Does the option: Option 1 Option 2 Option 3 Enhance Biosecurity • By preventing incidents 1 2 NA • By helping respond 3 NA NA Foster Lab Safety • By preventing incident 3 1 NA • By helping respond NA 1 NA Protect the environment • By preventing incidents NA 1 NA • By helping respond NA 2 NA Other considerations • Minimizing costs and burdens to stakeholders 2 2 3 • Feasibility? 2 1 3 • Not impede research 3 1 1 • Promote constructive applications 1 1 1
Homework Questions from Professor Jacobson
DNA Polymerase Error Rates and the Human Genome Error Rate of Polymerase: In biological synthesis, error-correcting polymerase has an error rate of approximately $1:106$. This is significantly more accurate than raw chemical synthesis, which has an error rate of roughly $1:102$. Comparison to the Human Genome: The human genome is approximately 3 billion base pairs ($3 \times 109$) in length. At an error rate of $1:106$, copying the entire human genome would result in roughly 3,000 errors per replication cycle. How Biology Deals with the Discrepancy: Biology utilizes specific enzymatic functions to manage and correct these errors to ensure genomic integrity. This includes 3’-5’ proofreading exonuclease activity and 5’-3’ error-correcting exonuclease functions that work alongside template-dependent primer extension to identify and remove incorrect bases. Coding for Human Proteins Ways to Code for an Average Human Protein: The average human protein is 1,036 base pairs long. Because the genetic code is redundant (multiple different codons can code for the same amino acid), there are an astronomical number of possible DNA sequences that can result in the same protein sequence. The sources highlight that biology must find a balance between this codon redundancy and diversity to maintain “fabricational complexity”. Reasons Some Codes Do Not Work: In practice, many DNA sequences that technically code for the correct protein are “impossible” or difficult to use for synthesis or expression due to several biological and mechanical factors: Secondary Structures: Sequences that form hairpins or inverted repeats can interfere with replication and transcription machinery. Extreme GC Content: Regions with very high (≥90%) or very low (≤10%) GC content are often unstable or difficult for polymerase to navigate. Repetitive Sequences: Long terminal repeats, tandem repeats, or clusters of repeats can lead to “slippage” and errors during synthesis. Homopolymers: Long runs of an identical base (e.g., more than 30bp of A) are particularly prone to errors. RNA Cleavage and Stability: Certain nucleotide combinations may inadvertently trigger RNA cleavage rules (such as targets for RNase III), leading to the degradation of the mRNA before it can be translated. Codon Optimization: Not all redundant codons are treated equally by the cell’s translational machinery; choosing the “wrong” codons can lead to inefficient protein production. Homework Questions from Dr. LeProust
Part 3. Chose Protein I chose glucokinase (GCK) because in my biochemistry classes I found it to be a very interesting enzyme due to its unique functions and its critical role as a glucose sensor.
According to the sources, what makes this enzyme particularly fascinating is that, unlike other members of the hexokinase family, it is not inhibited by its product(glucose-6-phosphate). This allows the enzyme to remain active even when glucose is abundant in the system.
PART 1.  artistic design using the GUI LINK: https://opentrons-art.rcdonovan.com/?id=98conne30870554
PART 2. ARTICLE “An Automated Versatile Diagnostic Workflow for Infectious Disease Detection in Low-Resource Settings” DOI: https://doi.org/10.3390/mi15060708
The article highlights how implementing Opentrons for automated workflows in hospital and clinical settings helps significantly reduce turnaround times and accelerates overall logistics. By increasing sample throughput and enabling the simultaneous processing of multiple samples, the system greatly enhances operational efficiency. Furthermore, automation reduces the risk of human error inherent in manual repetitive tasks and minimizes the possibility of sample contamination or compromising the diagnostic process, ensuring more reliable results.
Part A. Conceptual Questions
Why do humans eat beef but do not become a cow, or eat fish but do not become fish? This is because the genetic code acts as an algorithm that dictates how proteins are assembled specifically for each organism. When humans consume animal proteins, these are broken down into amino acids; subsequently, the body uses its own transcription and translation machinery to reorganize those amino acids according to its own DNA instructions, creating human-specific proteins rather than those of the animal consumed.
DNA ASSEMBLY
Phusion High-Fidelity PCR Master Mix Components
Phusion HF PCR Master Mix is provided as a 2X stock that is diluted to a 1X concentration in the final reaction. While the sources do not list every chemical ingredient, they indicate its purpose is to amplify specific DNA sequences (such as the amilCP gene and the mUAV backbone) with high accuracy.
Standard high-fidelity master mixes like Phusion typically contain:
Phusion DNA Polymerase: A highly accurate enzyme with proofreading activity to minimize mutations. dNTPs (Deoxynucleotide Triphosphates): The building blocks (A, T, C, G) used to synthesize the new DNA strand. Buffer: Maintains the optimal pH and ionic strength for the enzyme. $MgCl_2$: A necessary cofactor for the DNA polymerase to function. Factors Determining Primer Annealing Temperature
Part 1 HW
Intracellular Artificial Neural Networks (IANNs), also known as neuromorphic circuits, provide significant advantages over traditional genetic circuits that rely on Boolean (digital) logic:
a) Advantages of IANNs
Biological Substrate Compatibility:** While digital logic attempts to force binary “on/off” behavior onto cells, IANNs operate through analog computation, which is much closer to the natural language of biology. Handling Non-linear Complexity:** Biological systems naturally manage highly non-linear and complex input-output relationships that Boolean logic oversimplifies. IANNs allow for the capture of these non-linearities and non-monotonic behaviors more robustly. Precision in Decision Boundaries: Unlike digital logic, which only recognizes “high” or “low” thresholds, IANNs can be programmed to respond to specific analog relationships (for example, activating only when two inputs are equal or when a weighted combination exceeds a bias), allowing for much more exact classification of cellular states. Flexibility and Scalability: The behavior of the circuit can be adjusted simply by modifying the translation rates of the components, allowing decision boundaries to be shifted without needing to redesign the entire system. b) Useful Application: Cancer Cell Classifier A primary application for IANNs is the creation of high-precision cell classifiers for cancer immunotherapy. Because there is rarely a single “magic” biomarker to distinguish a cancer cell from a healthy one, a sophisticated program is required to evaluate multiple signals simultaneously.
Subsections of Homework
Week 1 HW: Principles and Practices
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
NA
• By helping respond
3
NA
NA
Foster Lab Safety
• By preventing incident
3
1
NA
• By helping respond
NA
1
NA
Protect the environment
• By preventing incidents
NA
1
NA
• By helping respond
NA
2
NA
Other considerations
• Minimizing costs and burdens to stakeholders
2
2
3
• Feasibility?
2
1
3
• Not impede research
3
1
1
• Promote constructive applications
1
1
1
Subsections of Week 1 HW: Principles and Practices
Week 1 HW: Principles and Practices
Below is the translation of the proposal for the biological engineering tool and its associated governance framework.
Description:
I propose the development of Bio-Hybrid Designer, an AI-powered BioCAD software specifically engineered to design and optimize hybrid biosynthetic pathways for complex natural drugs, such as taxanes (paclitaxel).
Why develop it?
Inspired by the “How to Grow (Almost) Anything” (HTGAA) philosophy and the need for sustainable pharmaceutical production, this tool addresses critical enzymatic “bottlenecks”. Currently, complete biosynthesis of complex drugs in microorganisms is hindered by poor expression of enzymes like cytochrome P450 in heterologous systems. The software would identify the optimal transition point where biological synthesis should stop (producing an advanced precursor like baccatin III) and where selective chemical synthesis should take over to finalize the drug.
2. Governance and Policy Goals
The primary goal is non-malfeasance (preventing harm) to ensure the ethical deployment of this technology.
Sub-goal A: Prevention of Harmful Dual-Use. Ensuring the AI cannot be used to design pathways for known toxins or pathogens, thereby protecting international security.
Sub-goal B: Global Equity and Sustainability. Preventing abrupt “economic displacement”. If lab production replaces natural harvesting (such as from the Pacific yew tree), a just transition must be ensured for local communities that currently depend on these natural resources.
3. Proposed Governance Actions
Action 1: Mandatory AI Screening for “Critical Bio-Parts” (Federal Regulators & Companies)
Purpose: Currently, access to many biological databases is open. This action proposes that AI BioCAD tools must incorporate mandatory filters to block the design of sequences structurally similar to biological threat agents.
Design: Federal regulators would require companies providing BioCAD services to implement automated detection protocols (similar to internet content filters or anti-plagiarism software).
Assumptions: It assumes that biological threats have predictable genetic or structural “signatures” that AI can accurately identify.
Risks of Failure & “Success”: It could fail if attackers use sequence “obfuscation” techniques. Excessive “success” might hinder legitimate research on rare medicines that happen to share chemical precursors with toxins.
Action 2: Incentives for Chassis with “Kill-Switches” (Academic Researchers & Funding Agencies)
Purpose: To move from passive observation to technical biosecurity. All research grants for synthetic drug production would require the use of host organisms (chassis) equipped with programmed “kill-switches” to prevent environmental survival in case of a leak.
Design: Funding agencies (such as the NIH) would act as the primary actors, tying financial support to the use of validated containment protocols like “GeneGuard”.
Assumptions: It assumes these kill-switches are evolutionarily stable and will not be deactivated by natural mutations within the lab.
Risks of Failure & “Success”: Failure occurs if the microorganism survives environmental filters through horizontal gene transfer. “Success” might create a technological monopoly over “safe” chassis.
Action 3: Bioeconomy Transition Fund (International Organizations & Big Pharma)
Purpose: To mitigate global inequalities. Similar to energy transition funds, a levy would be placed on profits from drugs produced via synthetic hybrid pathways.
Design: The UN or WHO would coordinate with pharmaceutical giants to fund economic reconversion programs in regions where natural harvesting is displaced by industrial production.
Assumptions: It assumes synthetic production will be significantly cheaper, generating enough surplus to fund compensation.
Risks of Failure & “Success”: The main failure would be a lack of political will to tax new technologies. “Success” could potentially create a dependency on subsidies rather than fostering a new, sustainable local economy.
Week 2 HW: Lecture Prep
Homework Questions from Professor Jacobson
DNA Polymerase Error Rates and the Human Genome
Error Rate of Polymerase: In biological synthesis, error-correcting polymerase has an error rate of approximately $1:10^6$. This is significantly more accurate than raw chemical synthesis, which has an error rate of roughly $1:10^2$.
Comparison to the Human Genome: The human genome is approximately 3 billion base pairs ($3 \times 109$) in length. At an error rate of $1:106$, copying the entire human genome would result in roughly 3,000 errors per replication cycle.
How Biology Deals with the Discrepancy: Biology utilizes specific enzymatic functions to manage and correct these errors to ensure genomic integrity. This includes 3’-5’ proofreading exonuclease activity and 5’-3’ error-correcting exonuclease functions that work alongside template-dependent primer extension to identify and remove incorrect bases.
Coding for Human Proteins
Ways to Code for an Average Human Protein: The average human protein is 1,036 base pairs long. Because the genetic code is redundant (multiple different codons can code for the same amino acid), there are an astronomical number of possible DNA sequences that can result in the same protein sequence. The sources highlight that biology must find a balance between this codon redundancy and diversity to maintain “fabricational complexity”.
Reasons Some Codes Do Not Work: In practice, many DNA sequences that technically code for the correct protein are “impossible” or difficult to use for synthesis or expression due to several biological and mechanical factors:
Secondary Structures: Sequences that form hairpins or inverted repeats can interfere with replication and transcription machinery.
Extreme GC Content: Regions with very high (≥90%) or very low (≤10%) GC content are often unstable or difficult for polymerase to navigate.
Repetitive Sequences: Long terminal repeats, tandem repeats, or clusters of repeats can lead to “slippage” and errors during synthesis.
Homopolymers: Long runs of an identical base (e.g., more than 30bp of A) are particularly prone to errors.
RNA Cleavage and Stability: Certain nucleotide combinations may inadvertently trigger RNA cleavage rules (such as targets for RNase III), leading to the degradation of the mRNA before it can be translated.
Codon Optimization: Not all redundant codons are treated equally by the cell’s translational machinery; choosing the “wrong” codons can lead to inefficient protein production.
Homework Questions from Dr. LeProust
The most commonly used method for oligonucleotide synthesis currently is phosphoramidite chemical synthesis on a solid support. This process follows a cycle involving deprotection, base coupling, capping, and oxidation to add nucleotides sequentially to a growing chain.
Direct synthesis of oligos longer than 200 nucleotides is difficult because the cumulative yield drops exponentially with each coupling step. Chemical synthesis has a high raw error rate of approximately 1:10², meaning errors are introduced frequently as the chain grows. Because the efficiency of adding each base is not 100%, the probability of obtaining a perfect, full-length product decreases significantly as the length increases.
You cannot make a 2000bp gene via direct synthesis primarily because the error rate of chemical synthesis would result in approximately 20 errors in a sequence of that length. Furthermore, the physical yield of a 2000bp molecule would be vanishingly small due to the cumulative losses over 2000 coupling cycles. Instead, genes are constructed using enzymatic assembly to join many smaller, sequence-verified oligonucleotides into a single long fragment. While advanced chemistry has pushed direct synthesis limits to 700 nucleotides, creating a 2000bp gene still requires the assembly of multiple fragments to maintain accuracy and throughput.
Homework Question from George Church
Command IA: [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Based on the list of essential amino acids and the provided lecture materials, here is an analysis of the “Lysine Contingency”:
The 10 Essential Amino Acids
According to general biological standards (verified via external search), the 10 essential amino acids that most animals (including humans) cannot synthesize and must obtain through their diet are:
Phenylalanine
Valine
Threonine
Tryptophan
Isoleucine
Methionine
Histidine
Arginine
Leucine
Lysine
In Professor Church’s slides, the standard genetic code chart identifies these amino acids by their single-letter codes (e.g., K for Lysine, L for Leucine, M for Methionine) as the building blocks for protein synthesis.
The “Lysine Contingency” and Biocontainment
The “Lysine Contingency” is a concept popularized by Jurassic Park, where engineered organisms are made unable to produce lysine, theoretically preventing them from surviving in the wild without human-provided supplements.
Knowing that lysine is already an essential amino acid for all animals significantly changes the view of this contingency:
Redundancy in Nature: Since wild animals (and humans) already cannot synthesize lysine, they must constantly find it in their environment (by eating plants or other animals). Therefore, a “lysine contingency” is not a robust biocontainment strategy because lysine is widely available in the natural world. An escaped organism would simply find lysine in the wild just as any other animal does.
Church’s Advanced Solution: Professor Church’s research proposes a much more effective version of this idea through Genomically Recoded Organisms (GROs). Instead of relying on a natural amino acid like lysine, Church’s team engineered organisms to be dependent on Non-Standard Amino Acids (NSAAs).
Synthetic Dependency: These NSAAs do not exist in the wild. This creates a true “metabolic isolation” or “biocontainment” because, unlike lysine, the organism cannot find these synthetic building blocks in the environment, effectively preventing survival outside a controlled laboratory setting.
In summary, while the “Lysine Contingency” is a natural reality for all animals, it is an ineffective tool for synthetic biology containment. Modern genetic engineering instead uses synthetic amino acid dependency to ensure organisms remain contained.
Week 2 HW: DNA read write and edit
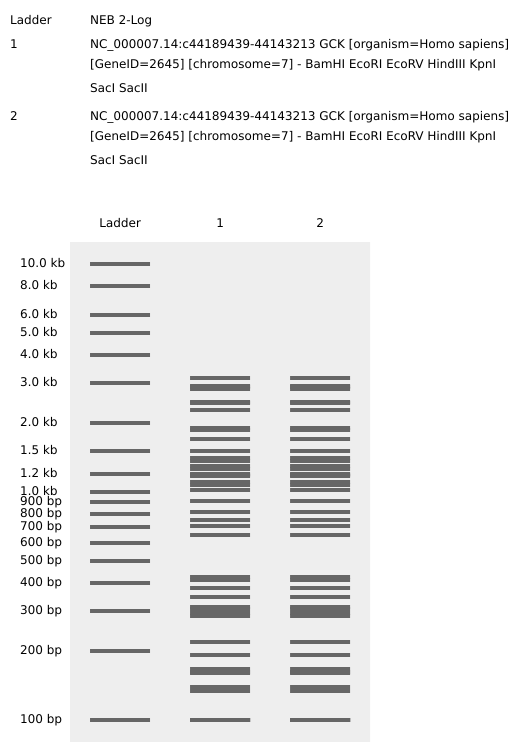
Part 3. Chose Protein
I chose glucokinase (GCK) because in my biochemistry classes I found it to be a very interesting enzyme due to its unique functions and its critical role as a glucose sensor.
According to the sources, what makes this enzyme particularly fascinating is that, unlike other members of the hexokinase family, it is not inhibited by its product(glucose-6-phosphate). This allows the enzyme to remain active even when glucose is abundant in the system.
Furthermore, glucokinase exhibits tissue-specific expression in the pancreas and the liver, which leads to different but equally vital functions:
In the pancreas, it is a key player in glucose-stimulated insulin secretion.
In the liver, it is essential for glucose uptake and its subsequent conversion into glycogen.
I also find it interesting from a clinical perspective, as mutations in this gene that alter enzyme activity are associated with several medical conditions, including Maturity-Onset Diabetes of the Young type 2 (MODY2) and hyperinsulinemic hypoglycemia. This demonstrates its fundamental importance in maintaining human glucose homeostasis.
Protein Sequence (Isoform 1 - Pancreatic):
The following sequence corresponds to the pancreatic isoform 1 (NP_000153.1 / UniProt P35557), which has a distinct N-terminus compared to liver isoforms.
3.2. Reverse Translation: Protein sequence to DNA sequence
The Central Dogma (DNA → RNA → Protein) allows us to work backward from a protein sequence to identify the corresponding DNA sequence. By using the NCBI Gene database (Gene ID: 2645), we identified the Coding Sequence (CDS) for Glucokinase.
In accordance with HTGAA conventions, the sequence is presented in the 5’ to 3’ coding strand** format, as found in GenBank or FASTA files.
DNA Sequence for Glucokinase (CDS - Variant 1):
This nucleotide sequence corresponds to the mRNA RefSeq NM_000162.5, which encodes the pancreatic islet beta cell isoform.
Based on the HTGAA course materials and the biological databases provided, here is the completion of your assignment for Glucokinase (GCK) in English.
3.3. Codon Optimization
Why do we need to optimize codon usage?**
Codon optimization is necessary because different organisms have distinct “preferences” or varying abundances of tRNA for the same amino acids. Since the genetic code is redundant (multiple codons can code for one amino acid), using the codons that are most frequently used by the host organism—and thus for which there are more available tRNAs—ensures that the translation process is more efficient and results in higher protein yields. Optimization also helps avoid technical synthesis issues such as extreme GC content, high repetition, or “Homo polymers” (long strings of the same nucleotide) that can cause errors during DNA printing.
a) Which organism have you chosen to optimize the codon sequence for and why?
I have chosen Escherichia coli_ (E. coli) as the target organism. According to the sources, E. coli is the preferred host for this course because it is the easiest and most accessible organism to work with in a laboratory setting.
Glucokinase DNA sequence with Codon Optimization (E. coli)
(The following sequence is a simulated optimization of the GCK coding sequence for E. coli, avoiding common restricted enzyme sites like BsaI or BbsI to ensure compatibility with Twist Bioscience synthesis tools.)
What technologies could be used to produce this protein from your DNA?
To produce Glucokinase from the optimized DNA sequence, two main approaches can be utilized:
Cell-dependent methods:** This is the most common approach, using E. coli_ as a “chassis”**. The DNA sequence is inserted into a circular DNA molecule called a plasmid and then introduced into the bacteria through a process called transformation (often using heat shock). The living bacteria then act as biological factories to produce the protein.
Cell-free methods: These methods involve using cell lysates (essentially the “guts” or internal machinery of a cell) to produce proteins without the need for a living organism. This allows for rapid prototyping and can even be incorporated into materials like textiles.
How the DNA sequence is transcribed and translated into your protein:
The production follows the Central Dogma of molecular biology:
a) Transcription: The process begins when an enzyme called RNA polymerase binds to the DNA template and “reads” the sequence to create a complementary strand of mRNA.
b) Translation:** This mRNA is then processed by a ribosome. The ribosome reads the mRNA in sets of three nucleotides called codons. Each codon corresponds to a specific amino acid. Transfer RNA (tRNA) molecules bring the correct amino acids to the ribosome, which links them together into a long chain that eventually folds into the functional Glucokinase protein.
What DNA would you want to sequence and why?
I would want to sequence the human GCK (Glucokinase) gene variants in patients with atypical metabolic profiles. Sequencing this DNA is critical for human health research because mutations in GCK are directly linked to conditions like Maturity-Onset Diabetes of the Young type 2 (MODY2)** and hyperinsulinemic hypoglycemia. By reading these sequences, researchers can characterize variant mechanisms and improve clinical diagnostics.
Technology and Details
a) Technology: I would use Illumina Sequencing (Next-Generation Sequencing), as it is the standard mentioned in the sources for high-throughput analysis of human samples.
b) Generation:** This is a second-generation technology. It is characterized by massive parallelism, allowing millions of DNA fragments to be sequenced simultaneously.
c) Input and Preparation:** The input is genomic DNA or cDNA. Preparation involves fragmentation of the DNA into smaller pieces, adapter ligation to attach specific sequences to the ends of fragments, and PCR amplification to create clusters for signal detection.
c) Essential Steps and Base Calling:** The technology uses “sequencing by synthesis” where fluorescently labeled nucleotides are added to the DNA template. During each cycle, the machine captures the fluorescence color (red, green, blue, or yellow) emitted as a base is incorporated, a process known as **base calling.
d) Output: The primary output is a FASTQ or FASTA file containing the strings of nucleotides (A, T, G, C) and their corresponding quality scores.
5.2 DNA Write
What DNA would you want to synthesize and why?**
I would want to synthesize an optimized expression cassette for human Glucokinase (GCK). This would be used for therapeutics and drug discovery, specifically to produce functional GCK protein in E. coli or mammalian cell lines to test new activators for diabetes treatment. I would use the codon-optimized sequence we generated previously to maximize protein yield.
Technology and Detllais
a) Technology:** I would use Silicon-based DNA synthesis provided by companies like Twist Bioscience.
b) Essential Steps: The process involves designing the sequence digitally in tools like Benchling, performing codon optimization for the host organism, and then using a silicon chip to print thousands of tiny DNA fragments (oligonucleotides) simultaneously using chemical synthesis (phosphoramidite chemistry). These fragments are then assembled into full-length genes.
c) Limitations:** The primary limitations include complexity; sequences with high GC content, repetitive regions, or long homopolymers (e.g., many ‘A’s in a row) are very difficult to synthesize and may fail during the printing process.
5.3 DNA Edit
What DNA would you want to edit and why?
In alignment with the goals of Colossal Biosciences, I would want to edit the genome of an Asian Elephant to include specific genes from the Woolly Mammoth. The goal of this “de-extinction” project is to restore historic animals to their ecological roles, which can help in nature conservation and the restoration of Arctic ecosystems. These edits would focus on traits like cold tolerance, hair growth, and fat distribution.
Technology and Details
Technology: I would use CRISPR-Cas9 technology, as it is the most precise and versatile tool for targeted genome editing discussed in class.
How it works: CRISPR-Cas9 acts as “molecular scissors.” A guide RNA (gRNA) is designed to match a specific target sequence in the DNA. The Cas9 enzyme then follows this guide to the exact location and creates a double-strand break. The cell then repairs this break, allowing us to delete or insert specific mammoth-like genetic information.
Preparation and Input: Preparation requires designing the guide RNA using digital tools to ensure specificity. The input includes the Cas9 enzyme, the gRNA, a DNA template for the desired mammoth traits, and the target host cells (elephant cells).
a) Limitations: Key limitations include efficiency (the edit may not happen in every cell) and precision (potential “off-target” effects where the enzyme cuts at unintended locations similar to the target).
PART 2. ARTICLE
“An Automated Versatile Diagnostic Workflow for Infectious Disease Detection in Low-Resource Settings”
DOI: https://doi.org/10.3390/mi15060708
The article highlights how implementing Opentrons for automated workflows in hospital and clinical settings helps significantly reduce turnaround times and accelerates overall logistics. By increasing sample throughput and enabling the simultaneous processing of multiple samples, the system greatly enhances operational efficiency. Furthermore, automation reduces the risk of human error inherent in manual repetitive tasks and minimizes the possibility of sample contamination or compromising the diagnostic process, ensuring more reliable results.
The system OpenTrons was demonstrated through a test for Neisseria meningitidis (meningitis) and consists of four integrated modules:
DNA Isolation: Utilizing magnetic beads to purify pathogen DNA.
DNA Amplification: Performing isothermal Recombinase Polymerase Amplification (RPA) at 37°C, which avoids the need for complex thermal cycling.
DNA Digestion: Using exonucleases to convert double-stranded amplicons into single-stranded DNA.
DNA Detection: Employing a vertical flow microarray (VFM) on paper, where gold nanoparticles create a colorimetric signal for visual results.
The automated process takes 110 minutes, making it approximately 18% faster than manual processing. Additionally, the cost is roughly $16 per sample, which is significantly more affordable than the estimated $94 for a standard PCR test
PART 3.
Final Project Automation Plan: “Bio-Hybrid Designer”
My plan to use automation to optimize hybrid biosynthetic pathways for complex drugs like paclitaxel. The goal is to identify the most efficient “transition point” where biological synthesis (producing precursors like baccatina III) should end and selective chemical synthesis should begin.
Cloud Lab Implementation (Ginkgo Nebula)
To achieve high-throughput screening of enzymatic variants and chemical cofactors, I will utilize the Ginkgo Nebula platform. The workflow will follow these steps:
Acoustic Liquid Handling: Use the Echo 525 to transfer nanoliter-scale droplets of DNA constructs and cofactors into destination plates.
Reagent Stamping: Use the Bravo system to stamp cell-free protein synthesis (CFPS) master mixes into 384-well plates.
Incubation: Seal plates with PlateLoc and incubate in Inheco modules at 37°C to express the enzymes.
Detection: After unsealing with XPeel, use the PHERAstar to measure fluorescence from biosensors, providing data for the next Design-Build-Test-Learn (DBTL) cycle.
Local Prototyping (Opentrons OT-2)
I will use the Opentrons OT-2 for initial protocol validation and mixing of chemical precursors. I will use a standardized layout:
Slot 5: Agar plate or reaction plate for the final output.
Slot 6: Source plate for bacterial cultures or chemical reagents.
Slot 9: Tip rack for the P20 pipette.
a) Python Script / Pseudocode Example (maybe use, I´m not sure right now)
My script will incorporate the dispense_and_jog function to ensure the pipette moves horizontally before dispensing vertically, preventing the tip from scratching the agar surface.
fromopentronsimportprotocol_apimetadata={'protocolName':'Bio-Hybrid Path Optimization','author':'User','description':'Automated mixing of chemical cofactors with biosynthetic precursors'}#defrun(protocol:protocol_api.ProtocolContext):## Load Labwaretips=protocol.load_labware('opentrons_96_tiprack_20ul','9')#plate=protocol.load_labware('corning_96_wellplate_360ul_flat','5')#reagents=protocol.load_labware('opentrons_24_tuberack_eppendorf_1.5ml','6')#p20=protocol.load_instrument('p20_single_gen2','right',tip_racks=[tips])## Automated serial dilution and mixing logicforiinrange(8):p20.pick_up_tip()p20.aspirate(5,reagents['A1'])# Implementation of vertical movement to protect labwarep20.dispense(5,plate.wells()[i])p20.mix(3,10,plate.wells()[i])p20.drop_tip()
Week 4 HW: Protein design part 1
Part A. Conceptual Questions
Why do humans eat beef but do not become a cow, or eat fish but do not become fish?
This is because the genetic code acts as an algorithm that dictates how proteins are assembled specifically for each organism. When humans consume animal proteins, these are broken down into amino acids; subsequently, the body uses its own transcription and translation machinery to reorganize those amino acids according to its own DNA instructions, creating human-specific proteins rather than those of the animal consumed.
Why are there only 20 natural amino acids?
The human body relies on 20 types of natural amino acids as fundamental building blocks. While the number of types is limited, their combinatory potential is vast: a chain of just 40 amino acids can generate more unique protein variations than there are total atoms in the known universe.
Can you make other non-natural amino acids?
Design some new amino acids. Yes, modern engineering allows for de novo design using non-natural materials to build biological systems from scratch. Using genome language models and AI tools, scientists can now explore “evolutionary spaces” not found in nature, designing building blocks with custom chemical properties for applications like drug delivery or new biomaterials.
Can you discover additional helices in proteins?
Yes, through what is called “helical arithmetic,” various complex structural organizations have been identified. In addition to simple helices, researchers have discovered coiled coils, coiled-coil trimers (in laminin), four-helical bundles (forming hydrophobic cores), five-helical bundles (in talin), six-helical bundles (in the coronavirus spike protein), and seven-helical bundles (in GPCR receptors).
Why do β-sheets tend to aggregate?
These structures have a natural tendency to aggregate because of their longitudinal bonds between different amino acids. This property makes them extremely rigid and resistant to tension, allowing for the formation of strong, extensive surfaces like silk fibers or the robust sheets found in various biological structures.
What is the driving force for β-sheet aggregation?
The primary driving forces are chemical bonds, specifically the hydrogen bonds that form between the amino acids. These bonds determine the intricate folding patterns and the stability of the secondary structure by allowing amino acid chains to adhere to each other in an orderly fashion.
Can you use amyloid β-sheets as materials?
Yes, proteins are considered extremely versatile biomaterials that can be designed with specific mechanical properties. Since beta-sheets provide exceptional strength (having inspired synthetic materials like Kevlar), they can be computationally designed and synthesized in a lab to create new functional biological materials, ranging from flexible to rigid.
Design a β-sheet motif that forms a well-ordered structure.
An effective design is the LS (Leucine-Serine) motif, identified as a critical and highly conserved component in the lysis proteins of various phages. A well-ordered design based on this motif would include: (1) a positively charged N-terminus, (2) a hydrophobic sequence rich in aromatic and beta-branched aliphatic residues, (3) the essential LS dipeptide for interaction, and (4) a phage-specific C-terminal domain.
Why are most molecular helices right-handed?
While nature predominantly favors right-handed (dextrorotatory) orientations, studying chirality is essential to understanding biological exceptions. For example, the work of Shuguang Zhang is recognized for contributing to the deciphering of left-handed RNA structures, challenging biological norms and expanding our understanding of the structural possibilities of macromolecules
Myoglobin is a classic all-helix protein; it is a ubiquitous type of protein, related to the heme group and has a low molecular weight, consisting of 150 amino acids. It is a protein present in cardiac and skeletal muscle. However, under destabilizing conditions, where it is found in a partially folded intermediate conformation, it forms amyloid fibrils. Additionally, it contains one heme group per molecule and has a structure similar to that of hemoglobin monomers
Another fact is that myoglobin is an iron-containing protein with a molecular mass of 18 kDa. It resembles hemoglobin, but it binds to one oxygen molecule instead of four
“Myoglobin is mostly formed of:
Leucine amino acids and, in smaller quantities, Alanine, Glutamate, Lysine
Globins are a family of small globular metalloproteins of about 150 amino acids in length and characterized by 8 α-helical segments (A-H) that exert a characteristic 3-over-3 folding, which is known as the globular structure.
The globin family includes eight different types of globins, but the best described and characterized in vertebrates are hemoglobin (Hb), myoglobin (Mb), cytoglobin (Cygb), and neuroglobin (Ngb).
Resolution: 2.07 Å
The solved structure contains three unique ligands: HEM, which is iron-containing Protoporphyrin IX; SO4, the Sulfate Ion; and NBN, or N-butyl Isocyanide. Regarding its structural classification family, according to RCSB, the protein belongs to the Oxygen Transport family.
Additionally, as established in our previous conversation, myoglobin is a member of the globin family, which is characterized by a specific alpha-helix folding that enables binding to the heme group.
Deep Mutational Scans
I use AI Gemini
Cpmand: Use ESM2 to generate an unsupervised deep mutational scan of myoglobin based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Result AI:
What is the identified pattern for the most significant mutation? The most significant log-likelihood ratio (LLR) was found for the mutation of Methionine (M) to Tryptophan (W) at position 1 (0-indexed position 0) of the human myoglobin sequence, with an LLR of -16.6641. This highly negative LLR suggests that the ESM2 language model predicts this substitution to be highly unfavorable or unlikely, possibly due to Tryptophan’s bulky nature disrupting local packing or stability at the N-terminus.
What are the summary findings from the myoglobin mutation scan? The unsupervised deep mutational scan provides a landscape of predicted mutational effects across the human myoglobin sequence. The heatmap visually represents LLRs for all possible single amino acid substitutions, where largely negative LLRs indicate positions strongly favoring the wild-type residue, and LLRs near zero or positive suggest higher mutational tolerance. Critical regions for function, such as the heme-binding pocket, are expected to show strong conservation (highly negative LLRs for substitutions).
How could the language model’s predictions be compared with experimental data? The language model’s predictions (LLR values) can be compared with experimental deep mutational scanning (DMS) datasets using several strategies:
Correlation Analysis: Calculate Pearson or Spearman correlation between LLRs and experimental fitness scores.
ROC/Precision-Recall Curves: Evaluate the model’s ability to classify deleterious or beneficial mutations based on LLRs.
Specific Hotspot Validation: Directly compare predicted LLRs for critical mutations (e.g., M1W) with their known experimental effects.
Structural Context: Analyze if predicted deleterious mutations align with residues important for 3D structure, heme coordination, or core packing.
Part D. Group Brainstorm on Bacteriophage Engineering
Individual proposal:
Project goals
Increased Toxicity (Hard): Optimize the lytic kinetics to trigger faster host cell lysis by bypassing the DnaJ-dependent “damping” mechanism.
Increased Stability (Easy): Redesign the N-terminal and transmembrane domains to prevent proteolytic degradation and ensure robust protein accumulation
. Generative Sequence Design (Tool: Evo 2)
Approach: Utilize the Evo 2 genome language model to generate a library of novel MS2 L variants. We will specifically prompt the model to design Lodj-like variants (L overcomes DnaJ) by truncating or modifying the N-terminal Domain 1, which normally slows down lysis through its interaction with host DnaJ.
Reasoning: Evo 2 has demonstrated the ability to navigate novel evolutionary spaces and generate viable phages with faster lysis kinetics than natural templates. This allows us to access sequence diversity beyond the 67 unique mutations identified in natural screens.
2. Sequence Stability Optimization (Tool: ProteinMPNN)
Approach: Use ProteinMPNN to perform inverse folding on the core transmembrane domain (TMD) of the generated candidates.
Reasoning: Many missense mutations in Domain 1 and the TMD lead to accumulation defects due to instability. ProteinMPNN can redesign sequences to fit the specific 3D backbone required for membrane insertion while optimizing for thermodynamic stability.
3. Functional Motif Tuning (Tool: ESM-2 / ESM-3)
Approach: Use the ESM-2/3 protein language models to extract embeddings and perform in silico mutagenesis on the essential Leu48-Ser49 (LS) motif.
Reasoning: The LS motif is the core of the essential protein-protein interaction domain. ESM models can identify which amino acid substitutions in the surrounding Domain 2 and Domain 4 preserve the critical hydrophobic and polar character necessary for function while maximizing toxic effect.
4. Oligomerization Verification (Tool: AlphaFold-Multimer)
Approach: Use AlphaFold-Multimer to predict the ability of designed variants to assemble into high-order oligomeric complexes (decamers or higher).
Reasoning: MS2 L must form large membrane-disrupting clusters (clusters of at least 10 monomers) to cause cytoplasmic leakage. AlphaFold-Multimer can validate if the designed mutations at the TMD interface promote or hinder this essential assembly.
II. Potential Pitfalls
The “Unknown Target” Problem: While we know MS2 L interacts with DnaJ, its definitive membrane-embedded host target is still unknown. Without a clear target structure, using tools like BindCraft or AlphaFold-Multimer to optimize heterotypic protein-protein interactions is speculative and relies on the assumption that L forms homomeric pores.
Toxicity vs. Titer Trade-off: Increasing toxicity (faster lysis) often leads to lower titers. If the L protein triggers lysis too early (e.g., 20 minutes earlier in Lodj mutants), the phage may not have enough time to assemble progeny virions, potentially making the therapy less effective overall.
III. Pipeline Schematic
[Input] Wild-type MS2 L Sequence (75 aa)
↓
[Step 1: Evo 2] Generate novel Lodj-style truncations/diversified C-terminal domains.
↓
[Step 2: ProteinMPNN] Redesign TMD for stability and robust membrane accumulation.
↓
[Step 3: ESM-2/3] Perform site-specific optimization of the LS motif and surrounding domains.
↓
[Step 4: AF-Multimer] Verify the variant’s ability to form high-order oligomeric clusters in the membrane.
↓
[Output] Top 5 Candidates for experimental synthesis and rebooting in E. coli C
The ipTM values observed serve as confidence markers from the AlphaFold model, representing the predicted reliability and strength of the binding interaction. Peptide 2, generated by PepMLM, is the most promising design with an ipTM of 0.82, which nearly coincides with the 0.85 score of the Known Binder. While none of the generated peptides technically surpass the known binder in this dataset, Peptide 2 demonstrates high potential for reducing mutant activity and could be a viable candidate for experimental validation, similar to the plaque assays used to test design effectiveness in the sources.
Peptide
Property
Prediction
Value
Unit
GIVEQCCTSICSLYQLENYCN
💧 Solubility
Soluble
1.000
Probability
GIVEQCCTSICSLYQLENYCN
🩸 Hemolysis
Non-hemolytic
0.099
Probability
GIVEQCCTSICSLYQLENYCN
🔗 Binding Affinity
Weak binding
5.322
pKd/pKi
GIVEQCCTSICSLYQLENYCN
📏 Length
—
21
aa
GIVEQCCTSICSLYQLENYCN
⚖️ Molecular Weight
—
2383.7
Da
GIVEQCCTSICSLYQLENYCN
⚡ Net Charge (pH 7)
—
-2.28
—
GIVEQCCTSICSLYQLENYCN
🎯 Isoelectric Point
—
4.05
pH
GIVEQCCTSICSLYQLENYCN
💦 Hydrophobicity (GRAVY)
—
0.21
GRAVY
Peptide two (AHSGAVALEHGP) is the most similar to the known binder among the generated sequences. It achieved the highest ipTM score of the designed peptides at 0.820, which is remarkably close to the 0.850 of the known binder. Furthermore, its Pseudo Perplexity was 12.849, significantly lower than that of the reference binder. This peptide was predicted to bind at a “shallow pocket” [user summary].
ipTM and Affinity Correlation
A higher ipTM score generally correlated with a stronger predicted binding affinity, which is consistent with the expected interpretation of the AlphaFold-Multimer ipTM metric used to evaluate these interactions. This metric serves as an important confidence marker for the reliability of the predicted binding.
Diverse Binding Locations
The generated peptides showed various predicted binding locations, including a shallow pocket, surface-bound, near the N-terminus (relevant to the aggressive A4V mutation), and an interface with a loop region. The ability to target diverse sites is critical for achieving the specificity required to inhibit the toxic aggregations of mutant SOD1.
PART C: L-Protein Mutants
Rank
Residue_Index
Wild_Type_AA
Mutation_AA
LLR_Score
Genome_Position
Notes
1
50
K
L
2.561464
989
High-impact mutation
2
29
C
R
2.395427
574
Multiple mutations at residue 29
3
39
Y
L
2.241778
769
High LLR candidate
4
29
C
S
2.043150
575
Multiple mutations at residue 29
5
9
S
Q
2.014323
173
Moderate-impact mutation
6
29
C
Q
1.997049
573
Multiple mutations at residue 29
7
29
C
P
1.971028
572
Multiple mutations at residue 29
8
29
C
L
1.960646
569
Multiple mutations at residue 29
9
50
K
I
1.928798
987
Alternative mutation at residue 50
10
53
N
L
1.864930
1049
Lower but relevant candidate
Week 6 HW: Genetic circuits part 1
DNA ASSEMBLY
Phusion High-Fidelity PCR Master Mix Components
Phusion HF PCR Master Mix is provided as a 2X stock that is diluted to a 1X concentration in the final reaction. While the sources do not list every chemical ingredient, they indicate its purpose is to amplify specific DNA sequences (such as the amilCP gene and the mUAV backbone) with high accuracy.
Standard high-fidelity master mixes like Phusion typically contain:
Phusion DNA Polymerase: A highly accurate enzyme with proofreading activity to minimize mutations.
dNTPs (Deoxynucleotide Triphosphates): The building blocks (A, T, C, G) used to synthesize the new DNA strand.
Buffer: Maintains the optimal pH and ionic strength for the enzyme.
$MgCl_2$: A necessary cofactor for the DNA polymerase to function.
Factors Determining Primer Annealing Temperature
The annealing temperature is critical for ensuring primers bind specifically to their targets. Key factors mentioned in the sources include:
Melting Temperature ($T_m$): The annealing temperature is generally set 2–5°C below the lower $T_m$ of the primer pair.
Binding Region Length: Primers should ideally have an 18–22 bp core binding region.
GC Content: The binding region should ideally have a GC content of 40–60%.
GC Clamp:Adding 1–2 G or C bases at the 3’ end promotes specific binding.
Secondary Structures: Avoiding strong hairpins or dimers ensures the primer is available to bind the template.
PCR vs. Restriction Enzyme Digests
Both methods generate linear DNA fragments but differ significantly in application:
Protocol: a)PCR uses thermal cycling (denaturation, annealing, extension) and DNA polymerase to synthesize millions of new copies of a specific region.
b)A restriction enzyme digest uses specific proteins to cut existing DNA at recognized sequences.
When to Use PCR: PCR is preferable when you need to introduce mutations (like the chromophore color changes), add specific overhangs for assembly, or amplify a gene from a template.
When to Use Restriction Digests: Digests are often used to linearize a circular vector or as a diagnostic tool (e.g., using DpnI to remove methylated template DNA) to verify fragment sizes on a gel.
Ensuring Appropriateness for Gibson Cloning
To ensure your digested and PCR-amplified sequences are ready for Gibson Assembly, you must verify:
Overlaps: Adjoining fragments must have 20–40 bp of sequence identity (overhangs) at their ends.
Orientation: Confirm that all fragments have the correct 5′→3′ orientation with matching overlaps to form a circular plasmid.
Purity: Use DpnI to digest the original methylated template plasmid. Then, perform DNA purification (silica adsorption) to remove enzymes and buffers.
Verification: Run a diagnostic gel to confirm the fragments are the correct size and measure concentration (ideally >30 ng/µL).
Molar Ratio:Use a 2:1 (insert:vector) molar ratio for the assembly reaction.
Plasmid Entry During Transformation
During transformation, the sources explain that the cell membrane is “opened up” by shocking the cells using either heat shock (abrupt temperature change) or electroporation (high voltage). This process creates pores in the bacterial cell wall. Once these pores are present, the plasmid DNA enters the E. coli cells by diffusion.
Alternative Assembly Method: Golden Gate Assembly
Golden Gate Assembly (GGA) is a molecular cloning method that allows for the simultaneous, seamless assembly of multiple DNA fragments.
It utilizes Type IIS restriction enzymes, which cut DNA at a specific distance away from their non-palindromic recognition sites. This unique feature allows designers to create custom 4-base overhangs that do not contain the original restriction site. Because the restriction sites are placed such that they are “cut off” during the reaction, the assembled product cannot be re-digested, driving the reaction toward the final circular plasmid. This makes GGA a highly efficient “one-pot” reaction where digestion and ligation occur simultaneously. It is particularly popular for modular cloning systems (like MoClo) because of its high fidelity and speed.
Define Parts: Select the backbone and the inserts. Benchling will automatically identify the 4-bp overhangs.
Verify Overlaps: Ensure the overhangs are complementary (e.g., the 3’ end of Part 1 matches the 5’ end of Part 2).
Finalize: Click “Assemble” to generate the new virtual plasmid and verify the sequence.
Asimov Kernel
Pending …
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Part 1 HW
Intracellular Artificial Neural Networks (IANNs), also known as neuromorphic circuits, provide significant advantages over traditional genetic circuits that rely on Boolean (digital) logic:
a) Advantages of IANNs
Biological Substrate Compatibility:** While digital logic attempts to force binary “on/off” behavior onto cells, IANNs operate through analog computation, which is much closer to the natural language of biology.
Handling Non-linear Complexity:** Biological systems naturally manage highly non-linear and complex input-output relationships that Boolean logic oversimplifies. IANNs allow for the capture of these non-linearities and non-monotonic behaviors more robustly.
Precision in Decision Boundaries: Unlike digital logic, which only recognizes “high” or “low” thresholds, IANNs can be programmed to respond to specific analog relationships (for example, activating only when two inputs are equal or when a weighted combination exceeds a bias), allowing for much more exact classification of cellular states.
Flexibility and Scalability: The behavior of the circuit can be adjusted simply by modifying the translation rates of the components, allowing decision boundaries to be shifted without needing to redesign the entire system.
b) Useful Application: Cancer Cell Classifier
A primary application for IANNs is the creation of high-precision cell classifiers for cancer immunotherapy. Because there is rarely a single “magic” biomarker to distinguish a cancer cell from a healthy one, a sophisticated program is required to evaluate multiple signals simultaneously.
c) Input/Output Behavior
Inputs: The circuit uses multiple intracellular biomarkers as inputs, commonly microRNAs (miRNA). For instance, a classifier can be configured to detect a profile where specific miRNAs (like miRNA-21) are high and others (like miRNA-141) are low.
Processing (Computation): The circuit utilizes elements called “sequestrons” which function as artificial neurons. These perform a weighted summation of inputs: positive signals increase the production of a messenger RNA (mRNA), while negative signals (using proteins like endoribonucleases) sequester that mRNA to prevent its translation.
Non-linear Activation: The circuit implements an activation function known as a ReLU (Rectified Linear Unit). If the negative weighted inputs exceed the positive ones, the output is zero; otherwise, the output grows linearly with the difference.
Output:** If the network classifies the cell as “cancerous” after processing these analog signals, it triggers the expression of a therapeutic agent, such as a killer protein or inflammatory cytokines (e.g., IL-12) to destroy the tumor.
d) Limitations of IANNs
Despite their potential, IANNs face several technical challenges:
Node Count Restrictions: There is a physical limit to how many synthetic proteins a cell can express without affecting its viability; most current designs are limited to 10 nodes or fewer.
Environmental Influences and Heterogeneity: External factors such as the immune system and the inherent variability between individual cells can alter circuit reliability and outcomes.
Modeling Complexity: Fully capturing biological realism (such as the spatial clustering of inputs) in computational models remains difficult and often requires multiple iterative cycles of experimentation to achieve accuracy.
Part 2 HW
a) Examples of Fungal Materials and Their Uses
Fungal materials, or biomaterials, are grown from microbes (primarily fungi) rather than being industrially manufactured. Some examples include:
Alternative Leathers Used in the fashion industry for “biocouture” and luxury garments.
Luxury Packaging and Insulation:** Due to their ability to grow on agricultural substrates (straw, wood chips), they are used as an alternative to polystyrene (Styrofoam).
Mycelium Bricks: These have been used to build architectural-scale structures, such as the “Hy-Fi” Pavilion at MoMA in New York.
Space Habitats: NASA is investigating their use to build structures on the Moon or Mars, taking advantage of the fact that fungi can be grown in situ.
b) Advantages and Disadvantages Compared to Traditional Materials
Advantages:
Superior Insulation:** These materials are thermally and acoustically insulating; blowtorch tests demonstrate that heat does not pass through the material even after a minute of direct exposure.
Lightweight and Sustainable:** They are extremely light and grown from agricultural waste, which significantly reduces the carbon footprint.
Space Logistics:** For space missions, it is far more efficient to transport a small vial of spores than tons of heavy construction materials.
Disadvantages:
Production Time: The growth process is not immediate; it can take weeks for the mycelium to colonize the substrate and months if the fungus is required to fruit to produce spores.
Fragility: While more resilient than some other biomaterials like bio-cement, some states of the material can be brittle before being processed or compressed.
Biological Complexity: Unlike bacteria, the fungi used for materials are genetically distant from traditional laboratory models, making their manipulation more difficult.
c) Genetically Modifiable Functions and Their Purpose
Synthetic biology seeks to imbue these materials with new properties that they do not naturally possess:
Physical Properties: Modifying fungi to make the material stronger, more flexible, or more resistant.
Biosensing: Altering the fungus to change color (for example, by producing melanin) in response to pollutants or sugars in the water.
Growth Control: Programming fungi to grow in specific shapes or predetermined patterns.
Extreme Adaptation: For space applications, researchers aim to make them radiation-resistant and capable of feeding on human waste or lunar soil (regolith).
d) Advantages of Synthetic Biology in Fungi vs. Bacteria
Structural Capacity: While bacteria are typically unicellular, fungi form complex networks (mycelium) that act as a biological glue to create solid objects and large structures, something bacteria cannot achieve on their own in the same way.
Neuromorphic Computation: The sources highlight that, unlike digital logic (typical in bacteria), one can implement Intracellular Artificial Neural Networks (IANNs). These allow for analog signal processing that is much more complex and closer to the natural language of biology.
Decision Complexity: With just 10 nodes of an intracellular neural network, a fungal system could perform extremely sophisticated biomonitoring classifications that surpass the simple Boolean logic systems commonly used in bacteria.
Diagram
flowchart LR
%% Inputs
X1["X1"]
X2["X2"]
%% Procesos externos
TX1["Tx"]
TX2["Tx"]
X1 --> TX1
X2 --> TX2
%% Célula
subgraph CELL[" "]
TI["Tl (Csy4)"]
TXm["Tx (Reporter mRNA)"]
SIGNAL["mRNA → Tl"]
%% Regulación
TI --| − | TXm
TXm --> SIGNAL
%% Activación positiva
TXm -->| + | SIGNAL
end
%% Salida
OUT["Fluorescent Protein"]
SIGNAL --> OUT