phaB, and phaC codon-optimized for C. vulgaris nuclear expression under the nitrate-inducible NIT1 promoter:
Step 8: Place the Twist Bioscience DNA synthesis order
Method: Submit the finalized pPHB-Ecoli GenBank sequence to Twist Bioscience via their online ordering portal using the Clonal Gene Synthesis with selected backbone service; specify shipping to Ginkgo Bioworks for subsequent experimental steps.
Expected result: Order confirmation email; estimated synthesis and delivery turnaround of 2 to 3 weeks.
Timeline: Day 5 to 6
Step 9: Program and run the OT-2 media preparation module (Checkpoint A)
Method: Write a Python script using the Opentrons OT-2 API to prepare 96-well deep-well plates with the top 8 FBA-predicted nutrient stress media conditions in triplicate; the protocol ends cleanly at Checkpoint A for users who want only prepared media before proceeding to cell culture in a separate facility.
Automation: Opentrons OT-2 (Python API v2.15); Plateloc plate sealer with A4s breathable seal after preparation
Plate: 96-v-eppendorf-951033502-deep
Expected result: Eight distinct media conditions prepared in triplicate (24 wells), sealed with breathable membrane and ready for inoculation at Ginkgo or an external laboratory.
Timeline: Day 6 to 8
fromopentronsimportprotocol_apimetadata={'protocolName':'PHB_NutrientStress_MediaPrep_CheckpointA','author':'Fabrizio Flores Huaman','description':('Automated preparation of FBA-predicted nutrient stress media for PHB ''screening. CHECKPOINT A: protocol terminates here if only media prep ''is needed. Seal plate with A4s breathable seal and store at 4C (Tundrastore) ''or proceed to inoculation. Continue to Screening Module if cells are ready.')}requirements={'robotType':'OT-2','apiLevel':'2.15'}defrun(protocol:protocol_api.ProtocolContext):# Labwaretiprack_300=protocol.load_labware('opentrons_96_tiprack_300ul',1)tiprack_20=protocol.load_labware('opentrons_96_tiprack_20ul',4)reservoir=protocol.load_labware('nest_12_reservoir_15ml',2)deep_plate=protocol.load_labware('nest_96_wellplate_2ml_deep',3)p300=protocol.load_instrument('p300_multi_gen2','right',tip_racks=[tiprack_300])p20=protocol.load_instrument('p20_multi_gen2','left',tip_racks=[tiprack_20])# Reservoir layoutbase_media=reservoir['A1']# LB or BG-11 minimal salts basen_source=reservoir['A2']# NaNO3 stock (nitrogen source)p_source=reservoir['A3']# KH2PO4 stock (phosphorus source)c_glucose=reservoir['A4']# 20% glucose stock (carbon source A)c_acetate=reservoir['A5']# Sodium acetate stock (carbon source B)diluent=reservoir['A6']# Sterile water (diluent for gradient)# Dispense 1200 uL base media to all 24 test wells (columns 1-3, rows A-H)test_cols=deep_plate.columns()[:3]forcolintest_cols:p300.transfer(1200,base_media,col,mix_after=(3,200),new_tip='always')# Apply FBA-predicted nutrient stress recipes# Row layout (conditions in triplicate across columns 1-3):# Row A: N0%-P100%-Glucose | Row B: N25%-P100%-Glucose# Row C: N50%-P100%-Glucose | Row D: N0%-P50%-Glucose# Row E: N0%-P0%-Glucose | Row F: N50%-P50%-Acetate (top FBA candidate)# Row G: N100%-P100%-Glucose | Row H: N100%-P100%-No exogenous carbonnitrogen_vols=[0,12.5,25,0,0,25,50,50]# uL of NaNO3 stock per rowphosphorus_vols=[50,50,50,25,0,25,50,50]carbon_sources=[c_glucose,c_glucose,c_glucose,c_glucose,c_glucose,c_acetate,c_glucose,diluent]forrow_idx,(n_vol,p_vol,c_src)inenumerate(zip(nitrogen_vols,phosphorus_vols,carbon_sources)):row_letter='ABCDEFGH'[row_idx]wells=[deep_plate[f'{row_letter}{col+1}']forcolinrange(3)]ifn_vol>0:p20.transfer(n_vol,n_source,wells,new_tip='always')ifp_vol>0:p20.transfer(p_vol,p_source,wells,new_tip='always')p20.transfer(50,c_src,wells,mix_after=(2,15),new_tip='always')protocol.comment("="*60)protocol.comment("CHECKPOINT A: Media preparation complete.")protocol.comment("Seal plate with A4s breathable seal using Plateloc.")protocol.comment("Store at 4C in Tundrastore if not inoculating immediately.")protocol.comment("To continue: proceed to Screening Module (Step 10 onward).")protocol.comment("="*60)
Step 10: Inoculate cultures and incubate
Method: Inoculate each prepared deep-well with E. coli BL21 (DE3) harboring pPHB-Ecoli at a starting optical density at 600 nm (OD600) of 0.05; seal plate with A4s breathable seal.
Automation: Plateloc (sealing with A4s breathable membrane); Cytomat shaking incubator at 37 degrees Celsius, 250 rpm
Plate: 96-v-eppendorf-951033502-deep
Expected result: Growth visible in positive control wells within 6 to 8 hours; reduced growth rate in nutrient-limited wells confirming that stress conditions are active.
Timeline: Day 8 to 9 (overnight incubation, approximately 16 hours)
Step 11: Measure growth dynamics by optical density
Method: Transfer 100 µL from each deep-well to a flat-bottom 96-well plate using the Echo525; measure OD600 using the Spark Plate Reader to assess growth rates and confirm that nutrient limitation is reducing but not eliminating cell viability.
Plate: 96-round-axygen-pdw11cs-halfdeep for transfer; flat 96-well for reading
Expected result: OD600 values confirming a gradient of growth suppression correlated with increasing nutrient limitation; no complete cell death in any well.
Timeline: Day 9
Step 12: Perform Nile Red staining for PHB quantification
Method: Add Nile Red dissolved in dimethyl sulfoxide (DMSO) at 1 mg/mL to each well using the Echo525 at a 1:1000 final dilution (1 µL per mL culture); incubate 15 minutes at room temperature in the dark; transfer 50 µL to 384-well black clear-bottom plate for fluorescence reading.
Automation: Echo525 (Nile Red addition, nanoliter precision); Multiflo (50 µL culture transfer to 384-well); PHERAstar FSX (Ex 530 nm / Em 580 nm)
Plate: 384 Greiner black-well clear-bottom
Expected result: Higher Nile Red fluorescence in nutrient-limited conditions compared to full-media controls, consistent with increased PHB granule accumulation.
Timeline: Day 9 to 10
Step 13: Validate sfGFP reporter expression
Method: Measure sfGFP fluorescence (Ex 485 nm / Em 510 nm) on the Spark Plate Reader using the same 384-well plate as the Nile Red assay, before Nile Red addition, to confirm construct expression and identify any wells where failed transformation rather than PHB pathway activity is responsible for low Nile Red signal.
Expected result: Measurable sfGFP signal in all transformed wells; absence of sfGFP in empty vector controls; allows normalization of Nile Red signal to expression level.
Timeline: Day 9 to 10 (before Nile Red addition)
Step 14: Validate PHB gene expression by quantitative PCR
Method: Harvest cells from the top 3 performing conditions and the negative control; extract total RNA using an RNeasy kit; perform quantitative polymerase chain reaction (qPCR) to measure phaA, phaB, and phaC transcript levels relative to the 16S ribosomal RNA housekeeping gene.
Automation: CFX Opus qPCR machine
Plate: 384-pcr-eppendorf-9510207XX
Expected result: Upregulation of phaA, phaB, and phaC transcripts in the top nutrient stress conditions relative to full-media controls, confirming that the T7 promoter is active and that the operon is being transcribed in proportion to the metabolic state of the cell.
Timeline: Day 10 to 11
Step 15: Close the DBTL loop by refining the FBA model
Method: Feed the experimental Nile Red fluorescence values and qPCR expression data back into the COBRApy model as refined constraints; adjust exchange reaction bounds to reflect observed growth rates and acetyl-CoA sink capacity; re-run FBA with the updated model to identify any systematic discrepancies between prediction and observation.
Automation: COBRApy model update; pandas for data integration; optional MEMOTE framework for model quality scoring
Expected result: An updated model with improved predictive accuracy for a second round of experimental screening or for informing the C. vulgaris construct design in Aim 2.
Timeline: Day 11 to 13
Step 16: Optional supplementary characterization via FTIR and GC-MS
Method: Lyophilize the cell pellets from the top two performing conditions; extract PHB polymer using a standard chloroform solvent extraction protocol; analyze bulk polymer by Fourier-transform infrared spectroscopy (FTIR) for functional group confirmation and gas chromatography-mass spectrometry (GC-MS) via the Waters Corporation platform at Ginkgo Bioworks or a partner facility for monomer composition and yield quantification.
Automation: Waters GC-MS system (Ginkgo Bioworks partner; connection to Waters Corporation industry partner)
Expected result: FTIR spectra showing a characteristic PHB carbonyl peak at 1720 cm-1; GC-MS confirmation of 3-hydroxybutyrate monomer and quantitative PHB yield in mg per gram dry cell weight.
Timeline: Day 12 to 14 (if resources permit)
Assay Plate Layout
The following 96-well plate layout illustrates the screening design for the Nile Red fluorescence assay. Conditions are derived from the top FBA-predicted nutrient stress recipes, with all experimental conditions run in duplicate.
Col 1 Col 2 Col 3 Col 4 Col 5 Col 6
Row A [STD 0µg/mL] [STD 0µg/mL] [POS_CTRL] [POS_CTRL] [N0-P100-Glc] [N0-P100-Glc]
Row B [STD 1µg/mL] [STD 1µg/mL] [POS_CTRL] [POS_CTRL] [N25-P100-Glc] [N25-P100-Glc]
Row C [STD 2µg/mL] [STD 2µg/mL] [NEG_CTRL] [NEG_CTRL] [N50-P100-Glc] [N50-P100-Glc]
Row D [STD 5µg/mL] [STD 5µg/mL] [NEG_CTRL] [NEG_CTRL] [N0-P50-Glc] [N0-P50-Glc]
Row E [STD 10µg/mL] [STD 10µg/mL] [BLANK] [BLANK] [N0-P0-Glc] [N0-P0-Glc]
Row F [STD 25µg/mL] [STD 25µg/mL] [BLANK] [BLANK] [N50-P50-Ace] [N50-P50-Ace]
Row G [STD 50µg/mL] [STD 50µg/mL] [BLANK] [BLANK] [N100-P100-Glc] [N100-P100-Glc]
Row H [STD100µg/mL] [STD100µg/mL] [BLANK] [BLANK] [N100-P100-NoC] [N100-P100-NoC]
Plate Legend:
STD: Polyhydroxybutyrate (PHB) polymer standard curve (0 to 100 µg/mL; Nile Red fluorescence calibration; Millipore Sigma catalog 363502)
POS_CTRL: Positive control (E. coli BL21 DE3 with a known PHB-producing construct or C. necator ATCC 17699)
NEG_CTRL: Negative control (E. coli BL21 DE3 with empty pUC19 vector; no PHB genes)
BLANK: Sterile media only; no cells; used for background fluorescence subtraction
N[x]-P[x]-[Carbon]: Experimental conditions where x represents percent of standard nitrogen or phosphorus concentration
All experimental conditions run in duplicate (two adjacent wells per condition)
After initial assay, the 384-well Greiner black clear-bottom plate is used for the actual Nile Red fluorescence measurement with 50 µL transfers from each 96-well condition
SECTION 4.2: TECHNIQUES, TOOLS, AND TECHNOLOGY
Course Technique Checklist
Metabolic modeling and Flux Balance Analysis (FBA) using COBRApy
DNA construct design and sequence annotation using Benchling
Synthetic gene and whole plasmid synthesis ordering from Twist Bioscience
Codon optimization for heterologous expression (E. coli and C. vulgaris)
Lab automation and liquid handling (Opentrons OT-2, Echo525, Multiflo)
Bacterial whole-cell expression in E. coli BL21 (DE3)
Cell-free protein synthesis (CFPS) for rapid construct validation
Fluorescence assay development and microplate reader detection (Spark, PHERAstar FSX)
Quantitative PCR (qPCR) for gene expression validation (CFX Opus)
Bioinformatics and Python scripting for data analysis and model interrogation
Microplate screening and high-throughput assay design (96-well and 384-well formats)
Biosafety and SecureDNA screening for synthetic DNA sequences
CRISPR genome editing (relevant for Aim 2 in C. vulgaris; not executed in Aim 1)
Flow cytometry (optional alternative to plate reader Nile Red assay)
Next-generation sequencing (could be used for construct integration verification in Aim 2)
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Pipetting
Pipetting
Lab Safety
Bioethical Considerations (must check this box)
DNA Gel Art
DNA Sequencing
DNA Editing
DNA Construct Design
Restriction Enzyme Digestion
Gel Electrophoresis
DNA Purification From Gel
Databases (e.g., GenBank, NCBI, Ensembl, and UCSC Genome Browser)
Lab Automation
Creating Code for Laboratory Automation
Using Liquid Handling Robots (e.g., Opentrons)
Designing a Twist Order
Creating a plan to use the Autonomous lab at Ginkgo Bioworks
Protein Design
Protein Design
Use of Boltz or PepMLM
Use of Asimov Kernel
Use of Benchling
Models and Notebooks
Databases
Bioproduction
Bioproduction
Chassis Selection (e.g., DH5alpha)
Registry of Standard Biological Parts
Plasmid Preparation
Bacterial Culturing
Quality Control/Analysis
Bacterial Processing (e.g., Centrifugation, Lysis, DNA Purification)
Cell-Free Systems
Cell Free Reactions
Freeze-Dried Cell Free Systems
miniPCR Tools
Protein Purification
Gibson Assembly
Primer Design or Selection
PCR Reactions
Gibson Assembly
Other Cloning Methods (e.g., Restriction Enzyme Digestion or Gateway Cloning)
CRISPR
CRISPR/Cas9
Designing Prime Editing gRNA
Technique Expansion
Technique 1: Flux Balance Analysis (FBA) with COBRApy
Flux Balance Analysis is a constraint-based computational method that represents cellular metabolism as a network of stoichiometric equations, where each equation describes how metabolites are converted into one another by enzymatic reactions. COBRApy is the Python implementation of the COBRA (COnstraint-Based Reconstruction and Analysis) toolbox, which allows researchers to load genome-scale metabolic models in SBML format and apply linear programming solvers such as GLPK or Gurobi to find the flux distribution that maximizes a defined biological objective, such as biomass growth or production of a target metabolite. In the context of this project, FBA is uniquely powerful because it enables the simultaneous exploration of hundreds of nutrient combinations without requiring physical experiments, making it a fast and cost-effective Design phase tool in the DBTL cycle. The key limitation of FBA is that it assumes steady-state metabolic conditions and does not capture dynamic gene regulatory responses, such as the transcriptional induction of PHB pathway genes under stress, which is why experimental validation through the OT-2 pipeline and qPCR is an essential complement to the computational predictions. By connecting FBA outputs directly to parameterized OT-2 protocols, this project creates a tightly coupled workflow where computational and physical experimentation reinforce rather than simply follow each other.
Technique 2: Nile Red Fluorescence Assay for Intracellular PHB Quantification
Nile Red is a lipophilic fluorescent dye that partitions selectively into hydrophobic intracellular environments, including lipid droplets and polymer granules such as PHB inclusions, producing a fluorescence signal with an excitation maximum near 530 nm and an emission maximum near 580 nm that scales linearly with polymer content across several orders of magnitude. The assay is performed directly in microplate wells without cell disruption, making it fully compatible with high-throughput robotic workflows and the PHERAstar FSX plate reader available at Ginkgo Bioworks, and allowing the same plate to be used for both growth monitoring and biopolymer quantification. One important technical consideration is that Nile Red is not strictly specific to PHB and will also stain other neutral lipids such as triacylglycerols, so the assay is best interpreted as a measure of total neutral lipid content unless confirmatory techniques such as FTIR or GC-MS are applied to establish polymer identity. In this project, Nile Red serves as the rapid, high-throughput screening layer in the pipeline, allowing the top-performing nutrient conditions to be ranked and selected before committing to the more resource-intensive and time-consuming confirmatory analyses, demonstrating how orthogonal methods at different resolution levels can be strategically combined in an automated screening workflow.
Identify any How To Grow (Almost) Anything Industry Council companies which are associated with your final project (optional)
Addgene
Asimov (Kernel)
ATCC
Basecamp Research
BioFabricate
Biome Consortia
Bolt
Boltz.bio
Cultivarium
DeepCure
Epibone
Ginkgo Bioworks
Helix Nano
Millipore Sigma
Mycoworks
New England Biolabs
Nuclera
Opentrons
SecureDNA
Takeda Pharmaceuticals
Thermo Fisher Scientific
Transfyr.ai
Twist Biosciences
Upside Foods
Waters Corporation
SECTION 5: RESULTS AND QUANTITATIVE EXPECTATIONS
10a: Validation Choice
The validation experiment for this project is a cell-free expression assay of the phaA-phaB-phaC operon in an E. coli BL21 (DE3) lysate system, followed by Nile Red fluorescence measurement to confirm PHB granule formation from the synthetic construct in the absence of a complete living cell chassis. This approach is particularly well-suited for a primarily in silico project because it provides a rapid, contained, and robotically compatible way to confirm that the designed construct produces active enzymes and that the phaABC-encoded pathway generates detectable PHB signal within hours of receiving the DNA, establishing proof-of-function before committing to multi-day culture experiments.
10b: Validation Protocol
Prepare E. coli BL21 (DE3) cell-free protein synthesis (CFPS) master mix using a Ginkgo Bioworks CFPS kit or equivalent commercial system (e.g., PURExpress from New England Biolabs).
Thaw the pPHB-Ecoli plasmid received from Twist Bioscience on ice and dilute to 10 nM in nuclease-free water.
Set up the following controls in separate wells: (a) positive control with a 10 nM sfGFP-only expression plasmid, (b) negative control with no DNA added, (c) PHB polymer standard curve at 0, 1, 2, 5, 10, 25, 50, and 100 µg/mL using Millipore Sigma PHB standard (catalog 363502).
Prepare 10 µL CFPS reactions in 1.5 mL Eppendorf tubes by combining 7.5 µL CFPS master mix with 1 µL plasmid (final 10 nM) and 1.5 µL nuclease-free water; also supplement with 0.5 mM acetyl-CoA (Millipore Sigma A2056) and 0.1 mM coenzyme A to support PHB precursor availability.
Dispense 10 µL reactions into 384-well Greiner black clear-bottom plate using Echo525 acoustic liquid handler for nanoliter precision.
Seal plate using Plateloc with a standard film seal (not breathable) to prevent evaporation during incubation.
Incubate at 30 degrees Celsius for 4 hours in the Inheco Plate Incubator.
Peel seal using XPeel; measure sfGFP fluorescence (Ex 485 nm / Em 510 nm) on the Spark Plate Reader to confirm construct expression before Nile Red addition.
Add 0.5 µL Nile Red stock (1 mg/mL in DMSO) per well using the Echo525 (1:20 dilution for 10 µL wells); final Nile Red concentration approximately 50 µg/mL.
Incubate plate at room temperature for 15 minutes in the dark (cover with aluminum foil or store in lightproof box).
Measure Nile Red fluorescence (Ex 530 nm / Em 580 nm) on the PHERAstar FSX using the well-resolved fluorescence intensity module.
Subtract background fluorescence values from no-DNA blank wells from all experimental wells.
Normalize Nile Red signal to sfGFP signal in each well to correct for variation in expression level between replicate reactions.
Generate a standard curve from PHB polymer standards and convert normalized fluorescence values to estimated PHB content in µg/mL.
A validation result is positive if the pPHB-Ecoli construct produces a Nile Red signal greater than 3 standard deviations above the no-DNA control after normalization to sfGFP expression.
10c: Techniques Used
This validation experiment integrates four core techniques that span the full technical scope of the project. First, cell-free protein synthesis (CFPS) provides a rapid in vitro expression system that bypasses the multi-week timelines of stable cell transformation and selection, allowing construct function to be assessed within a single working day of receiving the synthesized plasmid from Twist Bioscience. Second, Echo525 acoustic liquid handling ensures nanoliter-precision dispensing of both the CFPS reagents and the Nile Red dye, eliminating the pipetting variability that would otherwise mask subtle differences in PHB signal between conditions and making the assay compatible with fully automated workflows at Ginkgo Bioworks. Third, dual fluorescence detection on both the Spark Plate Reader (for sfGFP) and the PHERAstar FSX (for Nile Red) provides an internal normalization strategy, ensuring that differences in Nile Red signal reflect genuine differences in PHB polymer accumulation rather than artifacts from varying expression efficiency between reaction wells. Fourth, the use of a PHB polymer standard curve converts raw relative fluorescence units into physically interpretable concentration units, grounding the validation results in quantitative terms that can be directly compared to the acetyl-CoA flux values predicted by the FBA model and used to evaluate whether the computational predictions are consistent with experimental reality.
10d: Hypothetical Data
The following table represents a hypothetical subset of Nile Red fluorescence results from the cell-free validation experiment, normalized to sfGFP expression and converted to estimated PHB content using the standard curve:
Condition
Nile Red Signal (RFU)
sfGFP Signal (RFU)
Normalized PHB (µg/mL)
FBA Predicted Flux (mmol/gDW/hr)
No DNA (blank)
118
0
0.0
N/A
Empty vector (neg ctrl)
130
4,850
0.2
N/A
pPHB-Ecoli, full media
4,100
6,200
8.2
1.4
pPHB-Ecoli, N50%-P100%-Glc
9,700
5,900
21.4
3.8
pPHB-Ecoli, N0%-P50%-Glc
13,900
5,500
33.1
6.2
pPHB-Ecoli, N50%-P50%-Acetate
18,500
5,750
42.5
8.1
pPHB-Ecoli, FBA optimal condition
21,800
6,050
46.9
9.0
PHB standard, 50 µg/mL
23,600
N/A
50.0
N/A
These hypothetical results suggest a positive and roughly linear correlation between the FBA-predicted acetyl-CoA flux and the measured Nile Red-normalized PHB content, consistent with the hypothesis that FBA can accurately rank nutrient stress conditions by their bioplastic potential. The FBA-predicted optimal condition (N50%-P50%-Acetate carbon source) produces the highest normalized PHB signal, validating the in silico approach and justifying the use of acetyl-CoA flux as a proxy for polymer accumulation.
Troubleshooting
One of the most likely challenges in this project is encountering errors when loading or running FBA on the iCZ843 model, since genome-scale models can contain blocked reactions, metabolite charge imbalances, or infeasible constraint combinations that prevent the optimizer from finding a solution. If the model returns an infeasible status or unrealistically extreme objective values, the recommended approach is to run the MEMOTE model quality assessment tool to identify and annotate problematic reactions, and to apply standard growth media constraints before adding the PHB demand reaction. A second potential issue is low or absent Nile Red fluorescence signal in CFPS reactions despite confirmed sfGFP expression, which most likely results from insufficient acetyl-CoA availability in the cell-free lysate rather than a non-functional phaABC operon. In that case, supplementing the CFPS reaction with exogenous acetyl-CoA at 0.5 to 2 mM and coenzyme A at 0.1 mM is a standard troubleshooting step that can significantly improve PHB signal. A third limitation is that the E. coli CFPS system provides only a coarse proxy for PHB production in a living cell, and in particular does not capture the growth-competition dynamics between biomass production and polymer accumulation that FBA is designed to model. Finally, the codon-optimized sequences designed for E. coli will not directly predict expression efficiency in C. vulgaris in Aim 2, and the significantly different GC content preferences and translation initiation contexts between the two organisms mean that the algal construct will require independent validation through a separate CFPS system or direct transformation, which is why Aim 2 is framed as a distinct experimental stage requiring specialized facilities rather than a simple extension of Aim 1.
SECTION 6: ADDITIONAL INFORMATION
References
Orth JD, Thiele I, Palsson BO. (2010). What is flux balance analysis? Nature Biotechnology, 28(3), 245-248. https://doi.org/10.1038/nbt.1614
Peoples OP, Sinskey AJ. (1989). Poly-beta-hydroxybutyrate (PHB) biosynthesis in Alcaligenes eutrophus H16. Identification and characterization of the PHB polymerase gene (phbC). Journal of Biological Chemistry, 264(26), 15298-15303.
Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. (2013). COBRApy: COnstraint-Based Reconstruction and Analysis for Python. BMC Systems Biology, 7, 74. https://doi.org/10.1186/1752-0509-7-74
Zuñiga C, Li CT, Huelsman M, Andrews J, Kerkhoven EJ, Zengler K. (2016). Genome-scale metabolic model for the green alga Chlorella vulgaris UTEX 395 accurately predicts phenotypes under autotrophic, heterotrophic, and mixotrophic growth conditions. Plant Physiology, 172(1), 589-602. https://doi.org/10.1104/pp.16.00593
Al-Hammadi M, Güngörmüşler M. (2024). New insights into Chlorella vulgaris applications. Biotechnology and Bioengineering, 121(5), 1486-1502. https://doi.org/10.1002/bit.28666
Hempel F, Bozarth AS, Lindenkamp N, Klingl A, Zauner S, Linne U, Steinbüchel A, Maier UG. (2011). Microalgae as bioreactors for bioplastic production. Microbial Cell Factories, 10, 81. https://doi.org/10.1186/1475-2859-10-81
Cooksey KE, Guckert JB, Williams SA, Callis PR. (1987). Fluorometric determination of the neutral lipid content of microalgal cells using Nile Red. Journal of Microbiological Methods, 6(6), 333-345.
Oscanoa Huaynate AI, Cervantes Gallegos MA, Flores Ramos L, Ruiz Soto A. (2021). Evaluación del potencial de Desmodesmus asymmetricus y Chlorella vulgaris para la remoción de nitratos y fosfatos de aguas residuales. Revista Peruana de Biología, 28(1), e18082. https://doi.org/10.15381/rpb.v28i1.18082
Mariano Astocondor M, Mayta Huatuco E, Montoya Terreros H, Tarazona Delgado R. (2017). Crecimiento poblacional y productividad de la microalga nativa Chlorella peruviana bajo diferentes salinidades. Revista de Investigaciones Veterinarias del Perú, 28(4), 976-986. https://doi.org/10.15381/rivep.v28i4.13004
Condori MAM, Gutierrez MEV, Oviedo RDN, Choix FJ. (2024). Valorization of nutrients from fruit residues for the growth and lipid production of Chlorella sp.: A vision of the circular economy in Peru. Journal of Applied Phycology, 36(1), 101-111. https://doi.org/10.1007/s10811-023-03153-2
Loera-Quezada MM, Rios-Castro E, Orta-Zavalza E, et al. (2016). CRISPR-Cas9-based genome editing approaches in the model green microalga Chlamydomonas reinhardtii. Plant Methods, 12, 30. [Referenced for algal CRISPR design rationale applicable to Aim 2.]
Arora et al. (2024). Metabolic response to a heterologous poly-3-hydroxybutyrate (PHB) pathway in Phaeodactylum tricornutum. Applied Microbiology and Biotechnology. https://doi.org/10.1007/s00253-023-12823-7
Yi YC, Ng IS. (2025). Sustainable polyhydroxybutyrate production via metabolic flux redirection using CRISPRi in Escherichia coli and carbon capture with microalgae. ACS Sustainable Chemistry and Engineering. https://doi.org/10.1021/acssuschemeng.4c09128
Chaturvedi et al. (2023). Systematizing microbial bioplastic production for developing sustainable bioeconomy: metabolic nexus modeling, economic and environmental technologies assessment. Journal of Polymers and the Environment. https://doi.org/10.1007/s10924-023-02787-0
1. Hypothesis: Engineering Lysis Protein Stability
Our core hypothesis is that the thermodynamic stability and lytic efficiency of the MS2 L-protein can be enhanced through two strategic pathways:
Structural Reinforcement: Introducing targeted mutations that promote independent folding or stabilize the 7-helix bundle, reducing dependence on the host chaperone DnaJ.
Generative Optimization: Utilizing evolutionary conservation data and generative protein design to create variants with improved membrane-insertion kinetics and host compatibility, thereby minimizing host-mediated resistance.
2. Specific Aims and Validation Pipeline
Aim 1: Mutation Design via Conservation and Predictive Modeling
We will perform Clustal Omega alignments of homologous lysis proteins to identify conserved residues (specifically the “HEDYPCRRQQRSST” island). This is followed by:
In silico Mutational Scanning: Using ESM-2 embeddings and LLR scores to nominate stabilizing mutations.
Folding Assessment: Validation of fold accuracy via ESMFold and AlphaFold-Multimer to ensure independent folding propensity and multimeric pore symmetry.
Aim 2: Generative Design for Chaperone Independence
We propose using generative models (like ProteinMPNN or RFdiffusion) to optimize the soluble N-terminal domain. The goal is to redesign the interface to either:
Enhance co-folding with DnaJ under controlled structural constraints.
Enable “folding rescue” by alternative or orthogonal chaperones (e.g., DnaK or GroEL) to bypass host adaptation.
Aim 3: Evolutionary Analysis and Host Factor Integration
Using pBLAST to survey orthologs, we will reconstruct evolutionary trajectories of stability. Candidates will be screened against E. coli host factors to minimize proteotoxicity while maximizing the “aggressive” lytic potential identified through Genomic Language Models (GLMs).
3. Computational Tools and Workflow
Our design-build-test-learn (DBTL) framework utilizes the following stack:
Sequence & Conservation: Clustal Omega and pBLAST for “evolutionary grammar” analysis.
Mutational Analysis: ESM-2 (LLR scores) for high-speed structural feedback on the 7-helix bundle.
Generative Design: ProteinMPNN for sequence backbone optimization.
Structural Validation: AlphaFold 3 and AlphaFold-Multimer to ensure biophysical plausibility of the 8-chain pore assembly.
Contextual Gap: A lack of specific data regarding the host bacteria’s in vivo environment may lead to unexpected results despite positive simulations.
Functional Trade-offs: Mutations that improve structural stability might inadvertently perturb the membrane-interaction properties or the native lytic activity, leading to a loss of function.
Misfolding Risk: Compact lysis proteins are highly sensitive; even high-confidence predicted folds (pLDDT > 80) may aggregate or fail to insert into the membrane in a real biological system.
5. Expected Outcomes
If successful, this framework will yield L-protein variants with:
Increased Stability: Robust functionality under diverse environmental conditions.
Reduced Host Dependency: Decreased reliance on native DnaJ interactions, making the phage less vulnerable to host-dependent failure modes.
Optimized Lysis: Retention of a mature fold compatible with aggressive lytic activity, establishing a generalizable template for synthetic antimicrobial modules.
The study explains that the MS2 phage L protein is a 75 amino acid polypeptide that kills bacteria through a unique mechanism. Unlike other proteins like E or A2, which block cell wall synthesis, the L protein does not affect peptidoglycan production. Using a smart screening system with a lacZ reporter to filter out false positives, the researchers discovered that L depends entirely on the host chaperone DnaJ to function. Interestingly, a specific mutation in DnaJ called P330Q completely blocks lysis at 30°C. Through pulldown assays, they confirmed that DnaJ physically binds to the N-terminal “head” of the protein, which is full of basic charges and is actually dispensable for the killing process, serving instead as a control unit.
The final model proposes that this N-terminal domain of L acts as a biological brake that auto-inhibits the protein. The DnaJ chaperone acts like a key that unlocks this brake, allowing the hydrophobic tail of the protein to reach its actual target inside the cell. This was proven with Lodj mutants, which are versions of the L protein lacking the head. These mutants do not need DnaJ and kill the bacteria 20 minutes faster than the wild type. This system mirrors what happens with the E protein and its chaperone SlyD, suggesting that phages evolved these charged domains as a strategy to control lysis timing and ensure the virus has enough time to replicate before destroying the host.
The MS2 lysis protein (L) is a 75-amino acid polypeptide that triggers bacterial cell death without disrupting net peptidoglycan synthesis. Research reveals a conserved LS (Leu-Ser) dipeptide motif at residues Leu48-Ser49, which serves as the essential core for protein-protein interactions. While the N-terminal half of the protein is dispensable for lytic activity, the C-terminal domain is critical; specifically, the S49C mutation in the LS motif causes an absolute lysis defect. This motif is highly conserved across diverse phages, indicating it is a universal structural requirement for the lytic function in amurins.
The study suggests that the L protein interacts with a host membrane target through the LS motif and surrounding essential domains. The N-terminus functions as a regulatory domain that naturally inhibits this interaction, while the host chaperone DnaJ binds to the N-terminus to displace it from its inhibitory position. Interestingly, deleting the basic N-terminal domain allows the protein to bypass the need for DnaJ entirely. This confirms that the N-terminus acts as a regulatory gatekeeper, and DnaJ is the key that unlocks the protein’s ability to engage its cellular target.
The MS2 bacteriophage lysis protein (MS2L) facilitates host cell escape by punching holes in the bacterial wall through a dual-domain mechanism. It consists of a soluble HEAD domain and a transmembrane TAIL domain that anchors into membranes, behaving similarly to soap or micelles. A key finding is that the TAIL domain drives oligomerization, causing 10 or more proteins to clump into large complexes. CryoEM data confirms these clusters gather at specific spots to trigger a sequential rupture: first the outer membrane breaks, followed by the peptidoglycan layer, and finally the inner membrane, causing the cell contents to leak out.
The researchers identified the HEAD domain as a biological brake that regulates the timing of lysis. While the full MS2L protein is difficult to insert into membranes, removing the HEAD allows for relatively easy insertion, suggesting it functions as a timer to prevent premature cell death. Additionally, the helper protein DnaJ binds to MS2L but does not influence its membrane entry or oligomerization. From an engineering perspective, removing the HEAD domain could bypass this brake to achieve a “quicker kill,” a strategic goal for optimizing lytic toxicity in synthetic biology.
This paper explains how phages have evolved from a biological curiosity into a sophisticated therapeutic tool by focusing on their life cycles and resistance mechanisms. The review highlights that success in therapy depends on more than just injecting phages; it requires a deep understanding of pharmacokinetics and the patient’s immune response, as the body might neutralize the viruses before they reach the infection site. A key advancement mentioned is the use of genetic engineering to create “designer” phages that do more than just kill bacteria, such as degrading biofilms or working alongside traditional antibiotics to restore drug sensitivity. The future of the field points toward precision medicine where specific phages are selected or edited for each patient to overcome the regulatory and technical barriers that previously limited mass clinical use.
This text explores the historical evolution and the modern resurgence of phage therapy in response to the global antibiotic resistance crisis. It begins by reminding us that phages were used long before penicillin but were largely forgotten in the West due to a lack of standardized protocols and the convenience of broad-spectrum antibiotics. Currently, we are in a stage of “compassionate use” where phages are successfully applied in desperate cases of multi-drug resistant infections, which is driving new controlled clinical trials. The study concludes that the biggest challenge today is not just biological but also logistical and legal, as a global infrastructure is needed to collect and characterize phage libraries that can be quickly deployed against emerging pathogens. This marks a shift from general treatments to a completely personalized paradigm.
This research utilizes the Evo 1 and Evo 2 DNA foundation models to design functional biological systems at the whole-genome scale. Using the phiX174 lytic phage as a chassis, the AI successfully generated 16 viable phages with substantial evolutionary novelty. Some variants were highly distant from common natural sequences, proving that genomic language models (GLMs) can expand the known biological space. This is critical for phage therapy, as these AI-designed variants demonstrated a superior ability to overcome bacterial resistance in E. coli strains where natural phages failed.
The computational method employed taxonomic prompting (e.g., Riboviria) to guide the generative process toward specific viral realms. Novelty was rigorously validated using nucleotide BLAST against core databases to confirm the emergence of original sequences. This strategy offers a robust framework for creating diverse phage cocktails, a key requirement for modern antimicrobial treatments. By leveraging taxonomic labels and pretraining, the study establishes a “design-build-test” workflow for engineering complex, multi-gene systems beyond the limits of natural evolution.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
We will focus on increasing the structural stability of the L protein to ensure it remains functional under different environmental conditions.
We will also attempt to increase the toxicity of the lysis protein by optimizing its target regions to enhance bacterial cell wall disruption.
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
We will use ESMFold to perform in silico mutational scanning and identify target regions in the L protein.
We propose using Genomic Language Models (GLMs) to design and optimize sequences with higher lytic potential.
Finally, we will use AlphaFold-Multimer to validate the folding and stability of the engineered protein complexes.
Why do you think those tools might help solve your chosen sub-problem?
ESMFold allows for high-speed structural feedback, making it easier to test how mutations affect the 7-helix bundle.
GLMs are essential for capturing the “evolutionary grammar” of toxicity, helping to design proteins that are more aggressive than natural variants.
AlphaFold ensures that our computational designs are biophysically plausible and stable before any potential wet-lab implementation.
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Contextual Gap: There is a lack of specific data regarding the host bacteria’s environment, which might lead to unexpected results in vivo.
Misfolding Risk: The engineered protein might still misfold or aggregate in a real biological system despite having positive simulation results in the pipeline.
Include a schematic of your pipeline.
Here’s a short written schematic of our pipeline:
[Sequence Input] → [ESM-2 Mutational Scan] → [GLM Toxicity Optimization] → [AlphaFold Validation] → [Final Design]
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a
MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
L-Protein Engineering - Option 1: Mutagenesis
Step 1: Information Gathering
Here are the L-protein and Dnaj sequences
Lysis Protein Sequence (UniProtKB ID: P03609)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
Soluble N-terminal domain: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYV
TM domain: LIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Additionally, here’s a screenshot of the BLAST results for L-protein:
Lastly, these results were aligned using Clustal Omega, revealing a highly conserved “island” (HEDYPCRRQQRSST) at residues 24-38. These sites will be avoided during mutagenesis to preserve the critical interaction with DnaJ and overall biological function of the phage.
My approach is very straightforward: I combined computational LLR scores with experimental lab data using a copy of the HTGAA Colab. I filtered for mutations that showed “active lysis” (value 1) in the experimental spreadsheet and high positive LLR scores in the notebook.
Step 3: Filtering and Ranking
I used AlphaFold 3 to model the 8-chain assembly. This step was used to rank candidates that showed both positive computational scores and confirmed experimental activity, ensuring they don’t disrupt the pore’s symmetry.
Step 4: Final Mutated Sequences
These 5 mutations were selected because they are experimentally proven to maintain lysis (score 1) and show improved or stable computational scores.
Region
Mutation
LLR Score (ESM-2)
Experimental Lysis
Rationale
Soluble
S9Q
2.01
Active (1)
High computational confidence; replaces Serine with Glutamine to stabilize the N-terminal loop.
Soluble
C29R
2.39
Active (1)
One of the top scores; removing this Cysteine likely prevents incorrect disulfide bonding.
TM Domain
Y39L
2.24
Active (1)
High confidence score in the TM interface; optimizes hydrophobicity for membrane entry.
TM Domain
A45L
1.53
Active (1)
Consistent with experimental data; improves the hydrophobic core of the lytic pore.
TM Domain
N53L
1.86
Active (1)
Replaces a polar Asparagine with Leucine, significantly improving helix-helix packing in the multimer.
S9Q mutation 8-chain assembly:
C29R mutation 8-chain assembly:
Y39L mutation 8-chain assembly:
A45L mutation 8-chain assembly:
N53L mutation 8-chain assembly:
While AF3 structures were used to visualize the multimeric orientation, the ipTM scores remained low (~0.17) across all mutations. This is expected given the small, intrinsically disordered nature of the L-protein and the high flexibility required for its lytic function, which challenges standard multimeric confidence metrics.