Week 9 Review: Cell Free Systems
Cell-Free Systems
At a glance. Cell-free protein synthesis (CFPS) is transcription and translation in a tube — the molecular machinery a cell uses to read DNA and make protein, decanted into a defined buffer. Because the reaction is open and tunable from the moment you set it up, CFPS does things a living cell cannot: it expresses host-killing proteins, it incorporates non-canonical amino acids at scale, it can be freeze-dried into ambient-stable point-of-care diagnostics, and it can be encapsulated in lipid vesicles to build synthetic minimal cells from the bottom up. This page is a topic guide to the platform — what it is, when to reach for it, how it fails, and how the field has used it over the past decade to move from a lab curiosity to a clinical and field-deployable technology.
Course: HTGAA Spring 2026 · Author: Fiona Connolly, Committed Listener Lecture: Kate Adamala, Peter Nguyen, Ally Huang · Recitation: Ben Arias-Almeida, Ice Kiattisewee
Why go cell free?
For fifty years after Nirenberg and Matthaei cracked the first codon with an E. coli cytoplasmic extract, cell-free protein synthesis was a tabletop chemistry — useful, but only useful in a lab with –80 °C freezers, fresh lysate prep, and a trained operator. Then in 2014 the Collins lab freeze-dried a complete CFPS reaction onto a piece of cellulose paper, stored it at room temperature for over a year, reactivated it with a drop of water, and read out a programmable RNA-triggered color change by eye. That one paper changed what cell-free systems were for. In the decade since, the same architecture has been deployed as a Zika diagnostic in Brazilian clinics, as a SARS-CoV-2-detecting face mask, as an educational platform for K–12 classrooms, and as a freeze-dried payload on the International Space Station.
In parallel, the same technology is quietly running at GMP commercial scale. Sutro Biopharma manufactures antibody-drug conjugates with non-canonical amino acid sites in 4,500-litre cell-free reactions at Boehringer Ingelheim’s Vienna facility (Sutro / BI press release, January 2025; primary-literature anchor for linear scalability to ~100 L is Zawada et al. 2011, Biotechnol Bioeng 108: 1570–1578) — a class of biologic that would be essentially impossible to make in a living cell. Cell-free has stopped being only a research curiosity. It is now a production platform, a diagnostic platform, an educational platform, and the foundation for the bottom-up construction of synthetic minimal cells.
What it is not is a replacement for fermentation. Volumetric yields are still 10–100× lower per unit volume than a well-tuned bioreactor, and every cell-free reaction depends on machinery that came from a fermentation step upstream. CFPS expands the set of proteins we can make and the contexts we can deploy them in. It does not displace bulk biologics manufacturing.
Core concepts
Cell-free, in two axes
The field has two orthogonal distinctions. Get them straight before anything else.
The first is open vs closed. A living cell is closed — you put a gene in, you get a protein out, but the chemistry between is a black box you cannot manipulate while it’s running. A cell-free reaction is open — every component is on the bench in front of you, and you can add, remove, or titrate any one of them. That openness is the engineering win.
The second is lysate vs reconstituted. A lysate is what you get when you crack E. coli (or yeast, or wheat germ, or CHO cells) open and use the cytoplasmic extract. It contains ribosomes plus essentially every cytosolic protein — chaperones, the full tRNA complement, every aminoacyl-tRNA synthetase, every metabolic enzyme, and unfortunately also every nuclease and protease. It’s cheap, high-yield, and a little messy. A reconstituted system has every component purified individually and mixed in defined stoichiometry. The standard is PURE (Shimizu et al. 2001, Nat Biotechnol 19: 751–755), with about thirty-six protein components plus ribosomes plus tRNAs. Defined, expensive, lower-yield, and essential when you need to know exactly what’s in the tube.
These two axes are independent. PURE is open and reconstituted. Commercial E. coli CFPS kits like myTXTL and NEBExpress are open and lysate-based. A living cell is closed and lysate-equivalent.
What’s in the tube
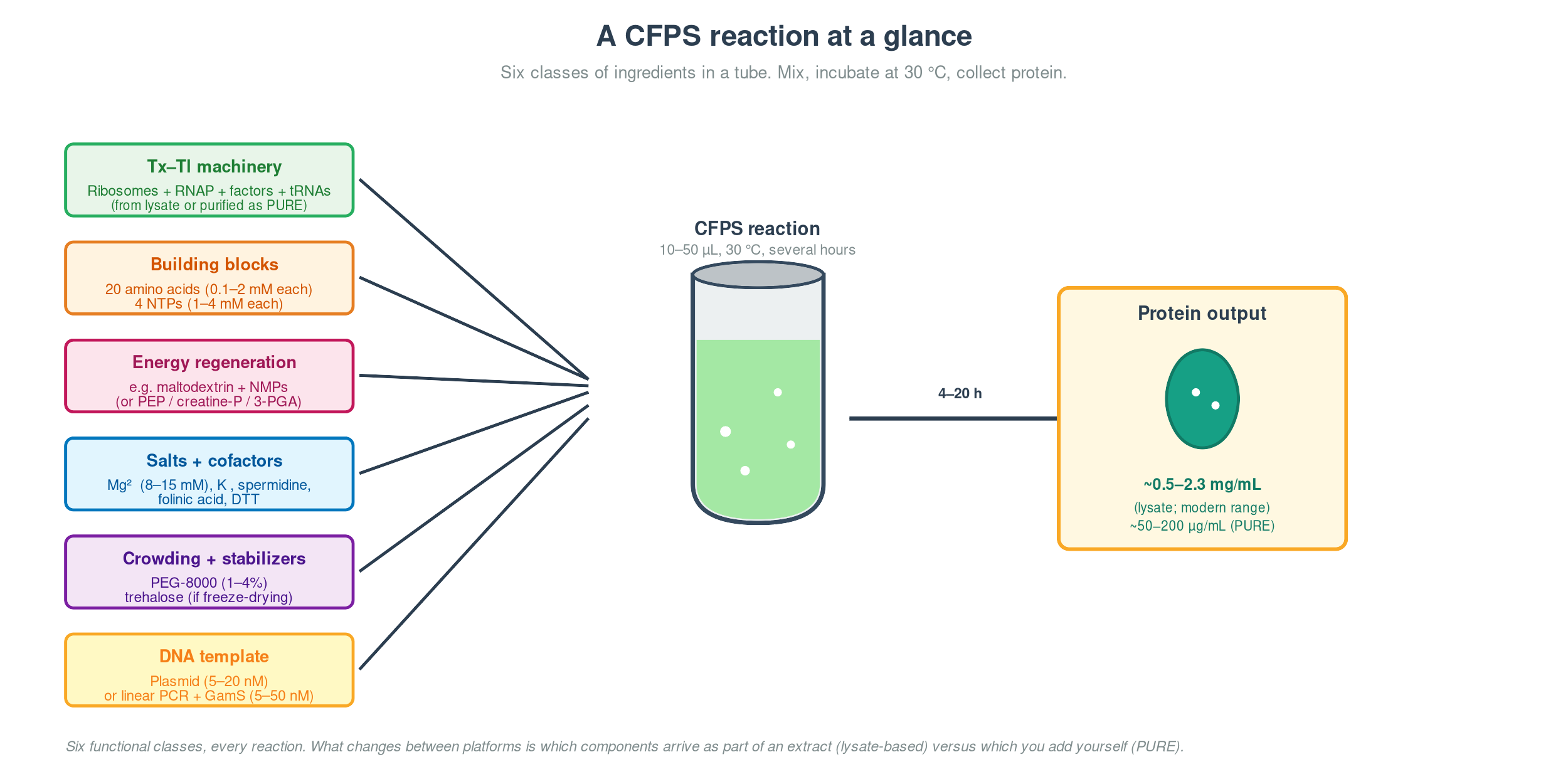
Every cell-free reaction, on any platform, contains the same six functional classes:
| Class | What’s in it | What it does |
|---|---|---|
| Transcription–translation machinery | Ribosomes, RNA polymerase, IFs, EFs, RFs, aminoacyl-tRNA synthetases, tRNAs | The expression pipeline — turns DNA into protein |
| Building blocks | 20 amino acids, 4 NTPs | Monomers for translation and transcription |
| Energy regeneration | Substrate (PEP, creatine phosphate, 3-PGA, or maltodextrin) + matching kinase | Replenishes ATP and GTP as translation burns them |
| Salts and cofactors | Mg²⁺ (8–15 mM), K⁺ (60–150 mM), spermidine, folinic acid, DTT | Mg²⁺ holds ribosomal subunits together and powers every phosphoryl-transfer step; the rest tune ionic strength, nucleic-acid charge, fMet-tRNA charging, and redox state |
| Crowding and stabilizers | PEG-8000, sometimes Ficoll-70 or trehalose | Mimics the ~300 mg/mL macromolecule density of the cytoplasm; trehalose protects through freeze-drying |
| DNA template | Plasmid or linear PCR product with promoter, RBS, gene, stop, terminator | The program — tells the machinery what to make |
The “machinery” line is what distinguishes platforms. In a lysate, classes 1 and most of class 4 arrive together — you crack open E. coli and you get ribosomes and the cytoplasmic salt mix for free, plus a lot of stuff you didn’t ask for. In PURE, you pay for the privilege of knowing exactly what’s in the tube by purifying ~36 protein components individually.
Picking a platform
The decision matrix is small. If you want maximum yield and tolerate background nucleases and proteases, use E. coli lysate. If you need every component characterized — for orthogonal translation, in-vitro evolution, or building a synthetic minimal cell from the bottom up — use PURE. If you need mammalian glycosylation, use a CHO lysate with ER microsomes. If you’re building a freeze-dried field diagnostic, use E. coli lysate. If you’re prototyping a genetic circuit, use TXTL with native sigma factors (Garamella et al. 2016, ACS Synth Biol 5: 344–355).
What’s actually in the PURE tube — for the curious reader. The ~36 protein components break down as: T7 RNA polymerase (1, for transcription); initiation factors IF1, IF2, IF3 (3); elongation factors EF-Tu, EF-Ts, EF-G (3); release factors RF1, RF2, RF3 plus the ribosome recycling factor RRF (4); methionyl-tRNA formyltransferase (1, sometimes EF-P as a 38th); aminoacyl-tRNA synthetases (20, one per amino acid); and four energy-cycle accessory enzymes (creatine kinase, myokinase, nucleoside diphosphate kinase, inorganic pyrophosphatase). Plus purified E. coli 70S ribosomes and total tRNA. Every one of these is purified separately by NEB or Genscript, which is why a 25 µL PURExpress reaction costs around $25 — about 50× more than the same volume of homemade lysate.
Methods
Energy regeneration — the silent killer
Translation is expensive. Each peptide bond costs about four high-energy phosphate bonds — two ATP for aminoacyl-tRNA charging, one GTP for EF-Tu·GTP delivery, one GTP for EF-G·GTP translocation — plus a GTP for initiation and one for release per protein, plus one NTP per transcribed nucleotide. A 300-residue protein burns through roughly 1,200 NTPs in translation alone.
A typical CFPS reaction starts with 1.2–4 mM total NTP and translates at 10–50 nM/s. Without regeneration, the NTP pool exhausts in 15–30 minutes and the reaction stops. The yield curve flatlines and from the outside it looks like the lysate died — but the lysate is fine. You just ran out of fuel.
There is a second, sneakier problem layered on top. Every ATP → ADP + Pi release dumps inorganic phosphate into the solution, and accumulated Pi chelates Mg²⁺ out of the 8–15 mM window the ribosome needs. By about 15–30 mM accumulated Pi the reaction stalls even if NTPs remain — which is why “I added more ATP and it didn’t help” is so common. The block isn’t the substrate; it’s the cofactor your substrate just sequestered. The energy-regeneration system you choose controls both how long you have ATP and how fast Pi accumulates.
The four canonical systems, with creative yield hacks for each:
| System | Substrate | Duration | Typical yield | Cost | Pi accumulation | Platform | Creative yield hacks |
|---|---|---|---|---|---|---|---|
| PEP / pyruvate kinase | Phosphoenolpyruvate | 1–2 h | ~0.5–1 mg/mL | $$$ | High | Both | Mid-run PEP spike at t = 45 min; add inorganic pyrophosphatase to clear PPi; run two-phase with creatine-P reservoir |
| Creatine-P / creatine kinase | Creatine phosphate | 1–3 h | 50–200 µg/mL (PURE); ~1 mg/mL (lysate) | $$ | Moderate | Default for PURE | Add GroEL/ES + DnaK/J/E + trigger factor chaperones; DsbA/C + GSH/GSSG for disulfides; drop RF1 + orthogonal aaRS for NCAA work (Sutro’s trick) |
| 3-PGA + endogenous glycolysis | 3-phosphoglycerate | 3–6 h | ~1–1.5 mg/mL | $ | Moderate | Lysate only | Pair with maltodextrin (Wang & Zhang 2009 hits 2.3 mg/mL via Pi recycling); add NAD⁺ to support GAPDH; switch K⁺-acetate → K⁺-glutamate |
| Maltodextrin + NMPs (Caschera–Noireaux) | Maltodextrin + maltose + NMPs | 10–20 h | ~2.3 mg/mL eGFP | $ | Low | Lysate only | Add bifunctional PPK2 from C. hutchinsonii (Wang 2019; 5× faster mRNA early); polyphosphate as bonus energy source; two-stage macromolecular crowding (20% Ficoll-70 during transcription, dilute for translation) |
Two honorable mentions for special use cases. Cytomim (Jewett & Swartz 2004, Biotechnol Bioeng 86: 19–26) exploits the fact that S30 lysate retains inverted membrane vesicles with a functional respiratory chain — supply glucose or pyruvate plus oxygen, and the vesicles do oxidative phosphorylation to regenerate ATP. It’s elegant biology, mimics native cytoplasmic energy metabolism, and gives clean reactions for 3–6 hours, but oxygen access is geometry-sensitive (thin films only) and the system is harder to make routine than the maltodextrin/NMP default. CECF (continuous-exchange CFPS; Spirin et al. 1988, Science 242: 1162–1164, plus modern microfluidic implementations from Niederholtmeyer 2013 and the Murray lab) replenishes substrates and removes Pi via dialysis through a semipermeable membrane, extending reactions to days at the cost of equipment complexity.
Cross-cutting hacks that apply to almost any system: add GamS (~3.5 µM) to protect linear PCR templates from RecBCD nuclease; keep PEG-8000 at 1–4% for crowding; DTT or 2-mercaptoethanol at 1–2 mM for reducing environment, swapping to GSH/GSSG for oxidative folding; folinic acid as a tetrahydrofolate source for fMet-tRNA charging.
How to pick:
flowchart TD
Start([Need to pick an<br/>energy regen system]) --> Q1{Defined-component<br/>requirement?<br/>e.g. SMC build,<br/>orthogonal translation}
Q1 -->|Yes — PURE system| PURE_E[Creatine-P + creatine kinase<br/><i>only practical option in PURE</i>]
Q1 -->|No — lysate OK| Q2{Goal?}
Q2 -->|Quick prototype<br/>under 2 h| PEP[PEP + pyruvate kinase<br/><i>legacy; only if rate matters more than yield</i>]
Q2 -->|High batch yield<br/>or long duration| Q3{Cost sensitive?<br/>i.e. scale-up,<br/>field deployment}
Q3 -->|Yes| Malto[<b>Maltodextrin + NMPs</b><br/>Caschera–Noireaux 2014<br/>~20 h, ~2.3 mg/mL, cheapest]
Q3 -->|Moderate| PGA[3-PGA + endogenous glycolysis<br/>Calhoun–Swartz 2005<br/>~6 h, ~1–1.5 mg/mL]
Q2 -->|Industrial preparative<br/>or kinetic study<br/>over days| CECF[Continuous-exchange CFPS<br/>Spirin 1988 + modern microfluidic<br/>indefinite duration]
Q2 -->|Want maximal<br/>biological fidelity| Cyto[Cytomim — oxidative phosphorylation<br/>Jewett–Swartz 2004]
PURE_E -.->|If yield too low,<br/>add chaperones<br/>+ extend with CECF| CECF
Malto -.->|If Pi still accumulates,<br/>extend with CECF| CECF
style Malto fill:#d4edda,stroke:#28a745,stroke-width:2px
style PURE_E fill:#cce5ff,stroke:#0066cc,stroke-width:2pxProkaryotic vs eukaryotic CFPS
The choice is the same one you’d make for in-vivo expression — match the lysate to the protein’s evolutionary context unless yield, cost, or a specific feature dominates.
| Dimension | Prokaryotic (E. coli, PURE) | Eukaryotic (wheat germ, CHO, insect Sf21, yeast) |
|---|---|---|
| Yield | 1–2.3 mg/mL | 0.05–0.5 mg/mL batch; WGE in continuous-exchange up to 1–9 mg/mL |
| Cost per reaction | $ | $$–$$$ |
| Reaction time to useful yield | 4–10 h batch; 10–20 h with maltodextrin/NMP | 6–24 h; overnight common |
| Translation machinery | 70S ribosomes | 80S ribosomes |
| Native chaperones | GroEL/ES, DnaK/J/E, trigger factor (lysate) | Hsp70/90, calnexin, BiP, PDI — only in CHO/insect with ER microsomes |
| Post-translational modifications | None natively; disulfides need DsbA/C + GSH/GSSG | N-glycosylation, signal cleavage, disulfides — only in CHO/insect with microsomes |
| Membrane-protein handling | Add detergent/nanodisc/liposome externally | CHO and insect lysates have native ER microsomes for co-translational insertion |
Note on common textbook simplifications. Three claims that travel from older slides and need updating: (1) “E. coli CFPS yields up to ~1 mg/mL” was the 2008-era ceiling; the current ceiling is ~2.3 mg/mL (Caschera–Noireaux 2014; Kwon & Jewett 2015) and Sutro routinely operates in g/L at GMP scale. (2) “Eukaryotic CFPS gives native folding, glycosylation, and phosphorylation” — only partly true; N-glycosylation only happens in CHO or insect lysates that retain ER microsomes, and wheat germ extract does not glycosylate. (3) “Prokaryotic CFPS is very fast — minutes to hours” describes time to first detection, not useful yield; reactions typically run 4–10 hours to plateau, longer with maltodextrin/NMP.
A concrete pair of picks, to make the choice tangible: for the anti-cholera-toxin VHH BL3.1 used in the Cholera Shield final project (Petersson et al. 2025, Nat Commun 16: 2722, doi:10.1038/s41467-025-57945-w), E. coli lysate is the right answer — ~15 kDa single-chain, no glycosylation required, one intra-domain disulfide handled with GSH/GSSG plus DsbA/C, codon-optimizable directly for E. coli, milligram-per-millilitre yields in four hours. For recombinant human erythropoietin, you cannot responsibly use a prokaryotic system — EPO carries three N-linked glycans at N24, N38, and N83 (mature-protein numbering; N51, N65, N110 in the preproprotein UniProt P01588), and removing them drops the in-vivo serum half-life from hours to minutes. Here CHO-based CFPS with ER microsomes (Brödel et al. 2014, J Biotechnol 178: 1–10) is the right call.
Synthetic minimal cells
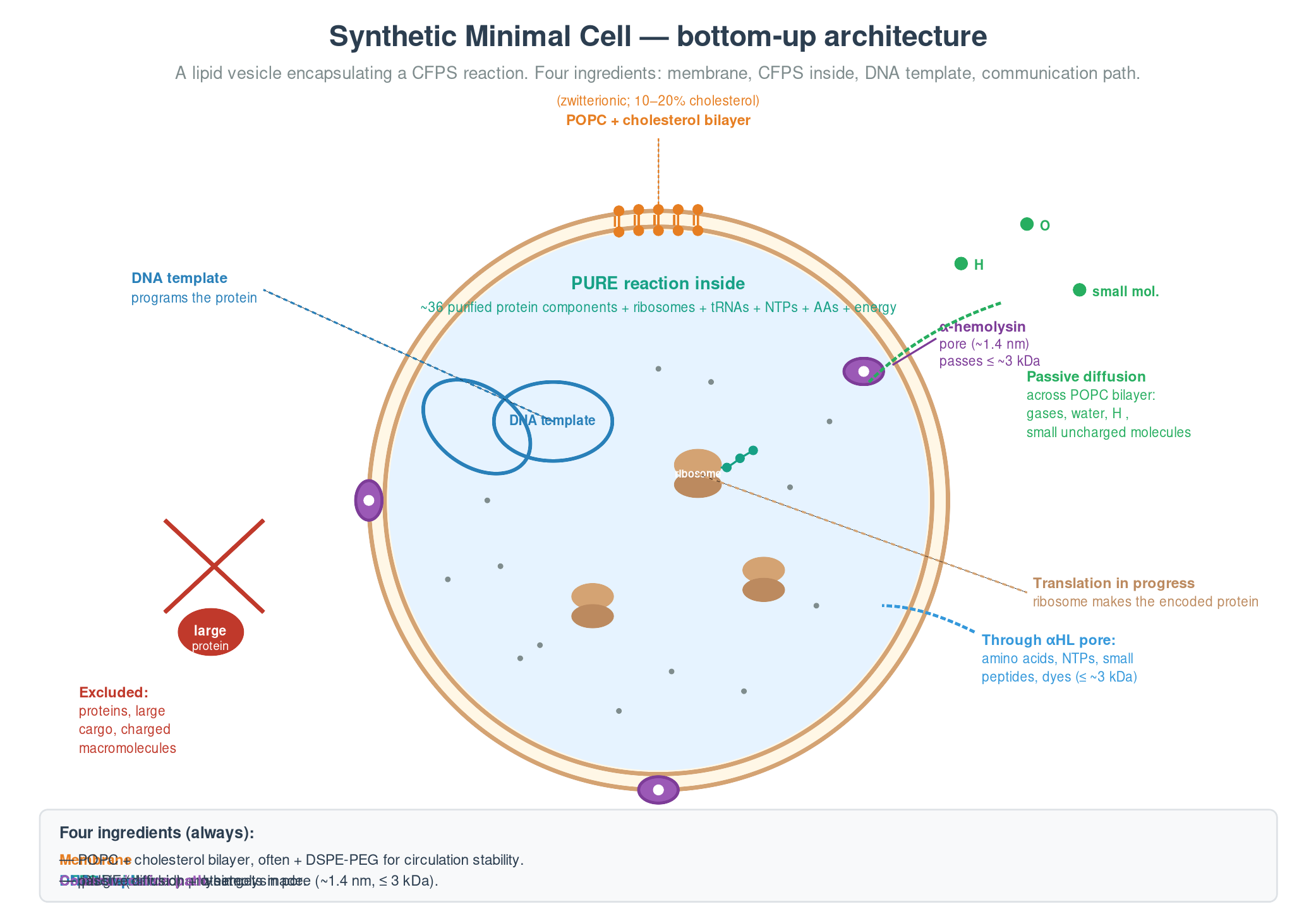
A bottom-up synthetic minimal cell is a lipid vesicle encapsulating a CFPS reaction. Four ingredients: a membrane (usually POPC, often with 10–20% cholesterol), a CFPS reaction inside (PURE is mandatory for defined-component construction), a DNA template, and a way to communicate with the environment (passive diffusion for small uncharged molecules, or a pore protein like α-hemolysin for everything below ~3 kDa).
The canonical worked example is Lentini et al. 2014 (Nat Commun 5: 4012) — a synthetic cell that senses theophylline (which E. coli can’t detect) via a riboswitch-controlled genetic inverter, and releases an output molecule (IPTG) that E. coli can read via its native Lac operon. The SMC acts as a chemical-language translator between two biology systems that couldn’t otherwise communicate, by re-encoding one small-molecule signal into another.
Bottom-up SMCs cannot yet self-replicate (Kuruma 2009 got ~50% of membrane components self-synthesized; ribosome biogenesis in a vesicle remains open), sustain themselves indefinitely without external feeding, or secrete protein at high yield. Within those constraints, they are an extraordinarily flexible engineering platform for chemical sensing, drug delivery, and origin-of-life research.
Freeze-dried CFPS — the 2014 inflection point
Three properties became simultaneously true for the first time in synthetic biology in 2014: shelf-stable (over a year at ambient temperature with no cold chain), abiotic (no living organism in the field, no biosafety containment, no environmental release concern), and instrument-free (visible colorimetric or fluorescent output read by eye). Pardee, Green, Yin, Takahashi et al. 2014, Paper-based synthetic gene networks (Cell 159: 940–954, doi:10.1016/j.cell.2014.10.004). Lyophilize a complete CFPS reaction onto cellulose paper, store at room temperature, reactivate with a drop of sample, and read the answer in colour.
The programmability came from a parallel breakthrough in the same group: toehold switches (Green, Silver, Collins, Yin 2014, Cell 159: 925–939). A toehold switch is an RNA hairpin in the 5′ UTR of a reporter gene that sequesters the ribosome binding site and the start codon. A complementary trigger RNA binds the exposed “toehold” at the 5′ end of the hairpin, strand-displaces through the stem, and exposes the RBS — switching translation on. The trigger sequence is design-flexible, so a custom switch can be built to detect almost any RNA target.
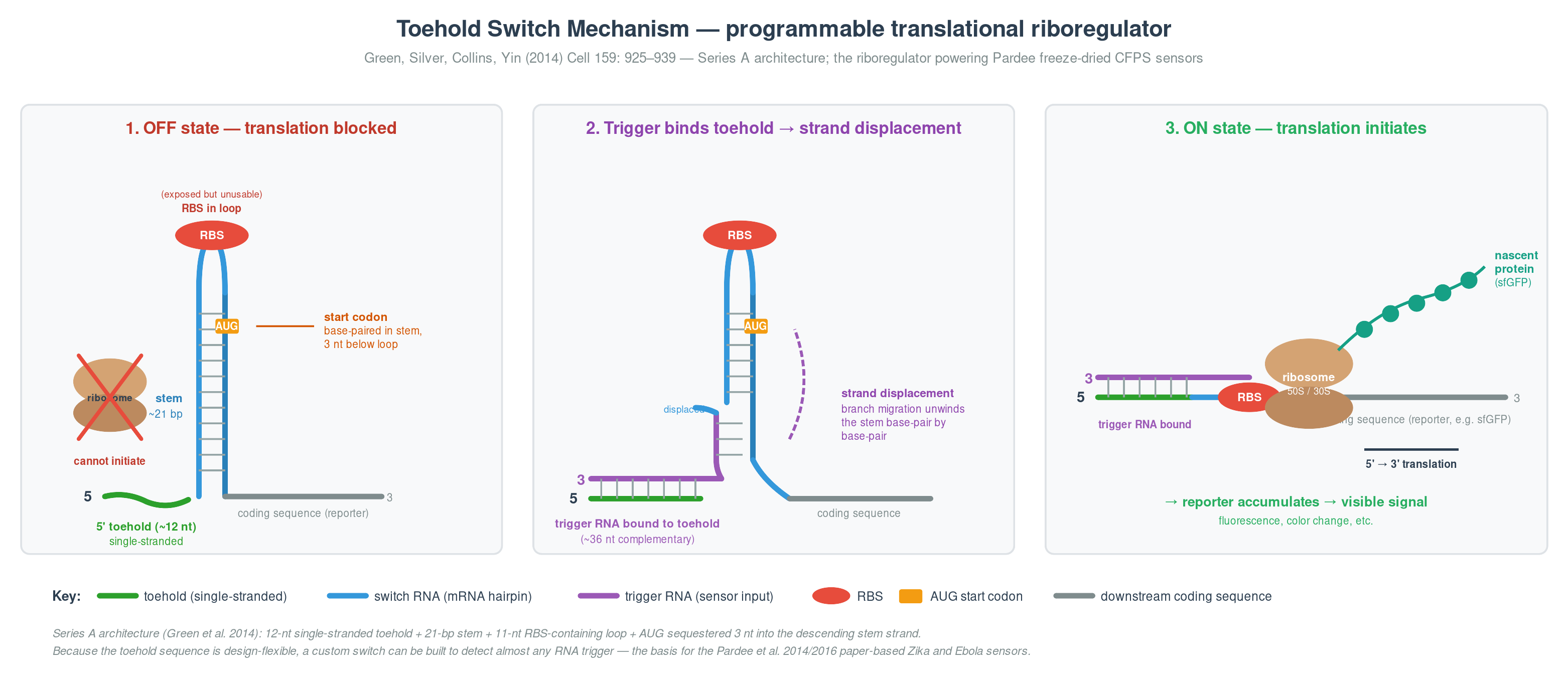
Figure 1. Toehold-switch architecture (Series A; Green et al. 2014). Left: in the OFF state, the RBS sits in the hairpin loop (exposed but unusable) and the AUG start codon is sequestered in the stem — the ribosome cannot initiate translation. Middle: the trigger RNA binds the exposed 5′ toehold and propagates through the stem by strand displacement. Right: in the ON state, the stem is fully unwound, the RBS and AUG are accessible, and translation produces the reporter protein.
The follow-up Zika sensor (Pardee 2016, Cell 165: 1255–1266) validated the platform on clinical samples from Brazil and Honduras: NASBA-amplified viral RNA → toehold switch → LacZ → CPRG colorimetric substrate, yellow-to-purple by eye, under $1 per assay, meeting WHO ASSURED point-of-care criteria. The same architecture now underpins BioBits K–12 classroom kits (Huang et al. 2018), the Huang 2024 ISS validation (Huang et al. 2024, ACS Synth Biol 13: 1922–1932), and the Wyss group’s wearable freeze-dried CFPS face-mask sensors for SARS-CoV-2 (Nguyen et al. 2021, Nat Biotechnol 39: 1366–1374). One paper, one paradigm shift, an entire downstream technology stack.
Membrane proteins
About 30% of the proteome is membrane-embedded (Wallin & von Heijne 1998, Protein Sci 7: 1029–1038, doi:10.1002/pro.5560070420) and roughly 60% of all approved drug targets are membrane proteins (Overington et al. 2006, Nat Rev Drug Discov 5: 993–996, doi:10.1038/nrd2199), and they are the hardest class to make in any expression system. CFPS is the platform where you have the most control: you can put a hydrophobic environment in the tube from t = 0, dial in chaperones, and run the reaction in a controlled redox state — but you have to make every decision deliberately. Default everything and you get aggregated, non-functional protein.
The five membrane mimetics, with the use case for each:
- Detergent micelles (DDM, LMNG) — cheap, well-established, but detergent above ~0.05–0.1% inhibits CFPS.
- Nanodiscs — MSP1D1 plus defined lipids form ~10 nm discoidal patches (Bayburt & Sligar 2003). Defined lipid environment; well-suited to GPCRs.
- Liposomes / proteoliposomes — pre-formed lipid vesicles; co-translational integration possible but directional insertion can be random.
- Native ER microsomes — only in eukaryotic lysates (CHO, insect); the only practical CFPS route to N-glycosylated mammalian glycoproteins.
- SMALPs / amphipathic polymers — extract native lipid disc without detergent; newer methodology.
A worked example to make this concrete: the β₂-adrenergic receptor is the canonical class A GPCR for CFPS optimization studies. Seven transmembrane helices, two structural disulfide bonds (C106–C191 and C184–C190 from the Cherezov 2007 crystal structure), three N-glycosylation sites (N6, N15, N187), and a fluorescent ligand binding assay with native Kᴅ ≈ 5 nM (BODIPY-FL-CGP12177). To express it in CFPS you want CHO lysate with ER microsomes for the native PTMs and chaperones, oxidizing buffer (5 mM GSH / 1 mM GSSG with no DTT) to allow the disulfides to form, a low-detergent fallback (LMNG at 0.005%) if microsomes are insufficient, 50 µM verapamil as a pharmacochaperone, 27 °C reaction temperature for better folding, a thin-film geometry for O₂ access, and a functional ligand-binding readout — not just SDS-PAGE. Typical functional yield: 50–200 µg/mL. The reference for E. coli-based CFPS GPCR expression is the Bernhard group (Klammt et al. 2007, J Struct Biol 158: 482–493).
Three worked examples
The next three sections — a logic-gated diagnostic in a vesicle, a freeze-dried sensor in a building material, and a freeze-dried sensor in space — together span the modern application landscape of cell-free systems. Each foregrounds a different CFPS strength: logic gating in synthetic minimal cells, materials integration via freeze-dried capsules, and remote field deployment via the lyophilized BioBits hardware stack. Together they make the case that the same underlying chemistry — a cell-free transcription-translation reaction in a small volume, with a designed RNA sensor and a colorimetric reporter — now sits at the centre of synthetic biology’s most ambitious application bets.
Worked example 1: a multi-biomarker prostate cancer diagnostic synthetic minimal cell
A pedagogically rich Adamala-style synthetic cell, designed to show what CFPS can do when you combine encapsulation with logic gating. The architecture is a design proposal — it has not been demonstrated in clinical samples — but every component has a published precedent.
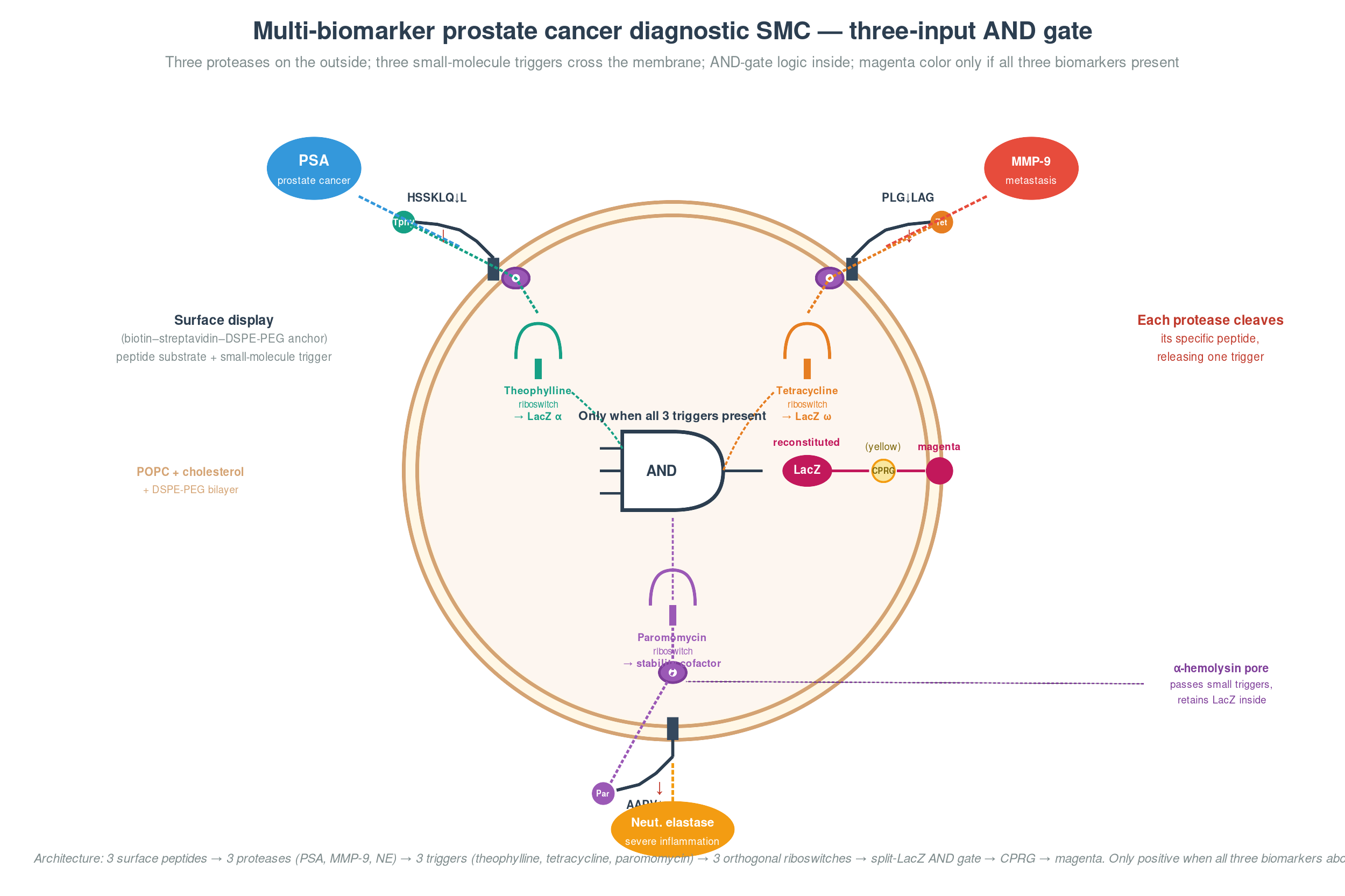
The concept. A synthetic minimal cell that simultaneously detects three biomarkers — PSA, MMP-9, and neutrophil elastase — and produces a visible yellow-to-magenta color change only when all three are present above their clinical thresholds. A 3-input AND-gate diagnostic for aggressive, metastatic, or inflamed prostate cancer, designed to improve specificity over single-marker PSA testing where benign prostatic hyperplasia drives roughly 30% false positives.
How it senses. Each of the three biomarkers is a protease, and each has a well-characterized peptide substrate displayed on the outer surface of the SMC via a biotin–streptavidin bridge to DSPE-PEG(2000)-biotin in the bilayer. When PSA cleaves its substrate (Mu-HSSKLQ↓L-theophylline, derived from the semenogelin cleavage map), it releases theophylline. When MMP-9 cleaves its substrate (PLG↓LAG-tetracycline, the canonical MMP-9 recognition motif), it releases tetracycline. When neutrophil elastase cleaves its substrate (MeOSuc-AAPV↓-paromomycin, the standard chromogenic NE recognition motif), it releases paromomycin.
How it computes. All three small-molecule triggers diffuse through α-hemolysin pores into the SMC interior, where each activates a different orthogonal riboswitch — theophylline (Jenison et al. 1994), tetracycline (Berens & Suess), and engineered neomycin/paromomycin (Weigand et al. 2008, RNA 14: 89–97). The triggers feed a three-input AND gate that gates β-galactosidase translation. The simplest published architecture for this is the α/ω LacZ complementation split (one fragment under riboswitch 1, the other under riboswitch 2), but extending it to three inputs cleanly in CFPS-in-vesicle is an open engineering problem — the third gate element here is a placeholder for a still-to-be-engineered component, drawing on the logic-gate framework demonstrated by Adamala et al. 2017 (Nat Chem 9: 431–439), who showed programmable AND/OR/NOT gates in populations of liposome-encapsulated CFPS reactions communicating via diffusible small-molecule signals. Only when all three triggers are present does intact β-galactosidase reconstitute and cleave CPRG into the visible magenta product.
How it reads out. A drop of urine on a freeze-dried SMC strip; 30–60 minutes; smartphone-camera RGB analysis if you want to quantify; visible by eye otherwise. Cost target under $1 per test.
Where it’s weak. Three honest caveats. The neutrophil elastase substrate cross-reacts with proteinase 3, so the “severe inflammation” signal is technically NE-or-PR3, which is biologically acceptable but a peer reviewer will flag it. The third riboswitch (paromomycin) has lower dynamic range than the first two — backup architectures using split-protein complementation gated by stability cofactors are the next iteration. And the entire architecture has not been demonstrated end-to-end in clinical samples; the closest published precedent is Adamala et al. 2017 (Nat Chem 9: 431–439) on SMC logic gates.
Worked example 2: a self-reporting mold-detecting wall paint
The Peter-Nguyen-style application question — cell-free systems integrated into a material — with a single focused use case in the architecture field.
The pitch. A wall paint containing freeze-dried cell-free biosensors that turns visibly purple when black mold is growing behind the wall — warning residents weeks before mycotoxin exposure causes illness.
How it works. Millions of microscopic capsules suspended in standard latex paint. Each capsule contains a freeze-dried CFPS reaction, an RNA aptamer-coupled toehold switch targeting trichothecene mycotoxins from Stachybotrys chartarum (the aptamer would need to be selected via SELEX as a development step — trichothecene-binding aptamers exist in the literature but are not as well-characterized as small-molecule riboswitches), and the β-galactosidase reporter gene. When mold grows behind a painted wall, the mycotoxins seep through and bind the aptamer, which strand-displaces the toehold switch and switches on the reporter. The enzyme cleaves CPRG into a purple pigment, and within hours the affected wall area visibly changes colour. No power, no batteries, no inspector required.
Why it matters. After every major flood or hurricane, tens of thousands of homes develop hidden mold inside walls. Current detection is expensive — air sampling, surface swabs, or destructive wall inspection at $400–800 per home and several days for lab results. Post-disaster, the people who need it most can’t afford it. A self-reporting paint would turn every newly painted room into its own continuous mold monitor for slightly more than ordinary paint.
How it handles CFPS limitations:
| Limitation | How the paint handles it |
|---|---|
| Water-activation is single-use | Millions of independent capsules per painted wall — local events activate only the capsule population at the site, not the whole sensor |
| Dried CFPS has to survive storage | Silk-fibroin matrices (Kaplan lab, Tufts) plus trehalose preserve activity for over a year at room temperature |
| Sensitivity is limited | Isothermal amplification inside each capsule (NASBA, RPA, or LAMP) boosts detection ~10⁵-fold so trace mycotoxin triggers a clear color change |
| Usually only one analyte at a time | Different capsule batches carry sensors for different molds (Stachybotrys, Aspergillus, etc.) with different colour outputs |
Honest caveats. The integrated wall-paint product doesn’t yet exist — every component has been demonstrated separately, but combining them into a manufacturable paint is the unsolved engineering step. The sensor reports past exposure but doesn’t remediate the mold. The color change is permanent by design (a record of exposure), which means re-painting is the only way to reset the sensor.
Worked example 3: a Genes-in-Space proposal — does microgravity accelerate horizontal gene transfer?
The Huang-style mock proposal, designed for the BioBits + miniPCR + P51 viewer hardware stack already validated for the ISS.
The question. Astronauts on long missions face a serious medical risk: if they get a bacterial infection, the bacteria may already be resistant to the antibiotics on board. Microgravity is known to change bacterial behaviour — increased virulence, thicker biofilms, activated stress responses. What we don’t know is whether microgravity also makes bacteria share antibiotic resistance genes with each other faster. The mechanism is horizontal gene transfer (HGT). Until now, measuring HGT in space required bringing bacterial samples back to Earth.
The molecular target. The mRNA of the tetA gene (tetracycline resistance) in recipient E. coli cells, transferred from a donor strain via conjugation.
How the target reports the question. Mix two E. coli strains — a donor that carries tetA on a transferable plasmid, and a recipient that does not. When the donor passes its plasmid to the recipient, the recipient starts expressing tetA mRNA. Detecting that mRNA inside recipient cells reports each successful HGT event. Use a paper-based toehold-switch sensor (the same architecture as the Pardee Zika test) that produces a yellow-to-purple color change when tetA mRNA is present. Compare in-flight color over time against identical ground-control cultures to quantify how much microgravity changes the HGT rate.
Hypothesis. Microgravity speeds up horizontal gene transfer between E. coli strains by 2 to 5 times compared to ground controls. Three independent observations from ISS bacterial-physiology studies predict elevated HGT: microgravity activates the SOS response (DNA-damage signaling, which upregulates conjugation genes); F-pilus assembly depends on membrane dynamics that behave differently in microgravity; and cell-envelope stress signaling is elevated in ISS-grown bacteria, and stress conditions are known to promote gene-sharing on Earth.
Experimental plan. Donor E. coli K-12 (F⁺, pBR322::tetA) and recipient (tetA⁻) co-cultured 1:1 in microfluidic chambers; sample at t = 0, 4, 8, 24, 48 h. Controls: ground-control parallel cultures, donor-only and recipient-only controls, heat-killed donor (rules out live transfer), pre-mixed plasmid (positive control for the sensor). At each timepoint, lyse a small aliquot, amplify tetA mRNA with NASBA, apply to a freeze-dried BioBits paper sensor, read color with the P51 viewer; confirm in parallel with miniPCR detection of the gene.
Pitfalls, controls, and how to know it worked
Most low-yield problems come from one of three failure modes — template degradation, the wrong Mg²⁺, or energy depletion plus phosphate accumulation. Check those three first, in that order. Earlier failure modes mask later ones; there’s no point optimizing chaperones if your template was being chewed up the whole time.
Beginner’s troubleshooting
Before every reaction: verify the template by Sanger sequencing (promoter, RBS, gene, stop, terminator); thaw the lysate aliquot only once; titrate Mg²⁺ on every new lysate batch; pick the energy module that matches your duration; add GamS (~3.5 µM) if you’re using a linear PCR template; spread the reaction in a thin film (~1 mm depth); and always run a GFP positive control in parallel — this single step saves more debugging hours than any other.
If yield is zero, check the GFP positive control first. If GFP is also dead, the lysate or reagents are dead. If GFP works but your target doesn’t, the problem is your template or target-specific.
If yield is low, the six failure modes in priority order:
| Step | What to check | Diagnostic | Fix |
|---|---|---|---|
| 1 | Is the template intact? | Plasmid vs linear parallel control | Add GamS for linear templates |
| 2 | Is mRNA being made? | RT-qPCR or denaturing gel | Fresh T7 RNAP + 1–2 mM DTT |
| 3 | Is Mg²⁺ right? | 6-point titration 6–16 mM | Adjust to peak |
| 4 | Does the reaction stall early? | Time-course at 15/30/60/120 min | Switch to maltodextrin + NMPs |
| 5 | Does the protein degrade over time? | Time-course Western shows degradation bands | Protease-deficient lysate (Lon⁻ OmpT⁻) + protease inhibitors |
| 6 | Total protein high, functional protein low? | Activity assay vs SDS-PAGE | Chaperones, lower temperature, oxidizing buffer with DsbA/C, codon-optimize |
flowchart TD
Start([Low or zero<br/>CFPS yield]) --> Q1{GFP positive<br/>control works?}
Q1 -->|No| LysateBatch[Lysate or reagent problem<br/>→ check Mg²⁺ titration,<br/>freeze-thaw history,<br/>reagent stock dates]
Q1 -->|Yes| Q2{Template type?}
Q2 -->|Linear PCR| Q2A{GamS added?}
Q2A -->|No| AddGamS[Add GamS<br/>~3.5 µM]
Q2A -->|Yes| Q3
Q2 -->|Plasmid| Q3{Reaction stalls<br/>before 60 min?}
Q3 -->|Yes| EnergySwitch[Switch to<br/>maltodextrin + NMPs<br/>+ pyrophosphatase]
Q3 -->|No| Q4{Total protein<br/>on SDS-PAGE high?}
Q4 -->|Yes — but not functional| Folding[Folding problem<br/>→ chaperones, 25–27 °C,<br/>GSH/GSSG + DsbA/C,<br/>codon-optimize]
Q4 -->|No| Q5{Time-course shows<br/>degradation bands?}
Q5 -->|Yes| Proteases[Protease problem<br/>→ Lon⁻ OmpT⁻ lysate<br/>+ PMSF / leupeptin]
Q5 -->|No| Q6[Run Mg²⁺ titration<br/>+ fresh T7 RNAP control<br/>+ check NTP stocks]
style LysateBatch fill:#ffe6e6,stroke:#c0392b,stroke-width:2px
style AddGamS fill:#d4edda,stroke:#28a745,stroke-width:2px
style EnergySwitch fill:#d4edda,stroke:#28a745,stroke-width:2px
style Folding fill:#fff3cd,stroke:#ffc107
style Proteases fill:#fff3cd,stroke:#ffc107One last piece of practical advice. A two-hour parallel-control panel at the start of any debugging session — plasmid versus linear template, plus a Mg²⁺ titration, plus a fresh-RNAP control — diagnoses most low-yield problems and saves a week of guessing. Run those three panels every time before you start changing the protein-specific conditions.
Recommended reading
Four primary-literature papers that together cover the modern shape of the field.
- Pardee, K., Green, A. A., Ferrante, T., et al. (2014). Paper-based synthetic gene networks. Cell 159: 940–954. doi:10.1016/j.cell.2014.10.004 — The paper that changed what cell-free systems were for. Freeze-dried CFPS, ambient stability, abiotic field-deployable diagnostics. The lineage every subsequent freeze-dried CFPS application traces back to.
- Caschera, F. & Noireaux, V. (2014). Synthesis of 2.3 mg/mL of protein with an all E. coli cell-free transcription–translation system. Biochimie 99: 162–168. doi:10.1016/j.biochi.2013.11.025 — The maltodextrin-plus-NMP energy module. Turned a two-hour assay into a twenty-hour assay and holds the current batch-yield record. The current default for lysate-based CFPS.
- Shimizu, Y., Inoue, A., Tomari, Y., et al. (2001). Cell-free translation reconstituted with purified components. Nat Biotechnol 19: 751–755. doi:10.1038/90802 — The PURE system. Defined CFPS, ~36 protein components plus ribosomes plus tRNAs. The platform required for bottom-up synthetic minimal cells and orthogonal translation work.
- Huang, A., Nguyen, P. Q., Stark, J. C., et al. (2018). BioBits™ Bright: A fluorescent synthetic biology education kit. Sci Adv 4: eaat5105. doi:10.1126/sciadv.aat5105 — The freeze-dried CFPS architecture translated into K–12 classrooms. Same hardware stack (with miniPCR and the P51 fluorescence viewer) used for the 2024 Huang ISS validation. The most visible demonstration that cell-free has matured from a research tool into a deployable technology.
Course resources
- Pardee 2016 Zika sensor — the canonical clinical-sample validation of paper-based CFPS. doi:10.1016/j.cell.2016.04.059.
- Huang 2024 ISS validation — CFPS in microgravity. doi:10.1021/acssynbio.3c00733.
- Nguyen 2021 wearable face-mask sensor — the wFDCF platform. doi:10.1038/s41587-021-00950-3.
- Garamella 2016 TXTL Toolbox 2.0 — modern lysate CFPS reference. doi:10.1021/acssynbio.5b00296.
- PURExpress (NEB) — commercial PURE system. neb.com.
- myTXTL / NEBExpress — commercial E. coli lysate CFPS.
- Avanti Polar Lipids — POPC, cholesterol, DSPE-PEG, E. coli polar lipid extract for SMC work. avantilipids.com.
- Addgene #133553 — pDG1730 backbone used for B. subtilis amyE integration in the Cholera Shield final project.
This page is a topic guide written as part of the HTGAA 2026 syllabus, intended as a stand-alone reference. The worked examples reflect design proposals; published demonstrations are cited where they exist, and design extrapolations are flagged honestly. Errors are mine — please point them out.