Week 4 Review: Protein Design Part I
Week 4 — Protein Design Part I
At a glance. This guide covers the amino-acid alphabet, secondary structure geometry, β-sheet aggregation and amyloid, and the modern ML protein design approaches and tools then applies f it to E. coli DHFR (Part B/C) and the MS2 L-protein engineering proposal (Part D) as worked examples. Written as an inegrated field primer.
Foundations of protein chemistry: alphabet, structure, and design
Part I is Part A of the Week 4 deliverable for HTGAA Spring 2026; Part II contains B, C, and D (sequence/structure analysis, ML protein design, MS2 L-protein engineering proposal) will be added as those sessions complete.
Course: HTGAA Spring 2026 Lecture (Tues, Feb 24, 2026): Thras Karydis, Jon Kaufman — Protein Design Part I Recitation (Wed, Feb 25): Allan Costa — Protein folding Author: Fiona (Committed Listener BioPunk)
Why should we study proteins so deeply?
Proteins do nearly all of the underlying functions in biology: catalysis, structure, transport, signaling, immunity. So being able to design a protein well essentially equates to ability to design a bespoke biological function, that could be a new therapuetic, a sensor , an activator or blocker.
For most of the field’s history, “designing” a protein meant rational point-mutation campaigns on a known scaffold. This was slow, limited in scope, and dependent on hard-won structural intuition. The last five years of advances in in silico protein design models has signficantly reduced that timeline. Modern protein language models read the fitness landscape from hundreds of millions of natural sequences; structure predictors fold sequences in seconds; inverse-folding networks rewrite sequences for any backbone. Together they make computational protein engineering tractable for problems that used to require multi-year wet-lab directed evolution experiments.
Before we get to those tools (Parts B, C, D), we need to full understand the substrates they operate on: the amino-acid alphabet, the geometric constraints on protein structure, and the thermodynamics of folding and aggregation. This is Part A. The conceptual questions in this section are drawn from Shuguang Zhang’s HTGAA prompt set, and answered as a connected story rather than an isolated Q&A, so you dear reader, landing on this page can follow it as a handy topic guide in itself.
Key theme to remember: running through everything: proteins are constrained polymers. Their building blocks are stereochemically restricted (only L-amino acids in nature, only ~20 of them, only one ribosomally-installable scaffold per residue). Those constraints set what proteins can do which anchors what synthetic biologists can engineer from these organic building blocks.
1. The amino-acid alphabet
1.1 The shared scaffold
Every amino acid except proline shares the same backbone scaffold: an α-carbon flanked by an amino group, a carboxyl group, a hydrogen, and a variable side chain (R). Peptide bonds link them via dehydration condensation; the resulting peptide bond is planar (partial double-bond character locks the C–N axis). That constrains the protein backbone to two free torsion angles per residue — φ (around N–Cα) and ψ (around Cα–C) — and the entire shape of secondary structure follows from the geometry of those allowed angles.
The 20 side chains span the chemistries biology needs: hydrophobic (A, V, L, I, M, F, W), polar uncharged (S, T, N, Q, Y, C), positively charged (K, R, H), negatively charged (D, E), and special-case residues — Gly (achiral, maximum flexibility), Pro (rigid imino acid that disrupts helices), Cys (forms disulfides). Proline is the structural exception: its side chain loops back to the backbone nitrogen forming a pyrrolidine ring, locking φ at roughly −60° ± 15°. That single deviation makes proline a helix-breaker, a Type II’ turn nucleator (when D-Pro is used), and the dominant residue of the polyproline II helix that builds collagen.
All 20 amino acids in life are L-stereoisomers. Glycine is achiral. This single geometric constraint is the most consequential one in protein structure: it’s why the α-helix in evolved proteins is right-handed, why D-amino-acid peptides invert handedness, and why every helical biological structure ultimately inherits the same handedness from its building blocks. We come back to this in Section 2.
1.2 The quantitative scale of protein chemistry
A useful intuition for protein chemistry comes from working out the throughput. If you eat 500 g of meat and treat it as pure protein with average residue mass 100 Da:
500 g ÷ 100 g/mol = 5 mol → 5 × 6.022 × 10²³ ≈ 3 × 10²⁴ amino acid residues.
Two caveats. Real beef is only ~20% protein by mass (~70% water, ~10% fat), so adjusted for actual protein content the number drops to ~6 × 10²³ residues — still close to Avogadro’s number. And these are residues incorporated in protein, not free amino acids; they’re released by digestive proteolysis before they enter the bloodstream. A single 500 g serving carries roughly Avogadro’s-number-worth of amino-acid monomers, which calibrates intuition for the throughput of protein chemistry on the human scale.
1.3 Why species identity is genomic, not dietary
A frequent question — why don’t humans become cows from eating beef? — has a clean answer: species identity is genomic, not dietary.
When you eat beef, digestive proteases such as trypsin hydrolyze cow proteins into free amino acids and short peptides before they cross into the bloodstream. Those amino acids enter a species-agnostic pool — a Phe is a Phe whether it came from a cow, a tuna, or a yeast cell. What happens next is the key: your ribosomes don’t read cow mRNA. They read your mRNA, encoded by your genome, and assemble your proteins from the recovered amino-acid pool. The cow contributed the building blocks; the assembly blueprint is yours.
Two qualifiers. Essential amino acids (Phe, Trp, Lys, Met, Thr, Val, Leu, Ile, and His — the last conditionally essential) are residues humans cannot synthesize in adequate amounts and must obtain from diet — diet matters for what you can build, not for what you become. And post-translational modifications (glycosylation, hydroxylation, etc.) are also genome-encoded — cow collagen and your collagen differ at the modification level too, not just at the sequence level. The principle is template-directed assembly: diet supplies monomers, identity comes from the blueprint.
1.4 Why ~20 canonical amino acids
All known life uses the same canonical 20 amino acids in ribosomal protein synthesis, with two genetically-encoded exceptions: selenocysteine (Sec) is encoded by recoded UGA in selenoproteins across all three domains; pyrrolysine (Pyl) is encoded by recoded UAG in some methanogenic archaea. No organism has ever been found using a fundamentally different ribosomal alphabet — strong evidence that 20 + Sec + Pyl is a deep evolutionary attractor, not just an unsampled corner.
Every amino acid shares the same backbone — α-carbon flanked by NH₂, COOH, H, and a variable side chain R. The peptide bond linking them is planar (partial double bond), leaving two free torsion angles per residue: φ (N–Cα) and ψ (Cα–C). Everything about secondary structure follows from which (φ, ψ) pairs are sterically allowed.

Why this particular set? Three mutually-reinforcing constraints filter the space:
- Prebiotic availability. Miller–Urey (1953) experiments and the Murchison meteorite (1969) both produced ~10 of the canonical 20 — Gly, Ala, Asp, Glu, Val, Leu, Ile, Pro, Ser, Thr — under abiotic conditions. The simpler half of the alphabet was available before life had to encode anything.
- Ribosomal compatibility. The peptidyl-transferase center of the ribosome has stereochemical and steric tolerances limiting which side chains it can incorporate accurately. β-amino acids and very bulky residues are translated poorly even today. Whatever life encoded had to fit through this geometric filter.
- Biosynthetic cost vs. chemical novelty. Once translation existed, selection favored adding amino acids that gave new chemistry not yet in the set, even at high biosynthetic expense. Trp and Tyr — the only sources of aromatic chemistry — are biosynthetically expensive and were almost certainly late additions. Trifonov (2000) reconstructs an inferred temporal order: small-and-cheap first, complex-and-functional later.
A fourth, complementary constraint is codon space: a 4-base, 3-position genetic code yields 64 codons; subtract three stops and you have 61 sense codons distributed across 20 amino acids with redundancy. Many more than ~20 would over-strain the ribosome’s codon-recognition machinery; many fewer would waste coding capacity.
These constraints together explain why these 20, but the relative weighting among them remains debated — Higgs & Pudritz (2009) emphasize prebiotic availability; Wong (2005) emphasizes codon-coevolution; Crick’s “frozen accident” (1968) holds that the specific 20 was partly arbitrary at the moment of code lock-in.
Engineered exceptions are real. Schultz, Chin, and the Church group’s Genomically Recoded Organism work (Lajoie 2013) have built orthogonal aminoacyl-tRNA synthetase / tRNA pairs that let engineered organisms incorporate over 200 non-canonical amino acids ribosomally. The ribosome is permissive enough; evolution simply didn’t go there.
So: 20 is not a physical limit. It is the biological-historical attractor produced by prebiotic supply, ribosomal compatibility, and selection-cost trade-offs, layered onto a finite codon space.
1.5 Designing non-canonical amino acids
Engineered ribosomal incorporation has produced over 200 non-canonical amino acids (NCAAs). The field is largely Peter Schultz’s (Scripps) and Jason Chin’s (MRC-LMB); the Church group’s Genomically Recoded Organism (Lajoie 2013) removed all UAG stop codons from E. coli to free that codon for NCAA assignment without translational crosstalk.
A novel amino acid must clear three filters to be installable:
- Stereochemical compatibility with the ribosome — must be α-amino (canonical backbone scaffold), with side-chain bulk roughly within the natural envelope. β-amino acids and very large side chains translate poorly.
- Orthogonal aminoacyl-tRNA synthetase / tRNA pair — a dedicated aaRS that doesn’t cross-react with native AAs, paired with a tRNA decoding a non-natural codon. Methanocaldococcus jannaschii TyrRS (Schultz) and archaeal pyrrolysyl-tRNA synthetase (Chin) are the workhorses.
- A free codon to assign — typically a reassigned stop codon (UAG amber, UGA opal).
Established NCAAs include p-azidophenylalanine (click chemistry), p-iodophenylalanine (X-ray phasing), photo-caged Lys/Tyr/Cys (light-activated control), and selenomethionine (heavy-atom for crystallography).
Two design proposals:
- Photoswitch-Phe (azoPhe). Phenylalanine analog with the para-H replaced by an azobenzene group. UV flips the azobenzene between trans (extended) and cis (~3 Å shorter); visible light reverses it. Use case: optically toggleable local geometry — caged active sites, photoswitchable PPI inhibitors, kinetic studies of folding intermediates. Real precedent: Beharry & Woolley (2011).
- Bipyridyl-Ala (BiPyAla). Alanine analog with a 2,2′-bipyridyl side chain replacing the methyl. Creates a bidentate metal-chelation site at a single residue. Drop Cu²⁺ or Fe²⁺ in and you have a redox-active or catalytic center installed on an arbitrary protein scaffold. A first-generation version exists in the Schultz lab; higher-affinity and pH-tuned variants remain open design space.
NCAAs are not the first tool for protein engineering — point mutations to canonical residues are simpler and the modern ML toolkit handles them well. NCAAs become useful when you want to covalently lock a fold (engineered disulfide, photo-crosslink) or install a sensor (FRET pair, EPR spin label) without disrupting the native sequence. Relevant for the L-protein engineering work later in the course.
2. Secondary structure and the chirality of life
Plot every residue’s (φ, ψ) torsions on a 2D plot — the Ramachandran plot — and only ~10% of the space is sterically allowed for L-amino acids. Most is forbidden by atomic clashes between the side chain and the backbone. Three allowed regions dominate: the lower-left (right-handed α-helix), the upper-left (β-sheet), and a small upper-right region (left-handed α-helix) tolerated only by glycine. Secondary structure is what you get when consecutive residues stack inside one of these allowed islands.
This single fact — the Ramachandran plot for L-amino acids — explains both the dominance of right-handed helices and the handedness of mirror-image polypeptides.

2.1 Why most molecular helices are right-handed
Organic life is homochiral, and the homochirality of biological building blocks fixes helical right- handedness across all macromolecular structures.
For protein α-helices specifically: living organisms use L-amino acids exclusively in ribosomal protein synthesis. L-side chains clash sterically with the backbone in any conformation other than the right-handed α-helix; the (φ, ψ) coordinates of a left-handed α-helix sit in a sterically forbidden region of the L-amino-acid Ramachandran plot. Result: every α-helix in every natural protein on Earth is right-handed, with rare local exceptions at glycine residues (Gly is achiral and tolerates either handedness).
The same homochirality argument generalizes beyond proteins. DNA uses D-deoxyribose; the canonical B-DNA double helix is right-handed (Z-DNA, a left-handed form, is a rare local conformation in special sequences). RNA uses D-ribose; the A-form helix is right-handed. Collagen is a striking edge case: each polyproline II chain is itself left-handed, but three such chains coil together into a right-handed triple super-helix. The chain-level handedness is still dictated by L-amino acid stereochemistry.
| Polymer | Sugar | Helix sense |
|---|---|---|
| Proteins (α-helix) | — (L-AAs) | Right-handed |
| B-DNA | D-deoxyribose | Right-handed |
| A-RNA | D-ribose | Right-handed |
| Collagen (each chain) | — (L-AAs, Pro-rich) | Left-handed chain → right-handed triple helix |
The deeper question — why life is homochiral in the first place — remains unsettled. Candidate explanations include parity violation in weak-force interactions producing tiny enantiomeric excess, asymmetric photolysis by circularly polarized light from neutron stars, and stochastic symmetry-breaking amplified by autocatalytic networks. Whatever the original cause, once L-amino acids and D-sugars were locked in by early biology, all derived helical structures inherited the corresponding handedness.
So molecular helices are right-handed in life because biological building blocks are homochiral, and that homochirality determines which Ramachandran (or analogous geometric) region is accessible.
2.2 D-amino acids and mirror-image proteins
If you build a polypeptide entirely from D-amino acids instead of L, the Ramachandran plot mirrors. The “L-allowed” lower-left region maps to the upper-right; helices wind in the opposite sense — left-handed.
The defining structural features of the α-helix are otherwise preserved: the i → i+4 hydrogen-bond pattern, the 3.6-residue periodicity, and the 5.4 Å pitch all hold. Only the handedness flips. Stephen Kent’s group (U. Chicago) chemically synthesized full-length D-VEGF — the D-amino-acid mirror of vascular endothelial growth factor — and crystallized it in the expected mirror-image structure with left-handed helices. Some natural antimicrobial peptides containing D-residues also exhibit local left-handed helical character.
Practical upside: D-peptides are proteolysis-resistant — natural proteases evolved for L-substrates.
A racemic polypeptide (mixed L and D residues) typically forms neither helix — consistent stereochemistry across consecutive residues is required to satisfy the Ramachandran constraints. For an accessible primer on racemic and mirror-image polypeptides, see Mirror Image Proteins (Mandal & Kent, 2017) and the Wikipedia article on Racemic crystallography.
A design-relevant insight from this: D-amino-acid peptides are proteolysis-resistant because natural proteases evolved to cleave L-peptides. Mirror-image therapeutic peptides are an active research area, particularly for oral or systemic delivery where stability is rate-limiting.
3. β-sheets, aggregation, and amyloid
A β-strand is an extended near-zigzag backbone with (φ, ψ) ≈ (−120°, +120°). Strands H-bond laterally to each other to form a β-sheet — parallel (strands run N→C in the same direction; H-bonds slant) or antiparallel (strands run opposite; H-bonds are roughly perpendicular and shorter, making antiparallel sheets slightly more stable). Side chains alternate above and below the sheet plane. Amphipathic β-strands (alternating hydrophobic-hydrophilic patterning at i, i+2) drive sheet formation in aqueous environments.
Two structural facts about β-sheets matter for everything that follows: edges are unsatisfied and faces can be hydrophobic. Both feed the aggregation thermodynamics in §3.1.

3.1 Why β-sheets aggregate
TLDR:
β-sheets have two structural vulnerabilities:
- Unsatisfied edge H-bonds — every sheet exposes a row of unpaired N–H donors and C=O acceptors along its edge. Recruiting another sheet satisfies them (~3–5 kcal/mol per H-bond, enthalpic).
- Hydrophobic faces — alternating hydrophobic side chains on the sheet face get buried when two sheets stack, releasing ordered water (entropic gain — the classical hydrophobic effect, Kauzmann 1959).
Together these drive sequences toward the cross-β amyloid fold — the deepest free-energy minimum on the folding landscape for many proteins.

Full Description
β-sheets aggregate because they are structurally incomplete on their edges. Each strand carries unpaired backbone hydrogen-bond donors (N–H) and acceptors (C=O), so a single β-sheet exposes a row of “lonely” hydrogen bonds along its edge. Adding a second β-sheet alongside lets those bonds pair up — the system’s energy drops and the configuration becomes more stable.
The driving force has two main components. Satisfying the unpaired hydrogen bonds (enthalpic): each new H-bond between sheets releases ~3–5 kcal/mol of heat. The hydrophobic effect (entropic): β-sheet faces often present alternating hydrophobic side chains; burying these against another sheet’s hydrophobic face releases ordered water molecules from around them, raising solvent entropy. This is the classical hydrophobic effect (Kauzmann 1959, Tanford 1962) — counter-intuitively, it’s entropy, not enthalpy, that drives most hydrophobic interactions in water.
There is one unfavorable contribution: the protein chain loses conformational entropy when it locks into a rigid stacked structure. This cost is outweighed by the gains above, but it’s why aggregation is concentration- and time-dependent rather than instantaneous.
Together, these forces drive sequences toward the cross-β amyloid fold — stacked β-sheets with strands oriented perpendicular to the fiber axis, side chains interdigitated between sheets, and all backbone hydrogen bonds satisfied along the fiber length. For many proteins, cross-β is the deepest free-energy minimum on the folding landscape, deeper even than the native fold. Once a sequence enters this minimum, escape requires aggressive denaturation — this is why amyloid is essentially irreversible under physiological conditions.
This also explains why amyloid-forming proteins span enormous sequence space: aggregation is driven by backbone properties (always present) plus generic hydrophobic contributions, not specific side-chain chemistry. Any sufficiently long, marginally soluble polypeptide can in principle form cross-β, given time and concentration. Knowles, Vendruscolo & Dobson (2014) is the canonical review.
3.2 Amyloid disease, propagation, and materials
Why cross-β architecture and aggregation drives diseases across tissues:
| Protein | Disease | Tissue |
|---|---|---|
| Aβ | Alzheimer’s | Brain |
| α-synuclein | Parkinson’s | Dopaminergic neurons |
| PrP^Sc | Prion disease (CJD/BSE) | Brain |
| Transthyretin | Cardiac amyloidosis | Heart |
| IAPP | Type 2 diabetes | Pancreas |
Disease-associated amyloidogenic proteins reflect the genericness of the cross-β attractor: Aβ in Alzheimer’s, α-synuclein in Parkinson’s, prion protein PrP in CJD/BSE, islet amyloid polypeptide in type 2 diabetes, transthyretin in cardiac amyloidosis, tau in tauopathies, huntingtin in Huntington’s. Different sequences, different organs, same cross-β architecture.
Pathology has two drivers. Toxic oligomeric intermediates — small soluble oligomers on the aggregation pathway, not the mature fibers — are the primary cytotoxic species. They permeabilize membranes, disrupt mitochondria, and trigger inflammation. Tissue mechanical damage comes from accumulated mature fibers, producing the histological plaques.
Seeding and templated nucleation
The progressive nature of amyloid disease is explained by seeding. Aggregation is bottlenecked by formation of the first cross-β nucleus; once seeded, monomers dock onto existing fibril edges and get templated into the same geometry. Aggregation becomes autocatalytic:
- Prion infectivity (Prusiner 1997 Nobel) — PrPSc converts native PrPC by template-directed refolding. A protein alone is the infectious agent.
- Cell-to-cell spread of α-synuclein, tau, and Aβ — pre-formed fibrils released from one neuron seed misfolding in the next, explaining anatomical-pathway progression over years.
- Secondary nucleation (Knowles 2009, Cohen 2013) — fibrils catalyze new nucleation on their surfaces, driving explosive late-stage growth.
Aging accelerates the cycle by weakening protein quality-control machinery (chaperones, proteasome, autophagy), letting rare nucleation events escape.
Therapeutic strategies — capping the chain
The same edge-pairing logic that drives aggregation suggests how to stop it. Five general approaches:
- N-methylated capper peptides. A short β-strand peptide docks onto the fibril edge but has methylated backbone N–Hs on its outward face — no donors for the next incoming monomer. Chain terminates. Demonstrated for Aβ, IAPP, and tau (Soto, Gazit, Eisenberg groups).
- Native-state stabilizers. Keep monomers out of the aggregation pool. Tafamidis (FDA-approved) stabilizes the transthyretin tetramer so it never dissociates into amyloidogenic monomers — the most successful clinical anti-amyloid strategy to date.
- Anti-oligomer antibodies target toxic intermediates. Aducanumab and lecanemab in Alzheimer’s; modest, contested efficacy but the mechanism is right.
- Anti-fibril-surface chaperones block secondary nucleation. The Brichos domain coats Aβ fibril surfaces; endogenous Hsp104 (yeast) and Hsp70/40 (mammals) disaggregate via ATP.
- Small-molecule disaggregators like EGCG, curcumin, and resveratrol bind hydrophobic stacking surfaces and remodel amyloid in vitro; clinical translation has been limited by bioavailability.
Amyloid as engineering material
The properties that make amyloid medically dangerous also make it materially valuable:
Engineering upside. Cross-β amyloid has a ~10 GPa Young’s modulus (silk-comparable), tensile strength rivaling steel by mass, resistance to proteolysis / heat / solvents, and spontaneous self-assembly from solution. Amyloid is an active engineering platform.
Natural functional amyloids show biology already exploits the architecture: curli fibers in E. coli biofilms, Pmel17 organizing melanin in melanosomes, fungal HET-s prion-like signaling, hormone storage in pituitary granules. Engineered applications include drug-delivery scaffolds, biosensors, conductive nanowires (cytochrome-c-amyloid hybrids, MIT Lu lab), and 3D-printable hydrogels. Recombinant silk proteins (e.g., AMSilk, Spiber) use the closely related β-sheet–crystallite architecture of natural silks, which is structurally distinct from canonical cross-β amyloid but exploits the same edge-pairing thermodynamics.
The elegant corollary: the same capping logic that prevents disease in vivo lets materials engineers tune fiber length and end-group chemistry in vitro.
Takeaway: Cappers are a key design parameter of engineering with β-sheets. The disease-relevant stability of cross-β is exactly the property that makes it useful if you can also engineer in control of capping where the chain stops.
4. Designing β-sheet motifs
A “well-ordered” β-sheet motif folds into a single intended sheet without unfolding or recruiting another sheet to aggregate. Four design rules govern the geometry to ensure this:
| Rule | Why it matters |
|---|---|
| Alternating hydrophobic-hydrophilic at i, i+2 | Creates the amphipathic face needed for strand pairing |
| Type I’ or II’ turn for antiparallel hairpins | Only turn types that fit the antiparallel geometry without strain |
| Edge-cap charged/aromatic residues | Blocks lateral H-bonding to a second sheet — most important anti-aggregation rule |
| 12–16 residues minimum | Smaller hairpins lack enough contacts to fold stably |
Turn geometry detail. Both Type I’ and II’ turns require positive φ at i+1 — sterically forbidden for L-amino acids. Only Gly (achiral) and D-Pro (locked positive φ) satisfy this. D-Pro-Gly is the strongest synthetic nucleator (Stanger & Gellman 1998).
Design Rules Explained
- Alternating hydrophobic-hydrophilic patterning at i, i+2 along each strand. The hydrophobic face must be buried (against another strand or core) or capped to prevent recruiting another sheet.
- Tight turns. β-hairpins (two antiparallel strands) require Type I’ or II’ turns — the only turn types that fit antiparallel topology without backbone strain. Asn-Gly is the most common natural Type I’ turn nucleator; D-Pro-Gly is the strongest synthetic Type II’ turn (Stanger & Gellman 1998).
- Edge capping. Place charged or aromatic residues on strand edges to disrupt lateral hydrogen bonding to other sheets — the single most important rule for preventing aggregation.
- Length and topology. β-hairpins (~12–16 residues) are the smallest stable motif. Larger antiparallel β-meanders (Greek-key, β-barrel) give thermostability.
Sidebar: β-turn types
A β-turn is a 4-residue motif that reverses the direction of the polypeptide chain — every β-hairpin is built on one. Turns are classified by the (φ, ψ) torsions of the two central residues (positions i+1 and i+2 of the four-residue stretch).
| Type | φ(i+1) | ψ(i+1) | φ(i+2) | ψ(i+2) | Required residues |
|---|---|---|---|---|---|
| I | −60° | −30° | −90° | 0° | flexible |
| II | −60° | +120° | +80° | 0° | Gly at i+2 |
| I’ | +60° | +30° | +90° | 0° | Gly at i+1 |
| II’ | +60° | −120° | −80° | 0° | Gly or D-Pro at i+1 |
The “prime” types are mirror images of the unprimed — same angles with signs flipped. Antiparallel β-hairpins force the chain to U-turn with specific handedness, so Type I’ and II’ turns are the natural fit; Types I and II produce slightly strained hairpin geometry. Both prime types require a positive φ at i+1 — sterically forbidden for L-amino acids (the same Ramachandran exclusion that forbids the left-handed α-helix in §2.1). Only glycine (achiral) tolerates positive φ in natural sequence, which is why every Type I’ turn has Gly at i+1. Type II’ turns have Gly or D-Pro at i+1; D-Pro forces a precise positive-φ geometry that even Gly can’t always nail. That’s why the strongest synthetic β-hairpin nucleator is D-Pro-Gly (Stanger & Gellman 1998).
Worked design example a stable β-hairpin
Starting scaffold: the GB1 hairpin (residues 41–56 of Streptococcus protein G B1 domain), GEWTYDDATKTFTVTE. This 16-residue peptide folds reversibly in water with two antiparallel β-strands joined by a Type I’ turn (D-D-A-T motif).
Designed: KEWTYDDATKVFTVTE
- Gly→Lys at N-terminal edge: positive charge suppresses lateral aggregation
- Retains Type I’ D-D-A-T turn; alternating W/Y/F/V hydrophobic / T/K/E polar strands
Maximum stability variant: KEWTY-(D-Pro)-G-AT-KVFTVTE
- D-Pro-Gly Type II’ turn is energetically optimal; requires solid-phase peptide synthesis

Verification pipeline: ESMFold sequence → align to PDB 1PGB (residues 41–56), target Cα RMSD < 1.5 Å, pLDDT > 80. Wet-lab confirmation: CD at 218 nm (β-sheet signal), ¹H-¹⁵N HSQC dispersion, thermal melt for two-state cooperativity.
Caveat: both ESMFold and AlphaFold are less reliable on short stand-alone peptides than on full domains, so for a 16-residue hairpin the computational round-trip is a sanity check, not a confirmation. Wet-lab validation is what closes the loop: CD spectroscopy (β-sheet signature at 218 nm), NMR ¹H-¹⁵N HSQC chemical-shift dispersion, and thermal melt for two-state cooperativity.
For a slightly larger and more thermostable motif, a four-strand antiparallel β-meander based on a Greek-key topology (~40 residues) is a good starting point. The standard test scaffold is ubiquitin’s β-grasp fold (PDB 1UBQ).
Recommended reading — 4 key papers
- Anfinsen, C. B. (1973). Principles that govern the folding of protein chains. Science 181: 223–230. — The Nobel-lecture statement of the sequence-determines-structure thesis. Every modern protein-ML model is a computational instantiation of this idea.
- Jumper, J. et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589. — The structure-prediction breakthrough. Read for the architecture (Evoformer + structure module) and the pLDDT/PAE confidence outputs you’ll use in Part C.
- Lin, Z. et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379: 1123–1130. — ESM-2 + ESMFold: how a language model alone (no MSA) can fold proteins. The engine inside the HTGAA Colab notebook for Part C.
- Knowles, T. P. J., Vendruscolo, M., Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15: 384–396. — The canonical review of cross-β as a deep free-energy minimum and its biological consequences.
Course resources & recordings
- Lecture recording (Karydis & Kaufman, Feb 24)
- Recitation recording (Costa, Feb 25)
- HTGAA Protein Design 2026 Colab notebook
Part II (B,C,D)
5. Part B — E. coli DHFR: sequence and structure
Why DHFR?
E. coli DHFR (UniProt P0ABQ4, gene folA) catalyzes:
DHF + NADPH + H⁺ → THF + NADP⁺
THF is the one-carbon carrier for dTMP synthesis. Block DHFR → DNA replication stops → cell dies. This is why it’s the target of methotrexate (cancer) and trimethoprim (antibiotics). The same essentiality makes it ideal for deep mutational scanning: every fitness-altering substitution shows up clearly.
Selected because: 159 aa (fast ESMFold), 2.1 Å crystal structure (PDB 1RX2), complete experimental DMS (Thompson et al. 2020), all 20 canonical AAs present — and the PLM pipeline used here transfers directly to the MS2 L-protein in Part D.
Sequence composition

Homologs: Hundreds of true homologs across all three domains of life. Human DHFR is ~35% identical — enough shared fold for methotrexate to inhibit both, different enough for trimethoprim selectivity. Type II DHFRs (R-plasmid–encoded, trimethoprim-resistant) are convergent: same reaction, completely different fold (homotetramer).
Family: Pfam PF00186, SCOP fold c.71 (DHFR-like) — a unique topology not shared with TIM barrels or Rossmann folds.
Crystal structure: PDB 1RX2
| Parameter | Value |
|---|---|
| Resolution | 2.1 Å (course threshold: 2.70 Å) ✅ |
| Deposited | January 11, 1997 |
| Ligands in ASU | Folate (substrate analog) + NADP⁺ (oxidized cofactor) |
| SCOP | Class c (α/β) → Fold c.71 → Superfamily: DHFR-like |
| Source | Sawaya & Kraut, Biochemistry 36: 586–603 (1997) |
1RX2 captures the closed conformation of the M20 loop (residues 9–24) — the catalytically productive state with both substrate and cofactor analogs bound. Best starting point for mutation analysis: active-site geometry fully resolved.
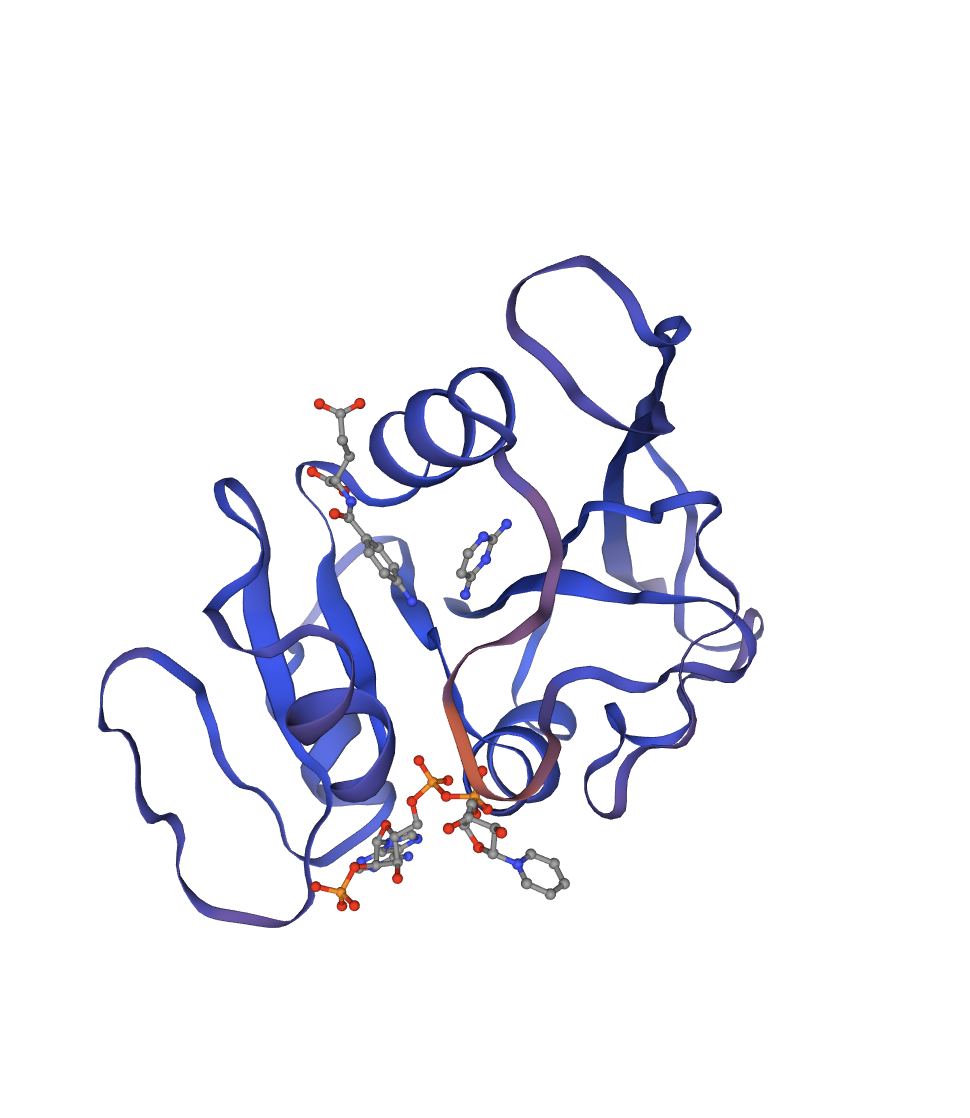 Reference crystal structure of E. coli DHFR (PDB 1RX2, 2.1 Å). Mixed α/β fold (8-strand β-sheet core, 4 α-helices). Active-site cofactors (NADPH, methotrexate) shown as sticks; blue-to-red colouring runs N- to C-terminus. Used as the fixed backbone for all ProteinMPNN inverse design runs.
Reference crystal structure of E. coli DHFR (PDB 1RX2, 2.1 Å). Mixed α/β fold (8-strand β-sheet core, 4 α-helices). Active-site cofactors (NADPH, methotrexate) shown as sticks; blue-to-red colouring runs N- to C-terminus. Used as the fixed backbone for all ProteinMPNN inverse design runs.
 AlphaFold2 prediction of the WT DHFR sequence (UniProt P0ABQ4). Uniform dark-blue colouring indicates very high per-residue confidence (pLDDT > 90) across the entire chain, consistent with a well-characterised, single-domain fold. Predicted topology matches the 1RX2 crystal structure.
AlphaFold2 prediction of the WT DHFR sequence (UniProt P0ABQ4). Uniform dark-blue colouring indicates very high per-residue confidence (pLDDT > 90) across the entire chain, consistent with a well-characterised, single-domain fold. Predicted topology matches the 1RX2 crystal structure.
Structural features
DHFR’s core is a central 8-stranded β-sheet (7 parallel + 1 antiparallel) flanked by 4 α-helices on each face. The fold is unique to the DHFR family.
- Core: Hydrophobic residues (Ile5, Leu28, Phe31, Ile94) pack the β-sheet interior
- Active site: One charged residue in the cleft — Asp27 — is the catalytic proton donor for N5 of dihydrofolate. Every other active-site contact is hydrophobic.
- M20 loop (residues 9–24): The “lid” of the active site. Cycles between closed (catalysis), occluded (product release), and open (substrate access) conformations. Highly dynamic → lower pLDDT in ESMFold.
6. Part C — The ML protein design stack applied to DHFR

Workflow run in the HTGAA Protein Design 2026 Colab notebook (GPU runtime). References: Lin et al. 2023 (ESM-2/ESMFold), Dauparas et al. 2022 (ProteinMPNN).
ESM-2 deep mutational scan
For DHFR’s 159 × 20 = 3,180 substitution heatmap:
- Most constrained: Asp27, Trp22, Arg52, Arg57 — strongly negative log-likelihood ratio (LLR) for all substitutions. ESM-2 learned from evolutionary conservation that these are untouchable.
- Most tolerant: C-terminal tail (positions 150–159), βC-βD loop — high divergence across bacterial DHFRs.
Focal residue — D27N: Asp27 → Asn removes the catalytic charge. ESM-2 predicts strongly negative LLR. Thompson et al. (2020) confirms: D27N fitness = −2 s.d. from WT median under trimethoprim selection. ESM-2 and experimental DMS agree for core/active-site positions (r ≈ 0.6–0.7); they diverge at surface positions where evolutionary pressure differs from the specific assay condition.
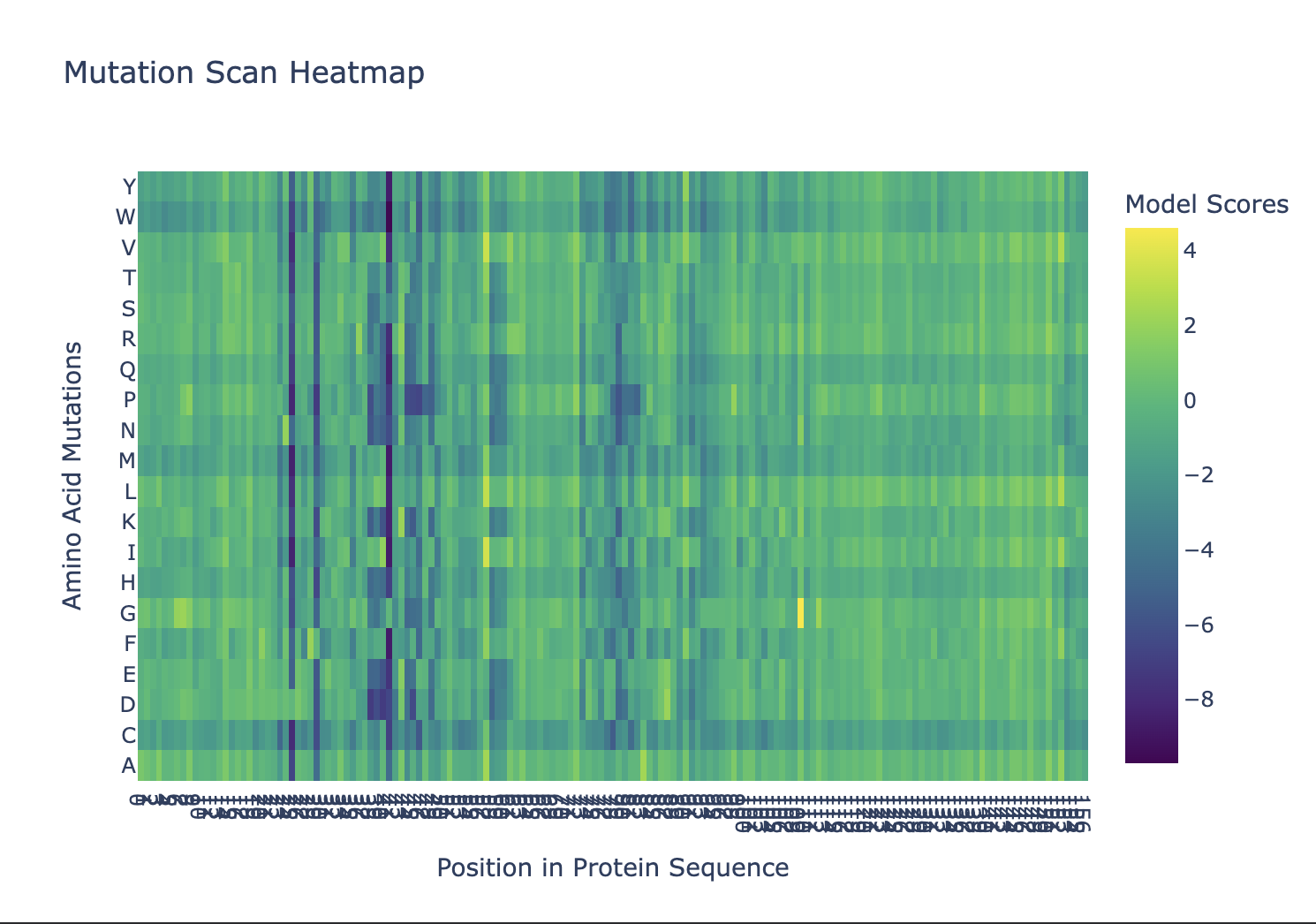 ESM-1v mutation scan across all 159 positions of DHFR. Colour scale: model log-likelihood score per substitution. Dark (low-score) columns mark positions under strong evolutionary constraint; bright (high-score) positions tolerate substitution. Positions that appear consistently low across all 20 substitutions identify the catalytic core.
ESM-1v mutation scan across all 159 positions of DHFR. Colour scale: model log-likelihood score per substitution. Dark (low-score) columns mark positions under strong evolutionary constraint; bright (high-score) positions tolerate substitution. Positions that appear consistently low across all 20 substitutions identify the catalytic core.
Latent space (UMAP embedding)
E. coli DHFR sits in the dense core of the gram-negative bacterial DHFR cluster — not at the family boundary. Trimethoprim-resistant type I DHFRs form a nearby sub-cluster. Type II DHFRs (convergent evolution, different fold) are distant. Human DHFR (P00374, ~35% identical) is well separated.
Figure needed (Colab): Run UMAP cell → export scatter → save
img/dhfr_umap_embedding.png. Label bacterial cluster, type II cluster, and human DHFR.
ESMFold structure prediction
Figure needed (Colab): Run ESMFold on WT DHFR sequence → pLDDT-coloured structure → save
img/dhfr_esmfold_plddt.png. Note M20 loop (residues 9–24, lower pLDDT).
- WT mean pLDDT > 85; Cα RMSD vs. 1RX2 < 2 Å for the β-sheet core
- M20 loop pLDDT ~60–70 — genuine disorder, not a model failure
Mutational resilience:
| Perturbation | ESMFold prediction |
|---|---|
| D27N (active-site) | Fold preserved; only local side-chain change |
| M20 loop deletion (res 12–20) | Substantial disruption; loop is integral to active-site lid |
| 20-residue core scramble (hydrophobic → polar) | Fold collapse; pLDDT < 50 across region |
ProteinMPNN inverse fold + round-trip check
Run on the 1RX2 backbone: ~40–50% sequence recovery overall. Highest at buried core positions (few alternative packing solutions). Asp27 → Asp at ~100% probability — the model has learned only an acidic residue fits the active-site geometry.
Round-trip gate: ESMFold the ProteinMPNN-designed sequence → target Cα RMSD < 2 Å vs. 1RX2. Any sequence failing this is discarded before synthesis.
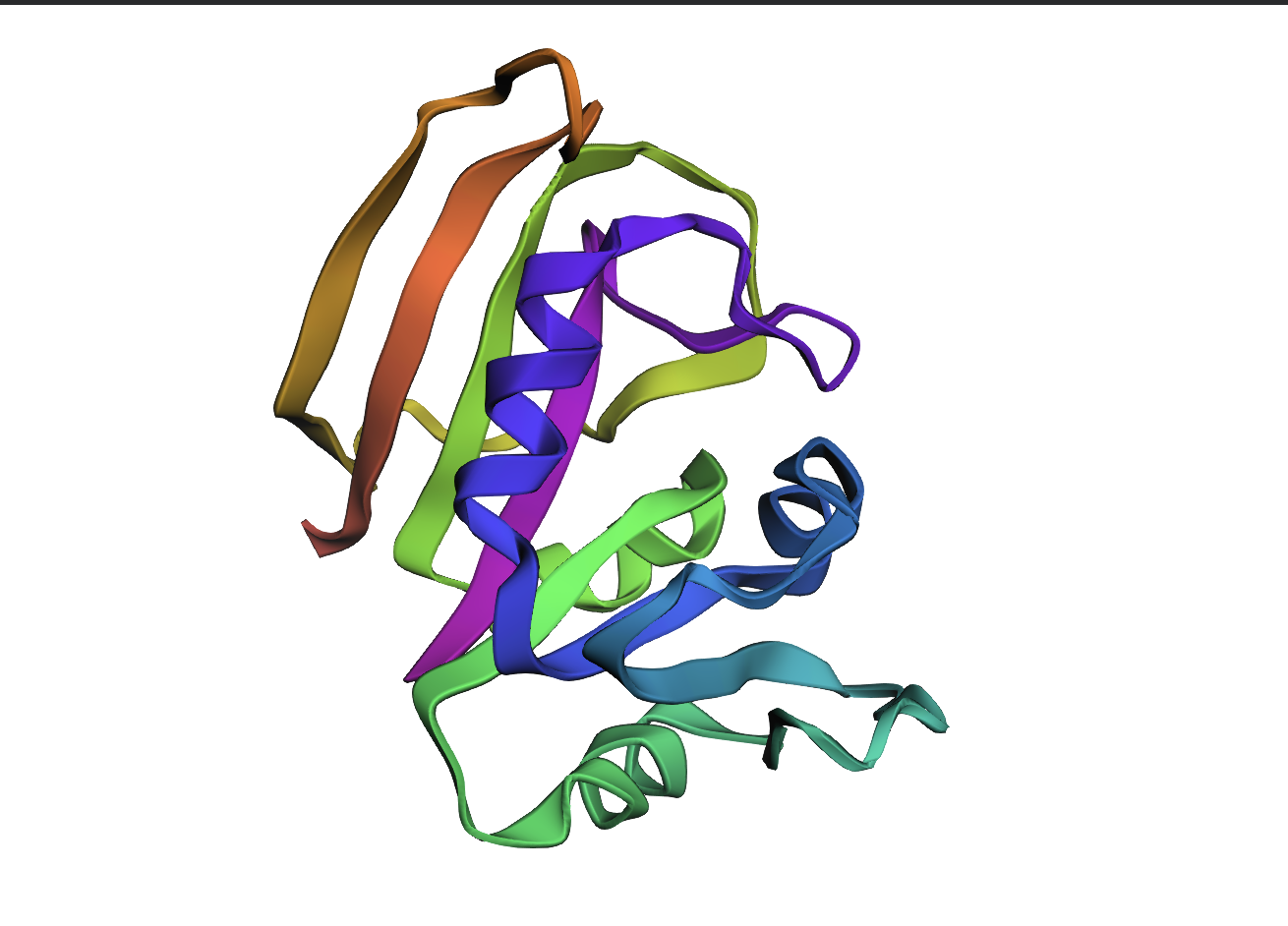 ESMFold structure prediction of the wild-type DHFR sequence (reference; ProteinMPNN log-likelihood score 1.4525). Rainbow colouring N (blue) → C (red). Used as the baseline topology against which inverse-design outputs are compared.
ESMFold structure prediction of the wild-type DHFR sequence (reference; ProteinMPNN log-likelihood score 1.4525). Rainbow colouring N (blue) → C (red). Used as the baseline topology against which inverse-design outputs are compared.
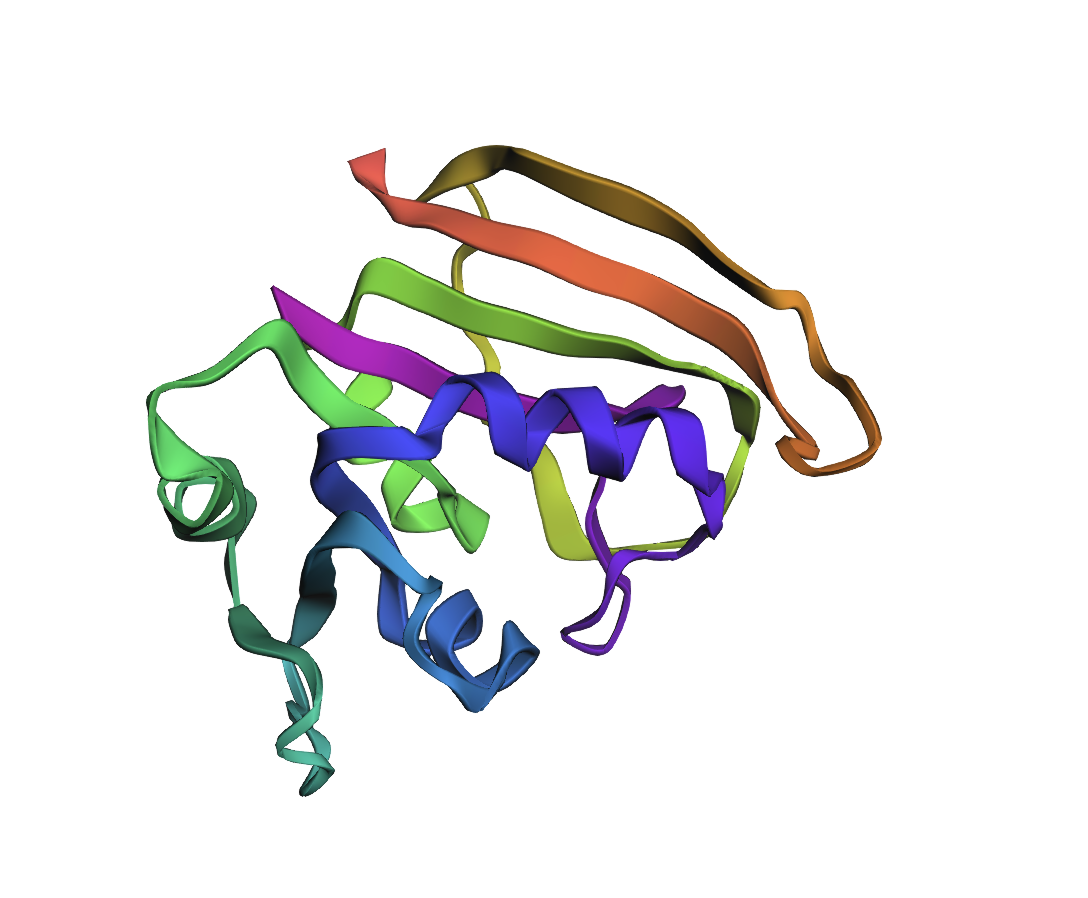 ESMFold fold prediction for inverse design #1 (ProteinMPNN T = 0.1, seed 0; score 0.7952, sequence recovery 50.3%). The α/β core topology is maintained, confirming the designed sequence is compatible with the DHFR scaffold. Deviations in loop regions reflect sequence divergence at non-core positions.
ESMFold fold prediction for inverse design #1 (ProteinMPNN T = 0.1, seed 0; score 0.7952, sequence recovery 50.3%). The α/β core topology is maintained, confirming the designed sequence is compatible with the DHFR scaffold. Deviations in loop regions reflect sequence divergence at non-core positions.
Design #1 is the closest to WT in overall compactness. The β-sheet core is well-packed and the helix arrangement mirrors the reference fold. The most visible deviation is a slight reorientation of the C-terminal helix bundle (the warm/red end of the rainbow)
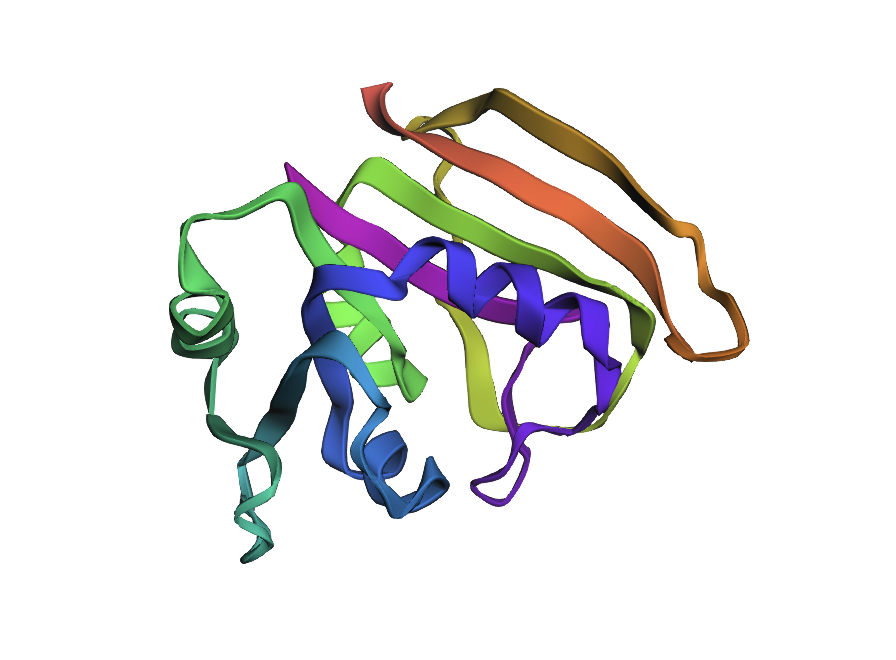 ESMFold fold prediction for inverse design #2 (ProteinMPNN T = 0.1, seed 0; score 0.8021, sequence recovery 52.8%). Highest sequence recovery of the three shown designs. Core β-sheet and helix arrangement preserved; slight reorientation of the N-terminal helix relative to the WT fold.
ESMFold fold prediction for inverse design #2 (ProteinMPNN T = 0.1, seed 0; score 0.8021, sequence recovery 52.8%). Highest sequence recovery of the three shown designs. Core β-sheet and helix arrangement preserved; slight reorientation of the N-terminal helix relative to the WT fold.
Design #2 (highest sequence recovery, 52.8%) shows a subtle but consistent outward shift of the N-terminal helix (blue region) relative to the sheet core. The β-strands themselves align well with WT geometry
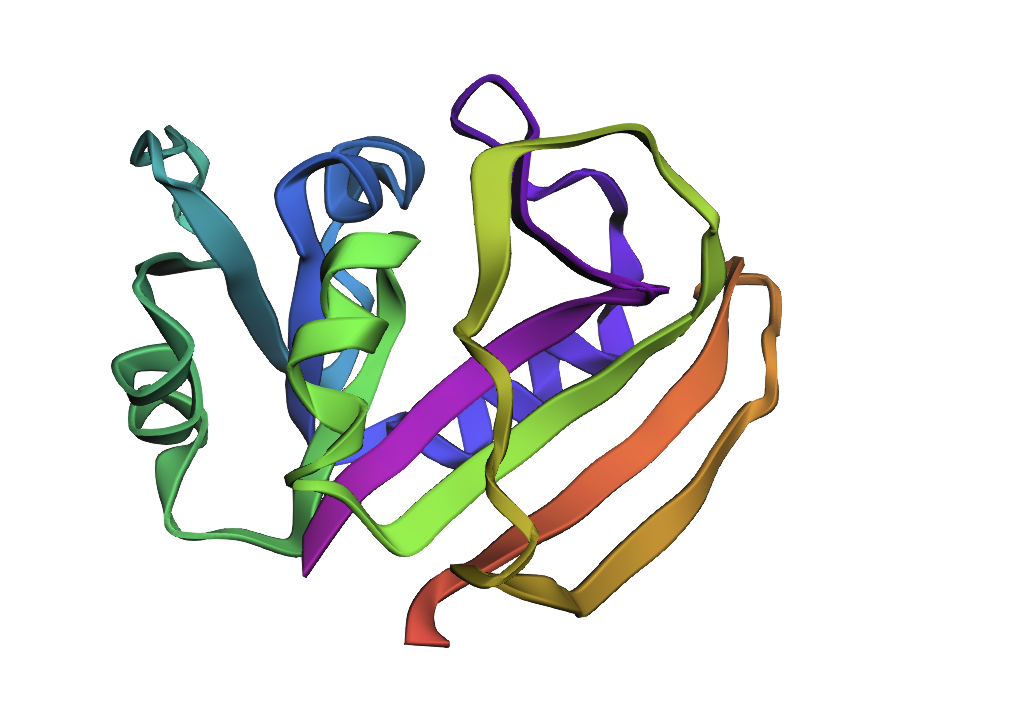 ESMFold fold prediction for inverse design #3 (ProteinMPNN T = 0.1, seed 0; score 0.8175, sequence recovery 50.3%). Core topology matches WT. The more pronounced rearrangement of surface loops compared to designs #1 and #2 likely reflects greater sequence divergence at solvent-exposed positions, which ProteinMPNN samples more liberally at low temperature.
ESMFold fold prediction for inverse design #3 (ProteinMPNN T = 0.1, seed 0; score 0.8175, sequence recovery 50.3%). Core topology matches WT. The more pronounced rearrangement of surface loops compared to designs #1 and #2 likely reflects greater sequence divergence at solvent-exposed positions, which ProteinMPNN samples more liberally at low temperature.
Design #3 is the most divergent. While the sheet topology is intact, the helical packing is noticeably looser: the α-helices appear to sit further from the β-sheet core, and the loop regions connecting secondary structure elements show more conformational scatter. This is consistent with its intermediate sequence recovery (50.3%) concentrated at core positions, leaving more freedom at helix-facing residues.
Takeaway: The β-sheet scaffold is robustly maintained across all three designs — exactly what you want if the goal is to preserve the DHFR fold while exploring sequence diversity. The increasing helix displacement from #1 → #3 is the structural signature of ProteinMPNN having sampled progressively more divergent residues at helix-packing and surface positions.
7. Part D — MS2 L-protein engineering proposal
Background
MS2 L-protein (UniProt P03609, 75 aa) is one of the smallest autonomous lytic agents. Unlike holins, it does not punch a membrane hole — it inhibits MurA (first committed step in peptidoglycan synthesis), causing osmotic lysis. Engineering challenge: L-protein is largely disordered in isolation, folding fully only upon MurA binding.
Goals (in order of computational tractability):
- Increased stability — primary target; more stable L accumulates to higher concentration before lysis, improving burst size
- Higher titers — downstream benefit of stability
Higher lysis toxicity— deprioritized (requires quantitative L:MurA interface modeling)
Computational pipeline
flowchart TD
A[MS2 L-protein\n75 aa · P03609] --> B[ESM-2 saturation scan\n75×20 = 1500 variants]
B --> C{LLR ≥ 0?\ntolerated?}
C -->|~50 pass| D[ESMFold\npLDDT + RMSD vs. WT]
C -->|Fail| Z[Discard]
D --> E{pLDDT ≥ WT±5\nRMSD < 2 Å?}
E -->|~20 pass| F[Rosetta cartesian_ddg\nΔΔG stability scoring]
E -->|Fail| Z
F --> G{ΔΔG < −1 kcal/mol?}
G -->|Pass| H[AlphaFold-Multimer\nL-variant : MurA complex]
G -->|Fail| Z
H --> I{ipTM ≥ 0.8\ninterface intact?}
I -->|Pass| J[Priority candidates\nfor wet-lab]
I -->|Fail| K[Flag: likely\nloss-of-function]Pitfalls
| Pitfall | Mitigation |
|---|---|
| Sparse phage PLM training data — ESM-2 has few MS2 L-protein homologs; LLR estimates are noisy | Relax threshold to LLR ≥ −0.5; use Rosetta ddG as primary rank, not ESM-2 |
| Intrinsic disorder — Rosetta ddG unreliable if WT pLDDT < 60 | Switch to ProteinMPNN log-probability as stability proxy if pLDDT is low |
| Toxic expression for wet-lab validation — L-protein is lethal to E. coli | Tight arabinose-inducible promoter (araBAD), low-copy plasmid, small volumes |
Recommended reading
- Anfinsen (1973). Principles that govern the folding of protein chains. Science 181: 223–230. — Sequence determines structure. The thesis every PLM is built on.
- Jumper et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596: 583–589. — Structure-prediction breakthrough; the pLDDT/PAE confidence metrics used throughout Part C.
- Lin et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379: 1123–1130. — ESM-2 + ESMFold; the core engine of the HTGAA Colab notebook.
- Thompson et al. (2020). Altered expression of a quality control protease in E. coli reshapes the in vivo mutational landscape of a model enzyme. eLife 9: e53476. — Experimental DHFR DMS used as ground truth for the ESM-2 comparison.
- Knowles, Vendruscolo & Dobson (2014). The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15: 384–396. — Cross-β as the deep free-energy minimum; canonical amyloid review.
Course resources
- Lecture recording — Karydis & Kaufman, Feb 24
- Recitation recording — Costa, Feb 25
- HTGAA Protein Design 2026 Colab notebook
*Last updated: 2026-05-23. * Section Guide:
- Part B — pick a protein, sequence and structural analysis: UniProt BLAST, RCSB structure quality, SCOP classification, PyMOL visualization (cartoon, ribbon, secondary structure / residue type coloring, surface pockets).
- Part C — modern protein-ML stack applied to the chosen target: ESM-2 unsupervised deep mutational scan; latent-space embedding; ESMFold structure prediction and mutational resilience; ProteinMPNN inverse folding with round-trip designability check.
- Part D — Bacteriophage MS2 L-protein engineering proposal: stability and auto-folding optimization using PLM-based in silico mutagenesis, AlphaFold-Multimer for L–DnaJ complex validation, and Rosetta/FoldX ddG scoring on top candidates.