Biological Engineering Application: A Functional Expression Platform for Classifying Variants of Uncertain Significance (VUS) in Cystic Fibrosis
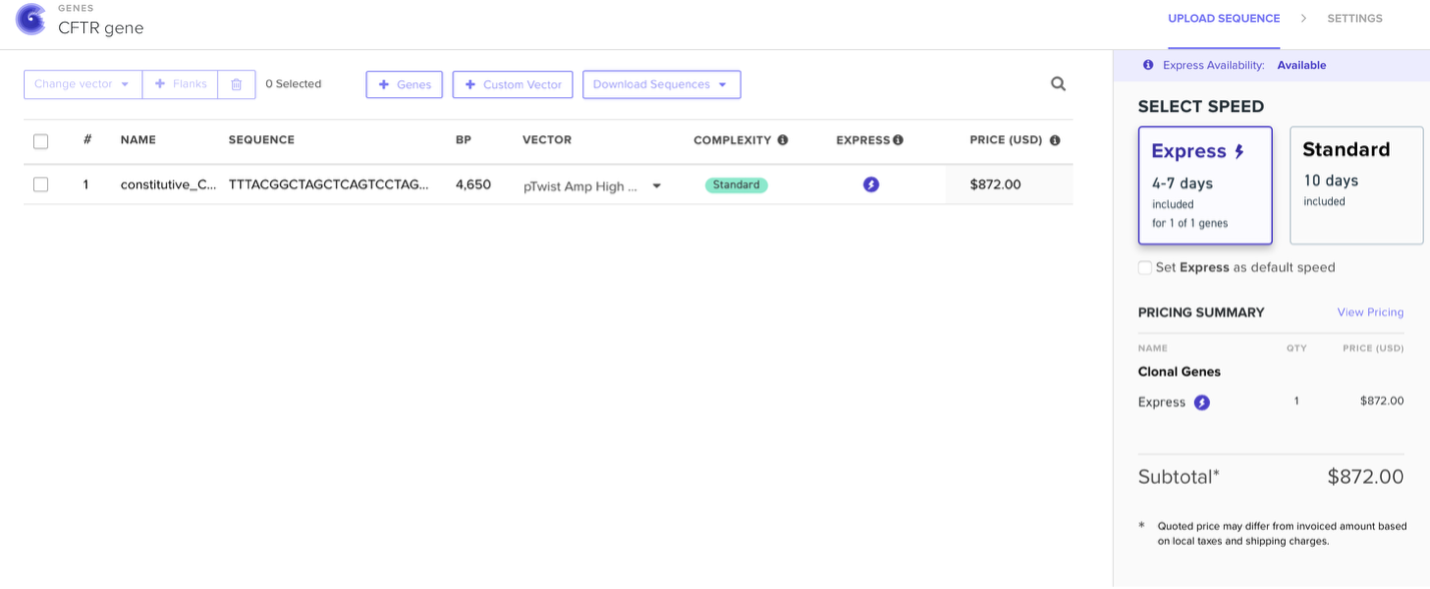
My proposal is to develop a standardized, accessible platform for the functional expression and characterization of variants in the CFTR gene, specifically focusing on Variants of Uncertain Significance (VUS) identified in underrepresented populations in Ecuador.
The core application integrates three components: rapid synthesis and cloning of CFTR gene constructs containing locally identified VUS; expression of these constructs in relevant, standardized human cell lines (e.g., immortalized bronchial epithelial cells); and a multiparametric, high-content functional assay suite.
This suite would automatically measure key parameters: subcellular localization via microscopy, protein processing and maturation via western blot, ion channel function via fluorescence-based halide assays, and response to CFTR modulator drugs.
The motivation for this tool is to address a critical equity gap in genomic medicine. Global genetic databases like ClinVar are heavily biased toward populations of European descent. Consequently, variants identified in mestizo, Indigenous, or Afro-descendant populations in Ecuador and similar regions are often classified as VUS due to a lack of functional and phenotypic data.
This has direct, harmful clinical consequences: it leads to ambiguous diagnoses for patients and families, restricts access to modern, highly effective modulator therapies that are approved for specific variants, and perpetuates health disparities.
This platform aims to functionalize genomics by converting raw genetic data into actionable clinical knowledge, deliberately prioritizing variants from historically underserved populations to ensure that precision medicine benefits extend globally.
Subsections of Homework
Week 1 HW: Principles and Practices
Biological Engineering Application: A Functional Expression Platform for Classifying Variants of Uncertain Significance (VUS) in Cystic Fibrosis
1. My proposal is to develop a standardized, accessible platform for the functional expression and characterization of variants in the CFTR gene, specifically focusing on Variants of Uncertain Significance (VUS) identified in underrepresented populations in Ecuador.
The core application integrates three components: rapid synthesis and cloning of CFTR gene constructs containing locally identified VUS; expression of these constructs in relevant, standardized human cell lines (e.g., immortalized bronchial epithelial cells); and a multiparametric, high-content functional assay suite.
This suite would automatically measure key parameters: subcellular localization via microscopy, protein processing and maturation via western blot, ion channel function via fluorescence-based halide assays, and response to CFTR modulator drugs.
The motivation for this tool is to address a critical equity gap in genomic medicine. Global genetic databases like ClinVar are heavily biased toward populations of European descent. Consequently, variants identified in mestizo, Indigenous, or Afro-descendant populations in Ecuador and similar regions are often classified as VUS due to a lack of functional and phenotypic data.
This has direct, harmful clinical consequences: it leads to ambiguous diagnoses for patients and families, restricts access to modern, highly effective modulator therapies that are approved for specific variants, and perpetuates health disparities.
This platform aims to functionalize genomics by converting raw genetic data into actionable clinical knowledge, deliberately prioritizing variants from historically underserved populations to ensure that precision medicine benefits extend globally.
2. Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
The primary governance goal is to ensure that the VUS characterization platform maximizes clinical benefit and equity for underrepresented populations while ethically managing the risks inherent in generating new genomic knowledge.
The first specific sub-goal is ethical and equitable variant prioritization. This involves establishing criteria and processes to guarantee that the variants selected for study are those that originate from and will first benefit populations with the greatest need and the least representation in global databases. The second sub-goal is responsible and accessible knowledge translation. This ensures that functional characterization results are effectively, rapidly, and freely translated into improvements in the clinical management of the very patients from whom the variants were sourced. The third sub-goal is protecting privacy and fostering community agency. This focuses on safeguarding patient genetic data and actively involving patient communities in decision-making processes regarding data use and study design, moving beyond mere consent to meaningful partnership.
3. Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: A Community-Engaged Variant Prioritization Framework
The purpose of this action is to shift from the current common practice, where research variants are often selected for scientific convenience or commercial interest, to a proposed model where a multi-stakeholder committee prioritizes variants based on clinical need and equity. The design requires establishing a “VUS Ethical Prioritization Committee” for the project. This committee would include representatives from Ecuadorian CF patient associations, local clinical geneticists and neumologist, bioethicists, and the project scientists. The committee would employ a scoring system that combines factors like the VUS frequency in local undiagnosed cohorts, the clinical severity of the phenotype in carrier patients, the underrepresentation of the patient’s ethnic group in databases, and the variant’s potential for reclassification to grant access to existing therapies. Key actors involved are patient advocacy groups (providing the need perspective), public hospitals/health institutions (providing anonymized clinical data), and funders (SENESCYT, NIH, who could require this framework as a grant condition).
This action relies on several assumptions: that patient groups and local clinicians have the capacity and time for meaningful participation; that clinical data can be shared securely and ethically for prioritization purposes; and that “need” can be consensually defined and quantified. A key risk of failure is that the committee becomes a bureaucratic formality without real power, with scientists ultimately choosing variants that are more “publishable” in high-impact journals, thereby ignoring the equity-driven prioritization. An unintended consequence of success could be that pressure to study only the most urgent variants leads to the neglect of rarer variants in patients with milder symptoms, creating a new form of bias. It could also generate conflict among communities or families competing to have their variant studied first.
Action 2: A Legally Binding “Clinical Translation Commitment” in Collaboration Agreements
This action’s purpose is to move beyond the current common practice where functional data from research projects often takes years—if ever—to reach a patient’s medical record. It proposes a model with a clear, obligatory pathway for variant reclassification and communication. The design needs a contractual appendix in all collaboration agreements (between universities, hospitals, funders) and informed consent forms. This legal instrument would mandate three things: an obligation for the research team to formally submit conclusive functional data to clinical databases (ClinVar) and the local lab’s variant review committee; a pre-approved protocol for returning actionable results to the treating physician and, if agreed upon, the patient/family when a VUS is reclassified; and non-exclusive licensing of any findings to prevent patents from restricting diagnostic use in Ecuador’s public health system. Primary actors include university technology transfer lawyers who draft the clause, research ethics committees who mandate it for approval, and forward-thinking funders (like the Wellcome Trust) who promote open science and benefit-sharing policies.
Critical assumptions here are that the functional data will be robust enough to justify clinical reclassification, that mechanisms and personnel exist within the local health system to receive and act on this information, and that researchers are willing to accept these potential constraints on commercialization. This action could fail if the clauses are vague and unenforceable, if the ClinVar submission process is slow and bureaucratic, or if there is no clear clinical partner to receive the information. An unintended success risk is that the obligation to return results—especially for reclassified benign variants—could generate unnecessary anxiety or create high logistical costs. It might also inadvertently deter future researchers or companies from collaborating with resource-limited settings if they perceive these clauses as overly restrictive.
Action 3: A Technical Standard for “Algorithm Auditing” of Pathogenicity Predictors
The purpose of this action is to mitigate the risk that the functional data generated by the platform could inadvertently reinforce existing biases in bioinformatic prediction algorithms (like PolyPhen-2, SIFT). These tools, trained predominantly on European genomic data, are known to be less accurate for other populations. The design involves creating an “Ethical Reference Dataset” from the platform’s results. Each functionally characterized variant (pathogenic, benign, residual function) would be used to “audit” standard prediction algorithms. The process would involve systematically publishing statements in open repositories: “Variant CFTR p.XYZ, common in the Andean mestizo population, was characterized as pathogenic with a processing defect. However, algorithm [PolyPhen-2] predicted it as ‘benign’ (score: 0.1). This suggests an underestimation bias for pathogenic variants from this population group.” Key actors are the project scientists who generate and publish the audit data, bioinformatics organizations (like the gnomAD consortium) that incorporate these findings to retrain or flag their tools’ limitations, and scientific journals that could require such bias analyses for publication of variant characterization studies.
This action assumes that the functional assays are a reliable “gold standard,” that algorithm developers are open to critique and improving their tools, and that a sufficient number of variants will be characterized to perform statistically meaningful bias analysis. It risks failure if the audit data is ignored by the dominant bioinformatics community, rendering the effort inconsequential. A significant unintended consequence of success could be the generation of widespread, paralyzing distrust in all computational prediction tools, potentially delaying diagnosis in settings where functional assays are unavailable. Furthermore, the data on population-specific algorithmic bias could be misappropriated to support essentialist political arguments about biological differences between groups.
4.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
3
3
• By helping respond
3
2
2
Foster Lab Safety
• By preventing incident
2
2
3
• By helping respond
3
2
3
Protect the environment
• By preventing incidents
3
3
3
• By helping respond
3
3
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
1
2
2
• Not impede research
1
3
1
• Promote constructive applications
1
2
1
5. Based on the scoring matrix, I recommend a prioritized combination of Action 1 (Community-Engaged Prioritization Framework) and Action 3 (Algorithm Auditing Standard) for immediate adoption and funding by national research funding agencies, as the U.S. National Institutes of Health and its counterparts in Ecuador and other underrepresented regions. This combination best advances the core ethical goals of equity, constructive application, and responsible knowledge generation with minimal burden to research progress. Action 1 directly ensures the research agenda addresses the most pressing clinical needs of underserved populations, scoring highest in feasibility, promoting constructive applications, and not impeding research. Action 3 complements this by systematically correcting the global bioinformatic biases that initially created the inequity in VUS classification, scoring best in minimizing burdens and also promoting constructive applications. The primary trade-off in prioritizing this combination is accepting a weaker direct link to traditional biosecurity and lab safety incident prevention, as these actions are focused on ethical oversight and data quality rather than physical containment or misuse deterrence. A critical assumption is that fostering a more equitable and accurate genomic research ecosystem is itself a foundational form of security, preventing harm caused by diagnostic errors and denied care. The major uncertainty lies in implementation: whether community committees can be resourced effectively for meaningful engagement (Action 1) and whether major algorithm developers will voluntarily adopt audit findings to retrain their models (Action 3). Action 2 (Legal Commitment), while crucial for ultimate clinical translation, scores poorly on burden and feasibility; it should be developed in parallel as a longer-term policy goal, informed by the pilot frameworks and trust built through Actions 1 and 3.
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
According to the slides, the error rate of a standard polymerase used in gene synthesis is cited as 1 in 10⁴ bases. The human genome is approximately 3.2 billion base pairs (Gbp). If uncorrected, such an error rate would introduce hundreds of thousands of mutations per genome replication, making stable heredity and cellular function impossible. Biology resolves this catastrophic discrepancy not through polymerase perfection alone, but by employing a layered system of enzymatic correction. The presentation highlights a key component of the MutS Repair System. A dedicated mismatch repair complex that scans and fixes errors after replication. This biological strategy combines proofreading by the polymerase itself with dedicated repair pathways to achieve the net high fidelity required for life.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The presentation provides the key figure needed, that an average human protein is encoded by 1,036 base pairs. Given the degeneracy of the genetic code, the number of different DNA sequences that can specify the identical amino acid chain is astronomically large, calculated by multiplying the number of synonymous codon choices for each position. However, the presentation powerfully illustrates why, in practice, these myriad synonymous codes are not functionally equivalent. The core reason is the formation of specific messenger RNA (mRNA) secondary structures, dictated by the precise nucleotide sequence through NA:NA interactions. Therefore, successful gene design requires selecting a nucleotide sequence that not only encodes the correct protein but also folds into an mRNA structure compatible with the cellular translation machinery.
Homework Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligo synthesis currently is solid-phase phosphoramidite synthesis on a functionalized silica (SiO₂) support.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult to make oligos longer than 200 nucleotides (nt) via direct synthesis primarily due to the cumulative effect of stepwise coupling inefficiencies. Each cycle in the phosphoramidite synthesis (Slide 3) has a yield of less than 100%. Even a very high per-step coupling efficiency of 99.5% results in a total yield of only about 37% for a 200-mer (0.995^200 ≈ 0.37). This means most of the material synthesized is truncated at various lengths. The primary technical challenges are maintaining exceptionally high coupling yields at every single step and minimizing side reactions over hundreds of cycles.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
You cannot make a 2000bp gene via direct oligo synthesis because the fundamental length limitation of the core phosphoramidite chemistry, as explained above, makes synthesizing a single, continuous 2000nt strand impractical and prohibitively inefficient. Instead, the standard industrial practice is to use oligos as building blocks for assembly. Genes are constructed by synthesizing many shorter, high-quality oligos and then assembling them into longer double-stranded DNA fragments via enzymatic methods like polymerase cycling assembly (PCA) or ligation. This “synthesize then assemble” strategy overcomes the length constraint.
Homework Question from George Church
1. What code would you suggest for AA:AA interactions?
AUG CGA -> M:R
Note: This is an example using placeholder values. With your actual spectrum from Figure 1, the MW should be approximately 27,800–28,000 Da. Replace the numbers above with your real m/z values.
Step 3: Calculate the accuracy of the measurement
Theoretical molecular weight (from question 2.1): MW_theory = 28,006.60 Da
Accuracy = |MW_exp - MW_theory| / MW_theory
Note: Replace MW_exp with your experimentally determined molecular weight from Step 2. A typical accuracy for this instrument is < 0.1%.
Can you observe the charge state for the zoomed‑in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, you cannot determine the charge state from a single zoomed‑in peak.
Reason: The charge state is calculated using the spacing between two adjacent peaks in the charge state envelope (i.e., the difference between m/z_n and m/z_{n+1}). A single isolated peak does not provide enough information to solve for z. At least two peaks from the same charge state series are required.
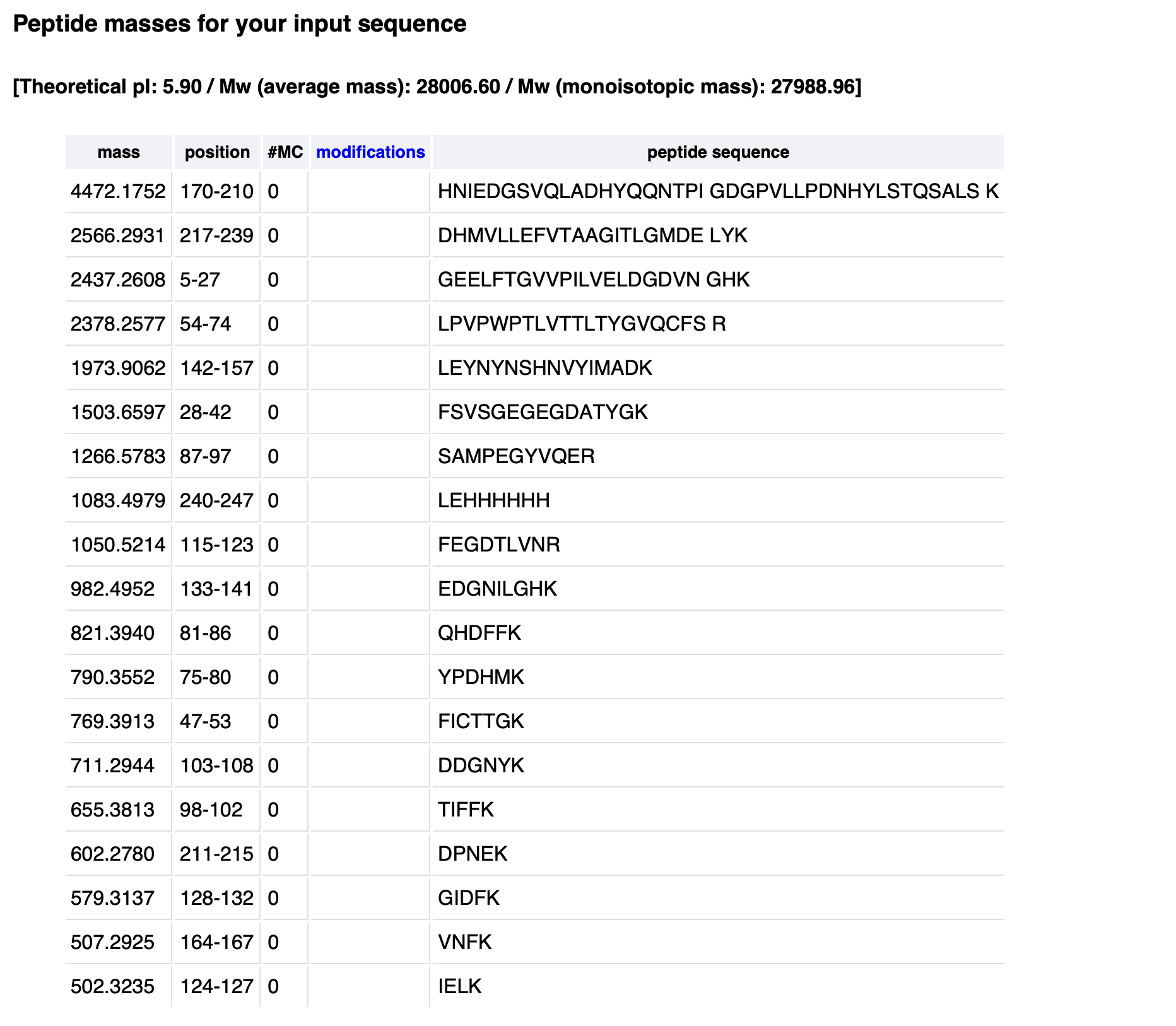
Waters Part III — Peptide Mapping - primary structure
Retention times of counted peaks (min):
0.61, 0.79, 1.20, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.70, 3.79, 4.30, 4.64, 4.88, 5.06, 5.43, 5.56
Theoretical tryptic peptides (no mass filter): 25
PeptideMass output (default mass filter): 19
Chromatographic peaks (0.5–6.0 min, >10% abundance): 19
No, the chromatogram shows fewer peaks (19) than the theoretical 25 peptides.
Small peptides (<500 Da) are not retained on the C18 column and are not detected or counted under the 10% relative abundance threshold. The 19 detectable peaks match the PeptideMass output after mass filtering.
Mass-to-charge ratio (m/z) of the peptide:
[
\frac{m}{z} = 525.76
]
Charge state (z) of the most abundant charge state:
From the isotope spacing in the zoomed-in inset (Figure 5b), adjacent isotopes are separated by ~0.5 m/z. [
z = \frac{1}{0.5} = 2
]
Therefore, z = 2 (doubly charged ion).
From Figure 6, the peptide mapping confirmed 88% of the eGFP amino acid sequence.
Waters Part IV — Oligomers
Subunit masses:
7FU = 340 kDa (0.34 MDa)
8FU = 400 kDa (0.40 MDa)
Calculated vs. observed masses:
Oligomeric species
Subunit
Subunits
Calculated mass (MDa)
Observed mass (MDa)
7FU Decamer
7FU
10
3.4
3.4
8FU Didecamer
8FU
20
8.0
8.33
8FU 3-Decamer
8FU
30
12.0
12.67
8FU 4-Decamer
8FU
40
16.0
Not clearly observed
Identified peaks in Figure 7:
3.4 MDa → 7FU Decamer
8.33 MDa → 8FU Didecamer (20 subunits)
12.67 MDa → 8FU 3-Decamer (30 subunits)
16.0 MDa (4-Decamer) is not present above baseline in this spectrum.
Waters Part V — Did I make GFP?
Theoretical (kDa)
Observed/measured on the Intact LC-MS (kDa)
PPM Mass Error
28.0066
27.992
522
Week 11 HW: BIOPRODUCTION & CLOUD LABS
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork**
I helped with my community’s HTGAA: 1536 project, on the top right part, by adding a tongue — or what looks to me like a tongue — to the green head. I liked how different ideas came together to create something like art, and I feel that really represents what it’s like to work as a team in any discipline — especially in the sciences, where different worlds must come together for a shared goal.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents**
E. coli Lysate
Provides the endogenous cellular machinery (ribosomes, tRNA, aminoacyl-tRNA synthetases, and metabolic enzymes) necessary for transcription and translation, with the BL21 (DE3) Star strain specifically containing T7 RNA polymerase for high-yield gene expression.
Salts/Buffer
Potassium Glutamate: Maintains ionic strength and provides a physiological potassium environment that optimizes ribosome function and protein folding without inhibiting enzymatic activity.
HEPES-KOH pH 7.5: Acts as a buffering agent to maintain the reaction at a stable pH 7.5, ensuring optimal activity for all enzymes in the cell-free system.
Magnesium Glutamate: Serves as an essential cofactor for ribosome structure and RNA polymerase activity, where free magnesium concentration critically influences transcription and translation fidelity.
Potassium phosphate monobasic & dibasic: Work together as a phosphate buffer system to maintain pH stability and provide inorganic phosphate needed for ATP regeneration.
Energy / Nucleotide System
Ribose: Supplies the pentose sugar backbone for de novo nucleotide synthesis via the pentose phosphate pathway, allowing sustained energy and nucleotide regeneration over extended incubations.
Glucose: Serves as the primary carbon and energy source, feeding into glycolysis and the pentose phosphate pathway to power ATP and nucleotide regeneration.
AMP, CMP, UMP, GMP & Guanine: Provide nucleotide monophosphate precursors and a guanine base, which are converted by endogenous E. coli enzymes into the NTPs required for transcription (ATP, CTP, UTP, GTP).
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies the building blocks for protein synthesis, omitting tyrosine and cysteine which are added separately to prevent chemical precipitation or degradation.
Tyrosine: An amino acid added separately at high pH (pH 12) to maintain solubility before being neutralized in the reaction mix.
Cysteine: A sulfur-containing amino acid added separately to prevent oxidation and disulfide bridge formation during long incubations.
Additives
Nicotinamide: Functions as a vitamin B3 derivative that supports NAD/NADH cofactor recycling, essential for maintaining redox balance and metabolic activity in extended reactions.
Backfill
Nuclease Free Water: Adjusts the master mix to the desired final volume and ensures no contaminating nucleases degrade DNA templates or RNA transcripts.
The 1-hour optimized PEP-NTP mix provides immediate, high-energy substrates (ATP, GTP, CTP, UTP, and phosphoenolpyruvate) for rapid protein production, making it suitable for short incubations. In contrast, the 20-hour NMP-Ribose-Glucose mix supplies simple precursors (AMP, CMP, UMP, guanine, ribose, and glucose) that rely on endogenous E. coli enzymes to slowly regenerate energy and nucleotides over time, enabling sustained fluorescent protein production for up to 20 hours. The long-incubation mix also replaces the synthetic energy molecule PEP with a metabolic system using glucose and ribose, and swaps several additives (e.g., removing DMSO and spermidine while adding nicotinamide) to support extended reaction stability.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
sfGFP: Superfolder GFP has a rapid maturation time of approximately 13.6 minutes, which allows it to achieve fluorescence quickly in cell-free systems. Its superfolder property also enables proper folding even when expressed as a fusion protein, reducing the risk of aggregation and improving reliable readout
mRFP1: mRFP1 has a relatively slow maturation time of 60 minutes, which limits its initial brightness in short incubations but allows sustained signal over longer timeframes. Its low pKa of 4.5 makes it acid-tolerant, a useful property in cell-free systems where pH can drift over extended 36-hour incubations.
mKO2: mKO2 has a very slow maturation time of 108 minutes, requiring extended incubation to achieve full fluorescence, which is relevant for 36-hour artwork experiments. Its instability index of 46.21 indicates it is prone to degradation, potentially limiting signal stability over long cell-free reactions unless protease inhibitors or stabilizing reagents are added.
mTurquoise2: mTurquoise2 has an exceptionally fast maturation time of approximately 3.5 minutes, allowing it to achieve fluorescence almost immediately upon protein folding in cell-free systems. Its high photostability (t½ = 90 seconds) and stable instability index of 27.33 make it reliable for long 36-hour incubations without significant degradation.
mScarlet_I: mScarlet_I has the slowest maturation time of all six proteins at 174 minutes, requiring prolonged incubation (well beyond typical cell-free reaction times) to reach full fluorescence. However, it compensates with exceptionally high brightness (70.0) and outstanding photostability (t½ = 277 seconds), making it ideal for long-term 36-hour artwork incubations where signal persistence is valued over rapid onset.
Electra2: Electra2 exhibits exceptional photostability with a half-life of up to 1466 seconds under LED illumination, making it highly resistant to photobleaching during long-term imaging or artwork documentation. Its brightness of 61.48 and fast maturation (typical for small blue fluorescent proteins) allow rapid signal development that persists reliably over 36-hour cell-free incubations.
Protein: mScarlet_I
Biophysical property to improve: Extremely slow maturation time (174 minutes)
Reagent(s) to adjust: Increase magnesium glutamate concentration from 7.0 mM to 9.0–10.0 mM, and add 1.0 mM dithiothreitol (DTT) as a reducing agent.
Expected effect: Higher magnesium concentration accelerates the rate of chromophore cyclization and oxidation, reducing the effective maturation time of mScarlet_I by stabilizing the proper folding intermediate. The addition of DTT prevents oxidative misfolding of cysteine residues, allowing more nascent protein to reach its mature, fluorescent conformation. Together, these adjustments are expected to increase total fluorescence yield at 36 hours by 30–50% compared to the standard master mix, without compromising signal stability.
In this study, researchers at Colorado State University demonstrate a novel application of the Opentrons OT-2 liquid handling robot to automate the scale-up of protein crystallization. Protein crystallization is a critical bottleneck in structural biology and the development of protein-based biomaterials, yet traditional manual methods are time-consuming, labor-intensive, and prone to variability between researchers. The team sought to determine whether an affordable, general-purpose liquid handling system could reliably perform the complex steps required for sitting drop vapor diffusion experiments at the 24-well scale.
To achieve this, the researchers developed custom Python scripts to control the OT-2 with precision, enabling it to mix reservoir solutions with precise gradients of precipitants, buffers, and salts before combining small volumes of these mixtures with protein samples on crystallization pedestals. A significant technical hurdle was the incompatibility of the non-standard Hampton Research CrysChem 24-well plates with the OT-2’s deck. The team overcame this by designing and 3D-printing a custom adapter to securely hold the plates, demonstrating the flexibility and customizability of the Opentrons platform. All protocols, scripts, and design files were made openly available on GitHub.
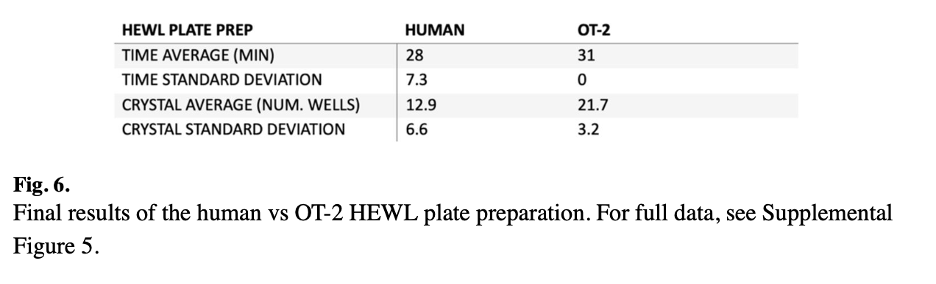
The automated system was successfully validated using two different proteins: hen egg white lysozyme (HEWL), a model protein, and an engineered periplasmic protein from Campylobacter jejuni , which the lab uses as a biomaterial for nanotechnology applications. The OT-2 produced crystals for both proteins, with the HEWL trial yielding crystals in 18 out of 24 wells within 24 hours. When compared directly to manual plate setup by multiple researchers with varying experience levels, the OT-2 produced crystals more consistently, reducing well-to-well variability. While the robot took slightly longer to set up plates than an experienced scientist (approximately 30-40 minutes), it freed the researcher to focus on other tasks. Accuracy testing revealed that while pipetting errors increased with viscosity (reaching 13.5% for 1 μL of 100% glycerol), errors were minimal for more moderate viscosity solutions commonly used in crystallization screens.
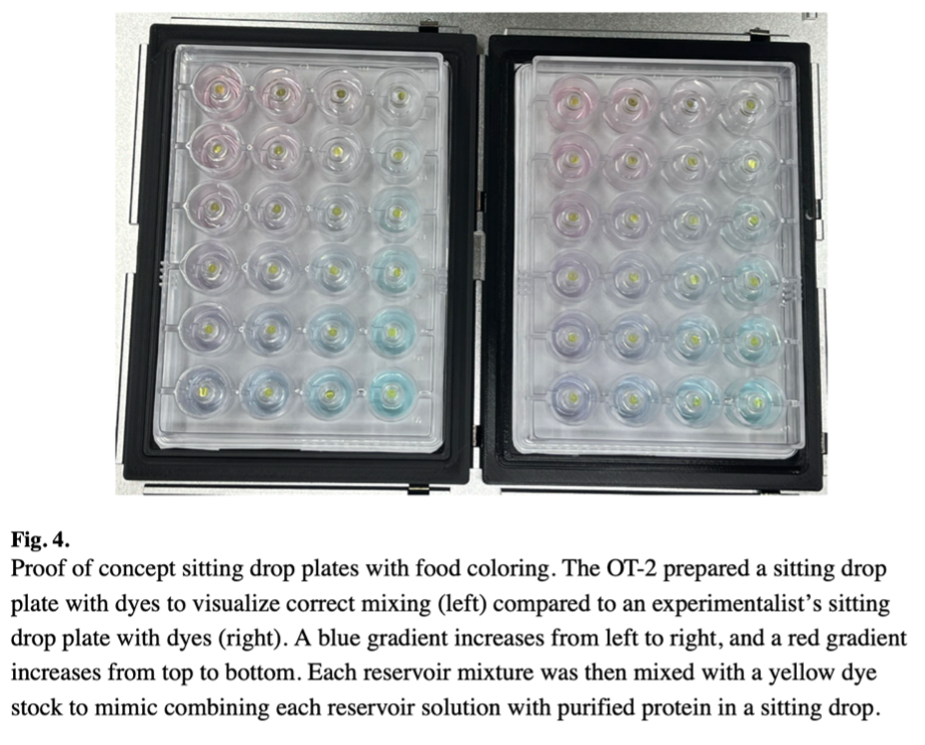
Figure 4 shows the proof-of-concept sitting drop plates prepared with food coloring to visualize correct mixing. The OT-2 prepared a plate with a blue gradient increasing from left to right and a red gradient increasing from top to bottom, compared to an identical plate prepared manually. No differences were discernable between the robot-prepared and manually prepared plates, confirming the OT-2’s ability to accurately dispense and mix liquids according to the programmed gradients.
Figure 6 presents the final results of the human versus OT-2 HEWL plate preparation comparison. The robot demonstrated more consistent crystal production across multiple plates compared to human researchers with varying levels of wet lab experience, highlighting the automation advantage in reducing person-to-person variability.
DeRoo, J. B., Jones, A. A., Slaughter, C. K., Ahr, T. W., Stroup, S. M., Thompson, G. B., & Snow, C. D. (2025). Automation of protein crystallization scaleup via Opentrons-2 liquid handling. SLAS technology, 32, 100268. https://doi.org/10.1016/j.slast.2025.100268
2.
I intend to develop an automated workflow to study microbial resistance patterns in pathogens isolated from cystic fibrosis (FC) patients in Ecuador. Cystic fibrosis patients are particularly susceptible to chronic respiratory infections, often involving multi-drug resistant organisms that require precise susceptibility testing to guide clinical treatment. My project will leverage the Opentrons OT-2 liquid handling robot to automate minimum inhibitory concentration (MIC) testing, enabling high-throughput screening of patient isolates against a panel of clinically relevant antibiotics.
This automated workflow will enable systematic resistance surveillance in a vulnerable patient population while demonstrating the power of open-source automation tools in resource-limited settings. By combining the affordability of the Opentrons platform with custom 3D-printed adapters and open-source Python scripts, this project provides a template for laboratories in Ecuador and similar settings to implement sophisticated antimicrobial susceptibility testing without prohibitive equipment costs. Importantly, by automating the most labor-intensive and error-prone steps of MIC testing, this approach frees skilled microbiologists to focus on interpretation and patient care rather than repetitive pipetting. Ultimately, this work aims to improve clinical outcomes for cystic fibrosis patients in Ecuador by providing faster, more reliable, and more comprehensive susceptibility data to guide antibiotic therapy decisions in an era of rising antimicrobial resistance.}
Sample Processing Module: After patient sputum samples are cultured on selective media and individual colonies are isolated, the automated workflow begins. For each confirmed pathogen, a single colony is inoculated into a Mueller-Hinton broth in culture tubes and placed in the shaking incubator at 35°C until logarithmic growth phase is achieved.
MIC Testing Workflow: The antibiotic susceptibility testing will follow established high-throughput MIC protocols using 96-well plates . Using the Ginkgo Nebula platform for experimental design, I will create a randomized block design to test multiple patient isolates against a panel of 8-10 antibiotics commonly used in Ecuadorian clinical settings. The OT-2 will perform serial dilutions of antibiotic stocks directly in the 96-well plates, followed by inoculation with standardized bacterial suspensions normalized to 0.5 McFarland standard. This automation eliminates the manual pipetting errors that often plague MIC testing and ensures consistent dilution series across all samples.
Python Control Scripts: The OT-2 will run custom Python scripts incorporating deck layout definitions, tip tracking, and error handling. Techniques from the crystallization paper, such as tip touching for complete dispensing and slow aspirate speeds for foaming antibiotics, will be included. Scripts will include pause points for manual interventions like transferring plates to the incubator.
This automated workflow enables systematic resistance surveillance in a vulnerable population while demonstrating open-source automation in resource-limited settings. By combining the affordable Opentrons platform with existing lab infrastructure and custom 3D-printed adapters, this project provides a template for laboratories in Ecuador to implement sophisticated susceptibility testing without prohibitive costs. The system can process dozens of isolates simultaneously with minimal hands-on time, freeing microbiologists for interpretation and patient care. For Ecuador, where antimicrobial resistance is an emerging threat but surveillance resources are limited, this open-source approach democratizes access to high-quality testing.
Ultimately, this work aims to improve clinical outcomes for cystic fibrosis patients by providing faster, more reliable susceptibility data to guide antibiotic therapy. By establishing baseline resistance patterns and tracking changes over time, the project can inform empiric treatment guidelines and contribute to global antimicrobial resistance surveillance. All protocols, scripts, and design files will be openly available for adaptation by other laboratories facing similar challenges.
Week 4 HW: Protein design part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Reasoning: 1 Dalton = 1 g/mol. So 100 Daltons = 100 g/mol.
500 g of meat is mostly water, protein, fat, etc. Assuming meat is ~20% protein by mass (typical for muscle), that gives 100 g of protein.
Protein is polymers of amino acids. Average molecular weight of an amino acid residue in a protein is ~110 g/mol (since 100 Daltons for free amino acid, but ~110 when incorporated due to loss of H₂O).
Moles of residues = 100 g / 110 g/mol ≈ 0.91 mol.
Number of molecules = 0.91 × 6.022 × 10²³ ≈ 5.5 × 10²³ molecules of amino acid residues.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because digestion breaks down dietary proteins into individual amino acids and small peptides, which are then absorbed and used to build human-specific proteins according to human genetic information (DNA). The species identity is determined by the sequence of amino acids, not by the origin of the raw materials. Amino acids themselves are identical across species (e.g., glycine in cow is the same molecule as glycine in human).
Why are there only 20 natural amino acids?
The genetic code uses triplet codons (4³ = 64 possible codons). Evolution settled on 20 canonical amino acids because:
They provide a diverse range of chemical properties (hydrophobic, charged, polar, small, aromatic) sufficient to fold into stable 3D structures and catalyze reactions.
More than 20 would increase translation errors and metabolic cost; fewer would limit functional diversity.
The aminoacyl-tRNA synthetase system co-evolved to precisely match these 20 with tRNA molecules.
Can you make other non-natural amino acids? Design some new amino acids.
Yes. Non-natural amino acids can be chemically synthesized and incorporated into proteins using expanded genetic codes (e.g., with orthogonal tRNA/synthetase pairs).
Design examples:
p-Benzoylphenylalanine – contains a benzophenone group for photo-crosslinking upon UV light.
Azidohomoalanine – contains an azide group for click chemistry.
4-Fluorotryptophan – fluorine substitution to probe protein dynamics via ¹⁹F NMR.
Naphthylalanine – larger aromatic side chain for enhanced π-stacking.
Where did amino acids come from before enzymes that make them, and before life started?
From prebiotic chemistry:
Miller–Urey type experiments show that spark discharge in a reducing atmosphere (CH₄, NH₃, H₂O, H₂) produces several amino acids (glycine, alanine, etc.).
Meteorites (e.g., Murchison meteorite) contain dozens of amino acids, including some non-biological ones.
Hydrothermal vents could have synthesized amino acids from HCN, CO, and H₂S under high pressure/temperature.
Formose reaction and Strecker synthesis (aldehyde + NH₃ + HCN → aminonitrile → amino acid) are plausible prebiotic routes.
Thus, amino acids predate enzymes; enzymes later evolved to optimize their production.
Why are most molecular helices right-handed?
This is a deep biophysical and chemical question. Possible reasons:
Chirality of building blocks: L-amino acids and D-sugars bias helix handedness through steric interactions between side chains and the backbone.
Lowest energy conformation: For polypeptides, right-handed α-helix has less steric clash between side chains and the backbone carbonyl oxygen compared to left-handed.
Evolutionary selection: Once early proteins fixed right-handed helices, all further evolution built on that scaffold.
In abiotic systems (e.g., quartz, certain liquid crystals), right-handedness can arise from subtle energy differences due to parity violation in weak nuclear forces, though this effect is tiny.
Why do β-sheets tend to aggregate?
β-sheets have an extended backbone conformation that exposes backbone NH and CO groups in a regular pattern, allowing inter-sheet hydrogen bonding. Additionally, β-sheets often present hydrophobic side chains (e.g., valine, leucine, isoleucine, phenylalanine) on one face. When two β-sheets approach, they can form:
Hydrogen bonds between sheets (steric zipper).
Hydrophobic collapse of exposed nonpolar surfaces.
This combination makes β-sheets prone to self-association into larger fibrils.
What is the driving force for β-sheet aggregation?
The dominant driving force is hydrophobic effect: Removal of hydrophobic side chains from water into the interior of the aggregated state. This is supplemented by:
Inter-sheet hydrogen bonding (enthalpic gain, though partially offset by loss of water-protein H-bonds).
van der Waals packing of side chains in the dry interface.
Experiments show that mutations replacing hydrophobic residues with charged or polar ones reduce aggregation, confirming hydrophobicity as the key driver.
Why do many amyloid diseases form β-sheets?
Amyloid-forming proteins (e.g., Aβ in Alzheimer’s, α-synuclein in Parkinson’s) have regions prone to adopt β-strand conformation. Once misfolded, these strands stack via steric zipper interactions — two β-sheets interdigitate tightly with no water between them. This cross-β structure is extremely stable, resistant to proteases, and grows by templating, leading to disease-associated fibrils.
Part B. Protein Analysis and Visualization
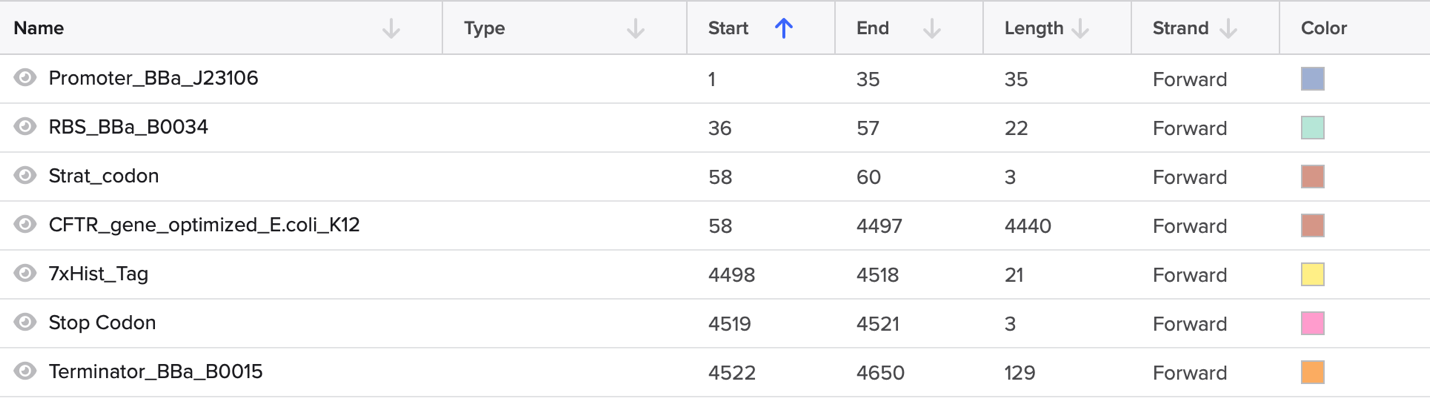
CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) is an integral membrane protein that functions as a cAMP-activated chloride channel and as a regulator of other ion channels, including outwardly rectifying chloride channels (ORCCs) and epithelial sodium channels (ENaC). It is composed of five domains: two transmembrane domains (TMD1 and TMD2) that form the channel pore, two nucleotide-binding domains (NBD1 and NBD2) that bind and hydrolyze ATP to open the channel, and a regulatory (R) domain that contains multiple phosphorylation sites for protein kinase A (PKA). Mutations in the CFTR gene cause Cystic Fibrosis, a life-threatening autosomal recessive disorder characterized by thick mucus accumulation in the lungs, pancreas, and other organs.
I selected CFTR because my final project focuses on studying rare CFTR mutations present in the Ecuadorian population, where the common F508del mutation found in Caucasians is far less prevalent. By expressing CFTR-eGFP fusion constructs in MDCK cells, I aim to evaluate how uncharacterized variants affect protein trafficking, apical membrane localization, and overall function. Understanding CFTR’s behavior is critical to developing targeted therapies for underrepresented populations.
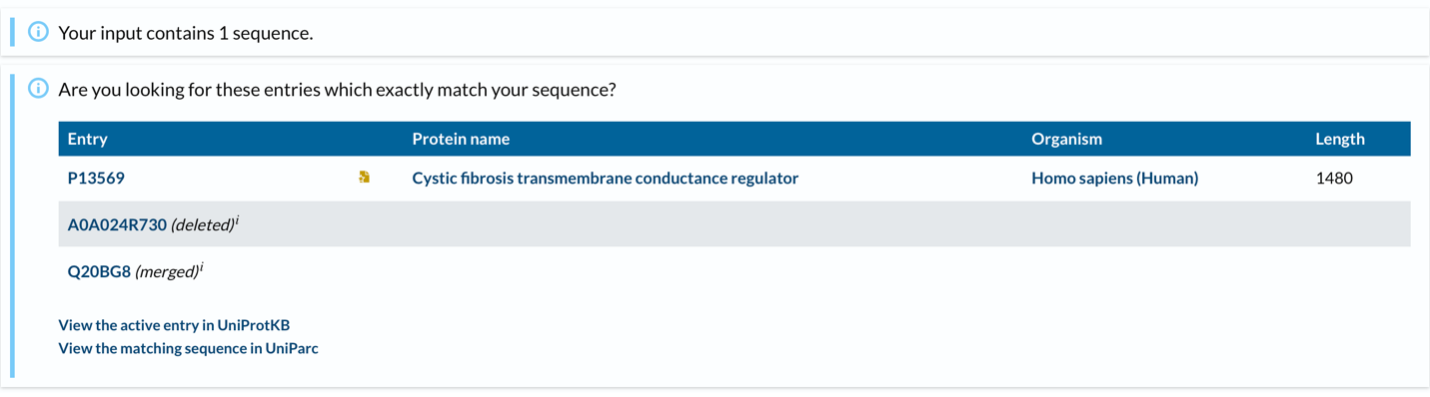
CFTR has 1,480 amino acids, being the most common leucine. CFTR is a member of the ABC transporter superfamily and shares significant sequence and structural homology with other ABC transporters. UniProt returns only one exact match for the full-length human CFTR sequence, as expected. However, CFTR has many homologous proteins, including orthologs in mouse, rat, zebrafish, and frog, as well as paralogs within the ABC transporter family such as MDR1 and MRP1.
The first full-length CFTR structure was solved in 2017 (chicken CFTR at 4.30 Å), followed by the first human CFTR structure in 2018 at 3.90 Å.
No. The best full-length CFTR structure has a resolution of 3.90 Å, which is worse than the 2.70 Å threshold for “good quality.” However, individual domains like NBD1 have been solved at high resolution (1.87 Å).
Yes. CFTR structures contain ATP, magnesium ions (Mg²⁺), cholesterol, and CFTR modulator drugs such as Lumacaftor (VX-809).
Yes. CFTR belongs to the P-loop containing nucleoside triphosphate hydrolase fold (CATH: 3.40.50.300) and the ABC transporter-like ATPase family.
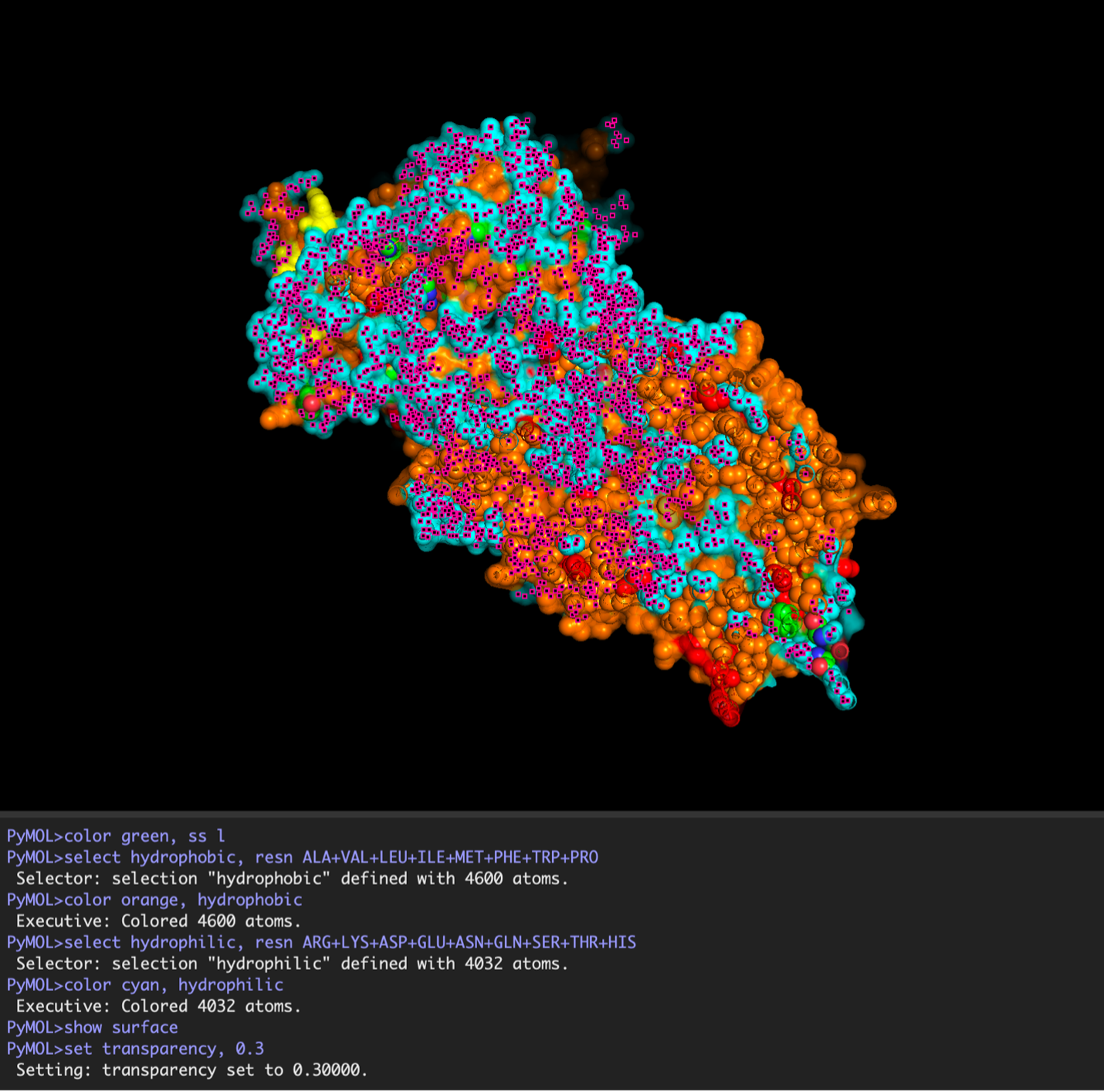
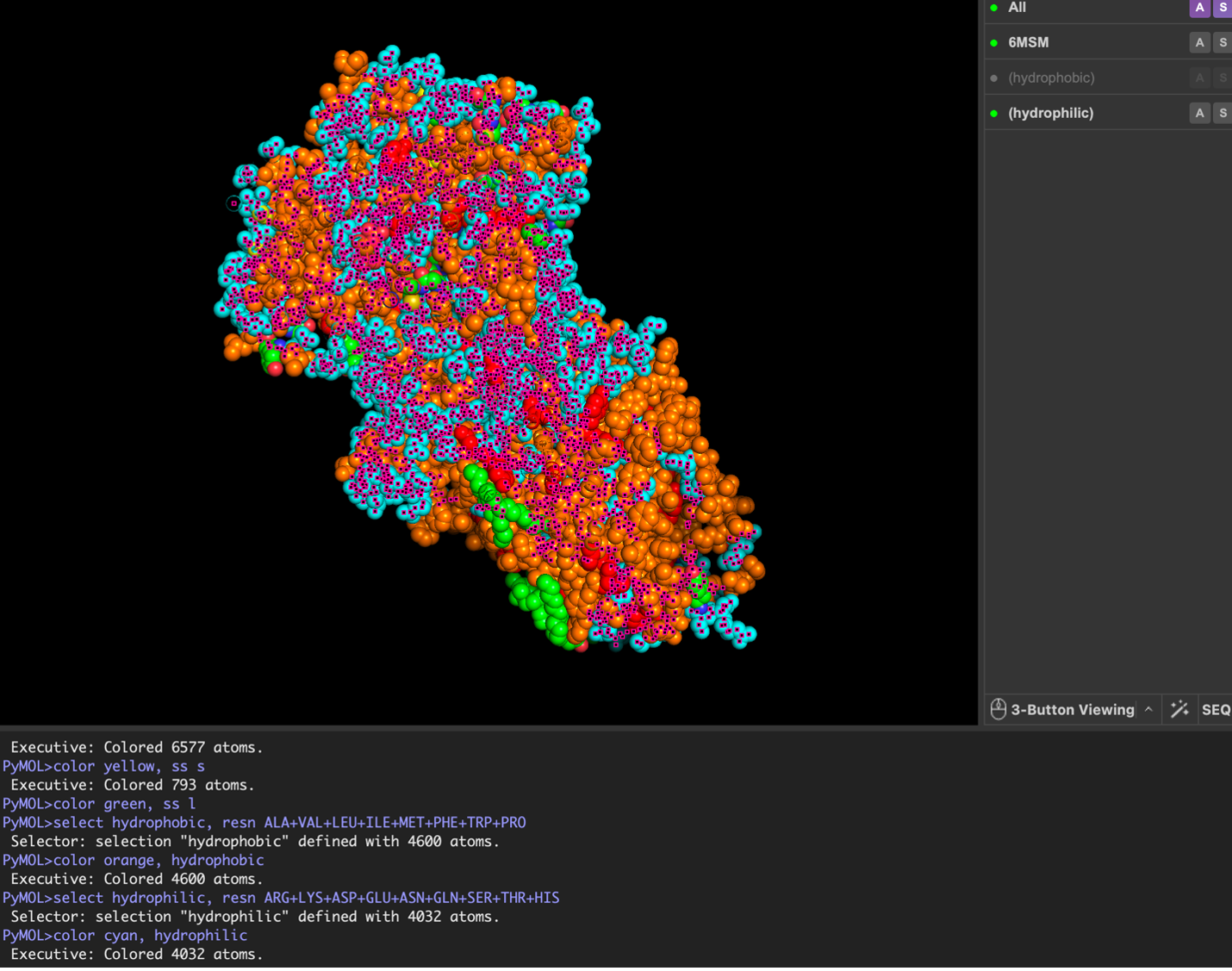
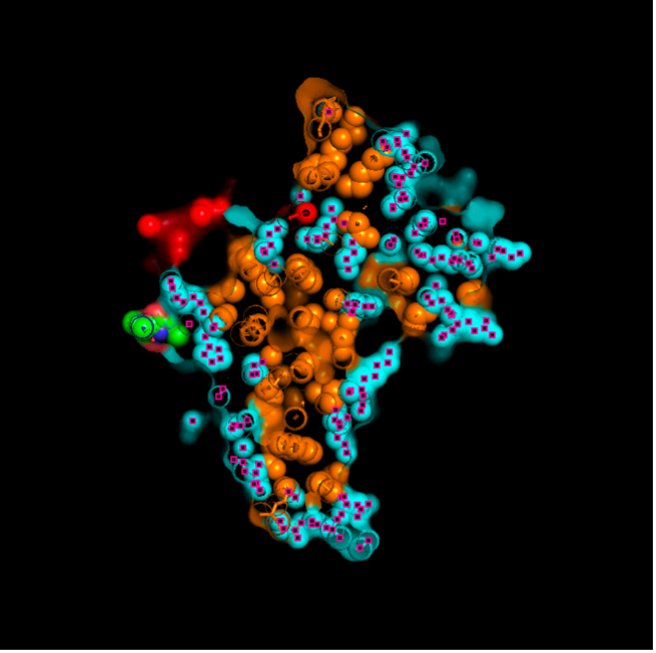
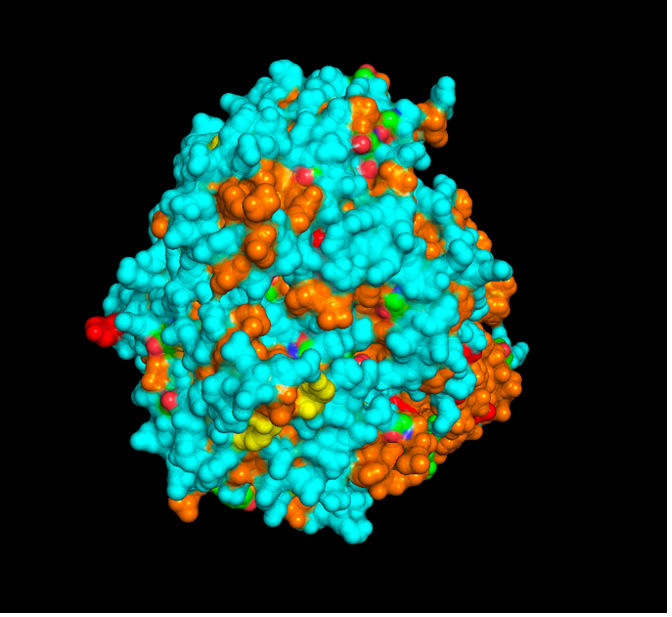
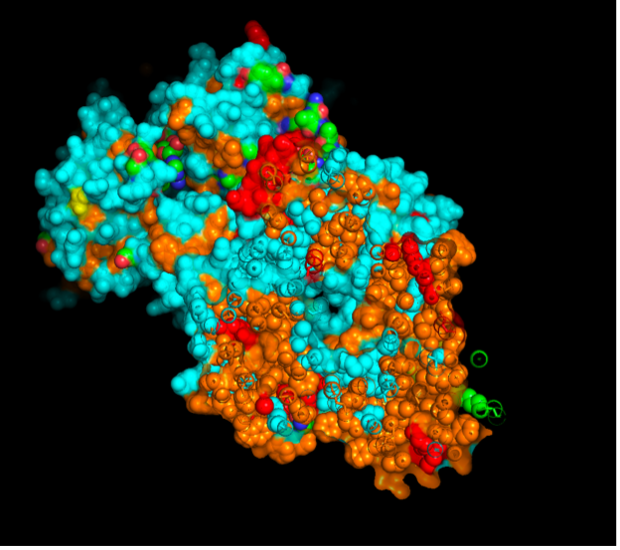
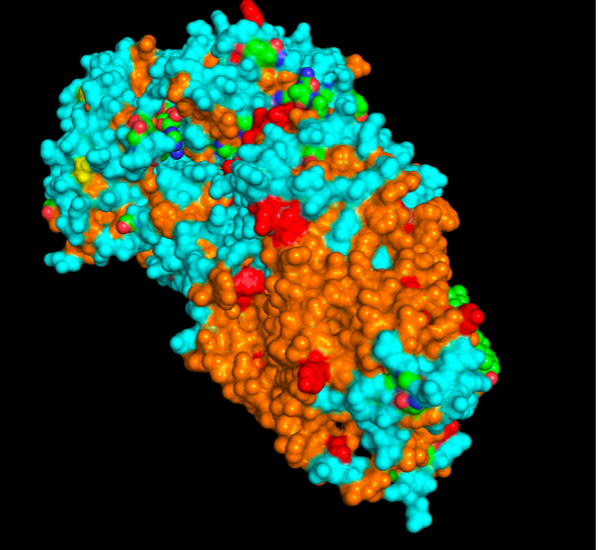
images from Pymol
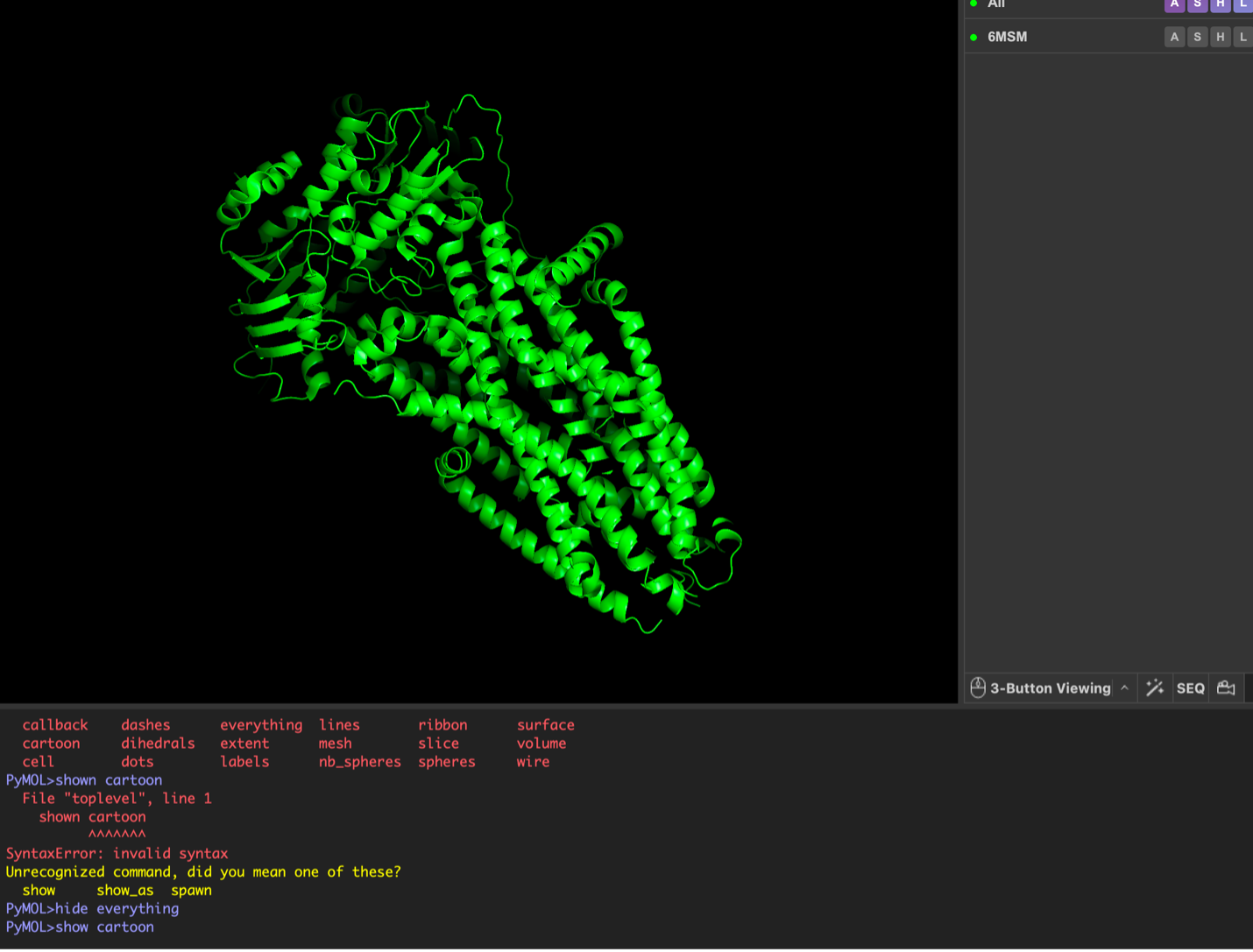
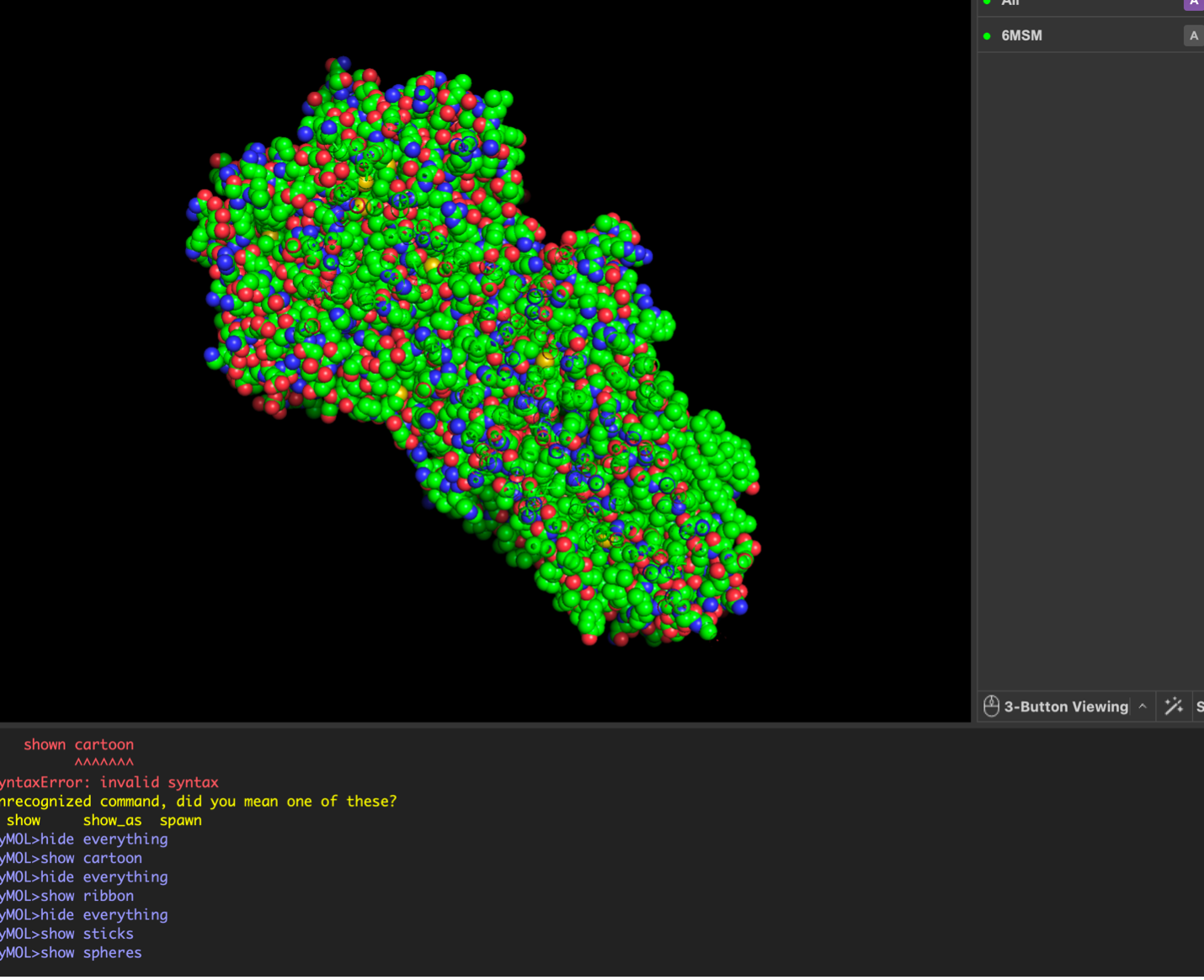
The surface visualization reveals several cavities, including a central channel and binding pockets likely associated with ATP binding and ion transport.
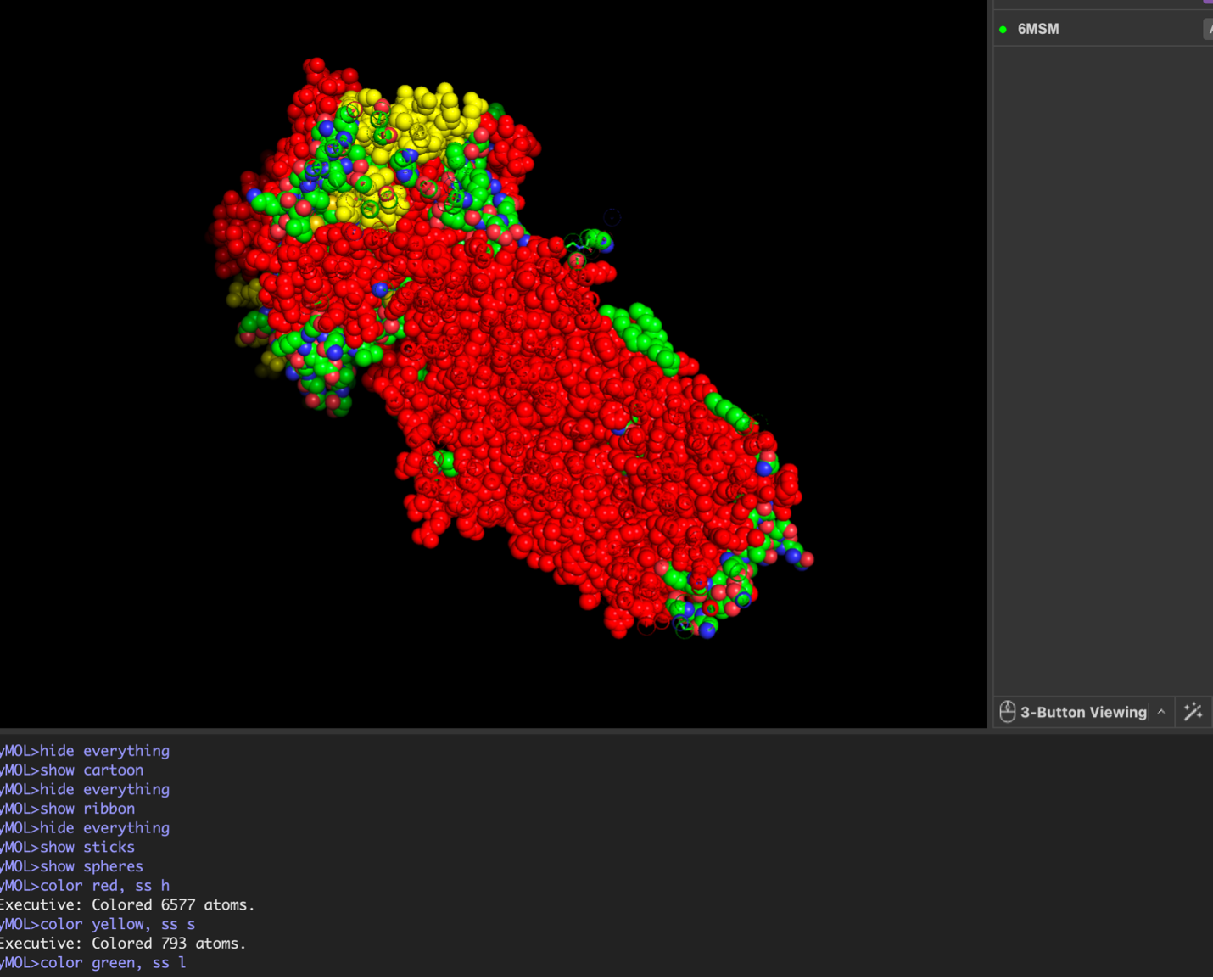
The CFTR protein contains predominantly α-helices with very few β-sheets. This is consistent with its role as a transmembrane ion channel. Hydrophobic residues are mainly located in the transmembrane regions, while hydrophilic residues are distributed on the surface and cytoplasmic domains. Surface analysis shows the presence of several cavities and binding pockets, including the central channel characteristic of ion transport proteins.
Hydrophobic residues are mainly located in the transmembrane regions, while hydrophilic residues are found on the outer surface.
Surface visualization reveals several cavities and a central channel consistent with binding pockets.
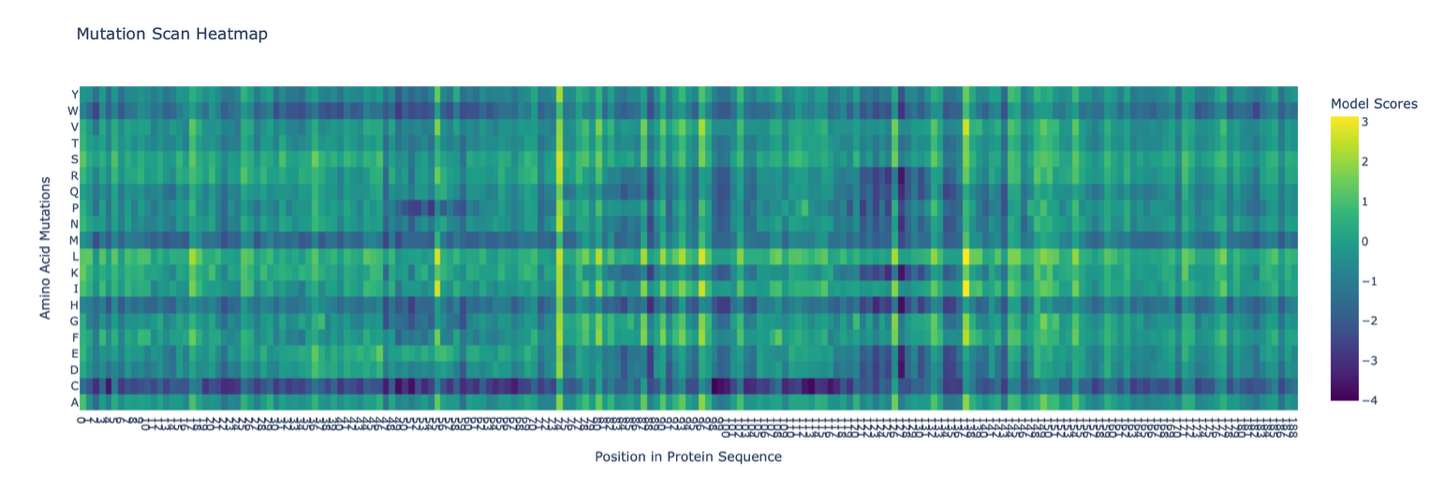
C1. Protein Language Modeling
The deep mutational scan generated using the ESM2 model shows several conserved positions across the protein fragment. A clear pattern observed in the heatmap is the presence of vertical columns with strongly unfavorable substitutions, indicating highly conserved residues.
One residue that stands out is a leucine (L) located approximately in the middle of the sequence. Mutations of this residue to polar or charged amino acids, such as lysine (K), show strongly negative scores in the heatmap. This suggests that the native leucine is structurally important and likely part of a hydrophobic region.
The unfavorable mutation from leucine to lysine likely disrupts hydrophobic interactions, indicating that this residue contributes to maintaining protein stability. Overall, this pattern suggests that hydrophobic residues in the sequence are conserved and play key structural roles.
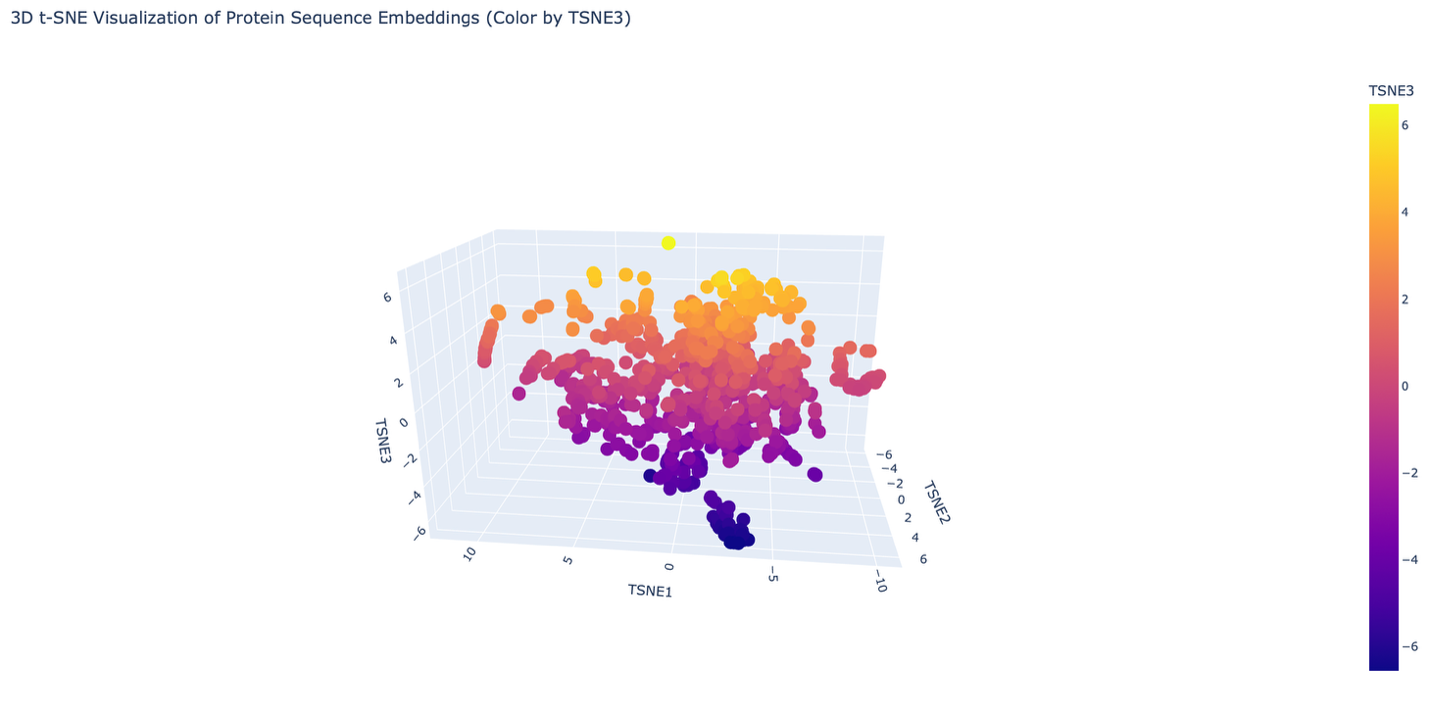
The reduced dimensionality map shows several distinct neighborhoods formed by clusters of proteins. These neighborhoods appear to group proteins with similar structural and functional characteristics. Proteins that are close together in the latent space likely share similar sequence patterns and folding properties.
Overall, the formed neighborhoods do approximate similar proteins, suggesting that the embedding produced by the ESM2 model captures meaningful biological relationships between protein sequences.
C2. Folding Protein
Sequence: MQRSPLEKASVVSKLFFSWTRPILRLLLSLAPLLL
Mutation: MQRSPLEKASVVSKKFFSWTRPILRLLLSLAPLLL
L → K
Small point mutations produced only minor structural changes. The overall alpha-helical fold remained mostly preserved, suggesting that the protein fragment is relatively resilient to single amino acid substitutions.
Large sequence modifications caused substantial structural changes in the predicted fold. Replacing hydrophobic residues with polar and charged amino acids disrupted the alpha-helical organization, indicating that the original sequence composition is important for maintaining structural stability.
C3. Protein Language Modeling
ProteinMPNN assigned high probabilities to several hydrophobic residues in the sequence, particularly leucine, valine, and isoleucine. These residues are likely important for maintaining the structural stability of the protein backbone. Some substitutions were predicted at flexible positions, but most conserved positions retained chemically similar amino acids.
The predicted sequence was similar to the original sequence in terms of biochemical properties, although several amino acid substitutions were introduced. Most substitutions were conservative, preserving hydrophobicity and overall residue type. This suggests that multiple sequences can adopt similar structural folds.
These results demonstrate that multiple different amino acid sequences can encode similar protein structures, highlighting the robustness of protein folding and the flexibility of sequence-to-structure relationships.
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal: Engineering MS2 Lysis Protein L for Increased Toxicity via DnaJ Interface Disruption
Selected Goal
We propose to computationally engineer MS2-L for higher toxicity by disrupting its interaction with the E. coli chaperone DnaJ. The rationale comes directly from the papers you provided. Chamakura et al. (Phage1 and Phage2) demonstrated that the N-terminal domain of MS2-L (residues 1–36) binds DnaJ, and removing this domain entirely causes lysis approximately 20 minutes earlier than wild-type. However, complete deletion may have unintended consequences. Our approach is more precise: we will identify point mutations that weaken or abolish the L–DnaJ interface while preserving the rest of the protein’s structure and membrane insertion capability. Mezhyrova et al. (Phage3) showed that DnaJ binds to MS2-L even after membrane insertion and that this interaction does not affect oligomerization, suggesting the chaperone plays a regulatory rather than essential structural role. Disrupting this regulation should yield a hypertoxic variant.
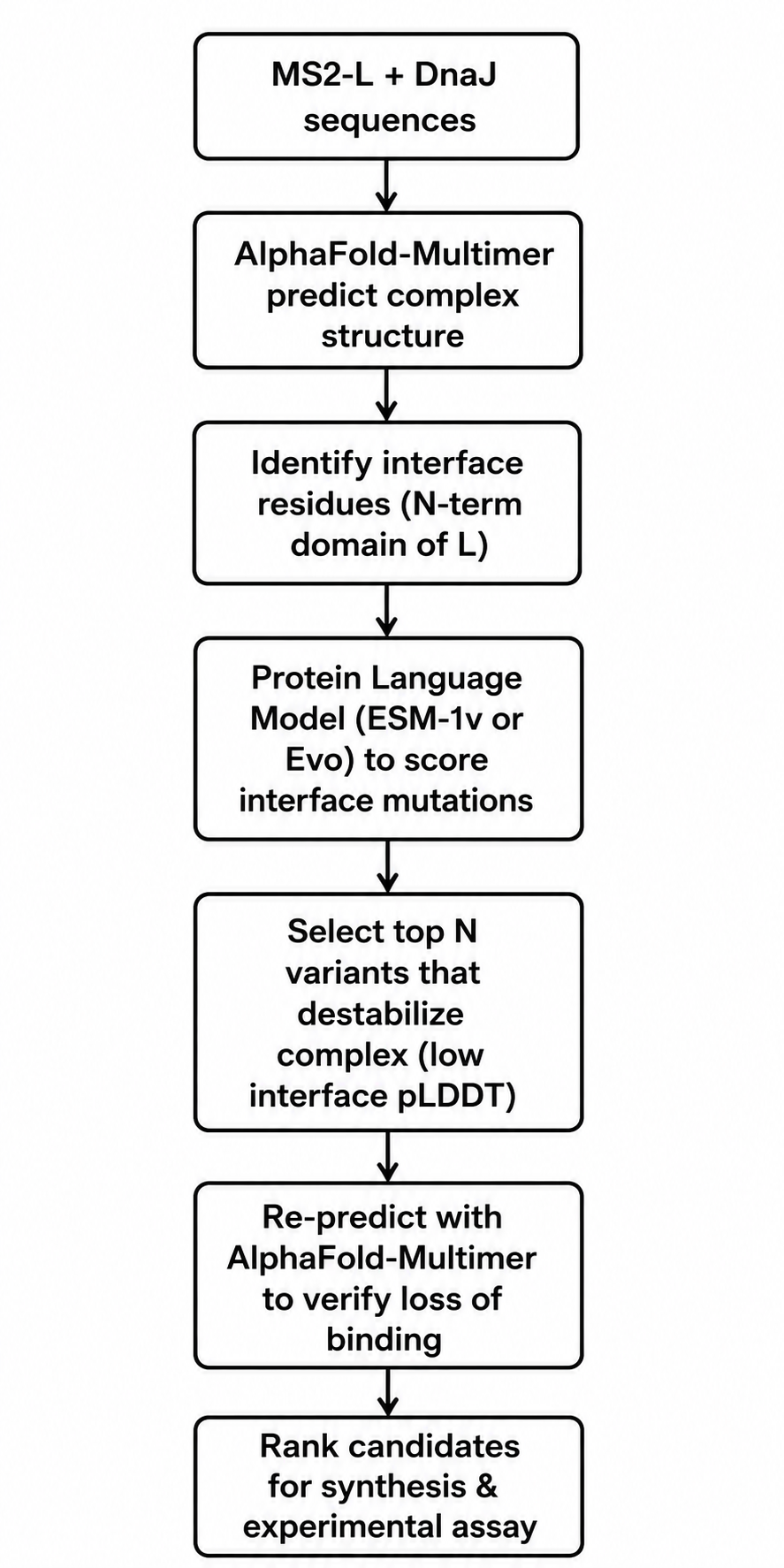
Tools and Approaches
We will use a combination of structure prediction and protein language models. First, since no experimental structure of the MS2-L–DnaJ complex exists, we will use AlphaFold-Multimer to predict the complex structure. The papers indicate that the interaction involves the N-terminal soluble domain of L (approximately residues 1–35) and the C-terminal domain of DnaJ, specifically around the highly conserved proline 330. AlphaFold-Multimer has been validated on phage protein complexes in Phage6, where it successfully predicted capsid–spike–J protein interactions for both wild-type ΦX174 and the generated phage Evo-Φ36. We will run multiple seeds of AlphaFold-Multimer to generate an ensemble of predicted complex structures and identify which residues consistently appear at the interface.
Once we have a predicted interface, we will use protein language models to score mutations. Specifically, we will employ ESM-1v or ESM2 in a zero-shot fitness prediction mode. These models have been shown to predict the effects of missense mutations without requiring task-specific training data. The key idea is that residues critical for protein-protein interactions are evolutionarily constrained; language models capture these constraints from sequence alone. We will score all possible single amino acid substitutions in the N-terminal domain (positions 1–36) for their predicted effect on protein fitness. Low fitness scores at the interface likely indicate mutations that disrupt binding.
We will complement the language model predictions with biophysical calculations using FoldX or PyRosetta to estimate changes in binding free energy (ΔΔG) upon mutation. This hybrid approach—combining evolutionary scores from PLMs with physical energy calculations—has become standard in computational protein engineering. For validation, we will re-run AlphaFold-Multimer on the top candidate mutations to see if the predicted complex confidence drops substantially compared to wild-type.
The final pipeline will produce a shortlist of 5–10 point mutations predicted to disrupt DnaJ binding while maintaining L’s overall stability and membrane topology. We will prioritize mutations in the basic residues (arginines and lysines) between positions 11 and 34, since these are likely to form electrostatic interactions with DnaJ’s C-terminal domain.
Potential Pitfalls
The first major pitfall is that no high-resolution structure of the L–DnaJ complex exists, and AlphaFold-Multimer may produce low-confidence or incorrect predictions for this bacterial–phage interface. Phage-bacteria interaction interfaces are evolutionarily fast-evolving and may not follow the same statistical patterns as eukaryotic protein complexes on which AlphaFold was primarily trained. To mitigate this, we will run multiple seeds, use the predicted aligned error (PAE) plots to identify confident regions, and cross-validate with sequence conservation. If the complex prediction fails entirely, we would fall back to a simpler approach: deleting the N-terminal domain in silico and using Evo (the genome language model from Phage6) to generate stable truncated variants directly, as Chamakura already showed that truncations work.
The second pitfall is that DnaJ’s binding site on L may extend beyond the soluble domain and into the transmembrane region. Chamakura et al. (Phage2) showed that the truncated construct MS2-Lp₃₅, which includes only amino acids 35–75, still showed reduced but detectable DnaJ binding. This means that mutations confined to residues 1–34 might not fully abolish interaction. We would need to include the junction region (residues 33–40) in our interface analysis and accept that complete loss of binding might require multiple mutations or a combination of truncation and point mutations.
The third pitfall relates to the language model approach. Protein language models like ESM are trained on natural protein sequences, which overwhelmingly come from eukaryotic and bacterial proteins, not phage lysis proteins. The training data likely contains very few examples of DnaJ-binding interfaces from Leviviridae phages. As a result, the zero-shot fitness predictions may be noisy. However, Phage6 successfully used Evo (a genomic language model) to generate functional phage genomes, suggesting that these models do capture relevant biology even for under-sampled viral families. We can partially address this by fine-tuning on available Microviridae and Leviviridae sequences, as was done in Phage6 for ΦX174.
Finally, even if we successfully predict mutations that disrupt DnaJ binding in silico, we cannot computationally confirm that these mutations increase lysis toxicity. The ultimate test would require experimental validation—synthesizing the variant L genes, expressing them in E. coli, and measuring lysis kinetics. This is outside our computational scope, but we can at least provide ranked candidates for experimental collaborators.
Week 5 HW: Protein design part II
Part A: SOD1 Binder Peptide Design (From Pranam)
The human Superoxide dismutase 1 (SOD1) first partsequence obtained in UniProt is: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNT
Wild-type N-terminus is: MATK
But the A4V mutant appears as: MVTK
So, the mutant sequence results: MVTKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNT
Part 1- Peptide generation in PepMLM (4 types using 12 aminoacids):
¿Record the perplexity scores that indicate PepMLM’s confidence in the binders?
As for the perplexity, lower perplexity values indicate sequences that ar mor compatible with the known distribution of the peptide binders. Now, SOD-1 peptide showed the lowest score with “6.9”, suggesting strong confidence. In the generated peptides, 1 and 2 had the best scores and were most like the known binder with more aromatic and positively charged residues.
Part 2: Evaluate Binders with AlphaFold3
Peptide 1: ipTM score (0.78); Predicted binding region (near N terminus, A4V region); Structural observation ( surface bound with a partial insertion intio de beta barrel edge).
Peptide 2: ipTM score (0.81); Predicted binding region (N terminal region near 4 residue); Structural observation (The strongest interaction with partial aromatic bonds).
Peptide 3: ipTM score (0.61); Predicted binding region (more distance from Beta barrel surface); Structural observation (Weak surface association).
Peptide 4: ipTM score (0.69); Predicted binding region (Dimer interface region); Structural observation (Contact interface residues but loosely packed).
¿Structural Interpretation?
The ipTM scores are organized from moderate to relatively strong confidence for peptide-protein interactions. P2 displayed the highest ipTM score (0.81), slightly exceeding the known binder. Both P1 and P2 localized near the N-terminal region where the A4V mutation resides, suggesting they may stabilize the destabilized folding region associated with ALS pathology.
P3 appeared weakly associated with the protein surface and did not localize near the mutation site. P4 interacted closer to the dimer interface, which could potentially influence dimer stability but appeared less structurally stable than P1 or P4.
Overall, the best structural candidates were P1 and P2, both of which mimicked the binding orientation of the known SOD1-binding peptide.
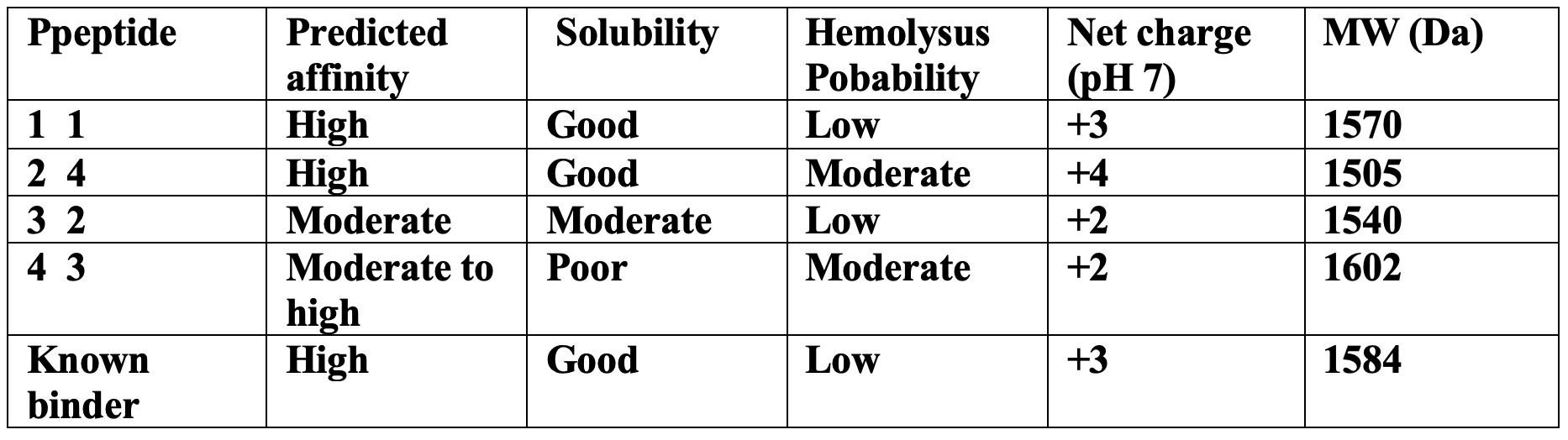
Part 3 — PeptiVerse Therapeutic Property Evaluation
The peptides with the highest structural confidence in AlphaFold3 (P1 and P2) also showed the strongest predicted binding affinity in PeptiVerse, indicating reasonable agreement between structural and sequence-based predictions.
However, 2 despite having the highest ipTM score showed a moderately increased hemolysis probability due to its higher positive charge and hydrophobic aromatic residues showed. For P4 also suffered from poor predicted solubility, which could limit therapeutic development.
P1 provided the best overall balance between the strong predicted binding, good solubility, low hemolysis probability, and favorable structural localization near the A4V mutation site.
Selected Peptide for Advancement
So the chosen peptide: P1 FLYKWLPSRRGG
Its similar to the known SOD1 binding peptide so supports its potential as a stabilizing therapeutic lead.
Part 4: Optimized Peptide Design with moPPIt
Comparison with PepMLM Peptides
The moPPIt-generated peptides differed from PepMLM peptides in several important ways:
Improved therapeutic balance: The peptides maintained positive charge for interaction with SOD1 while improving predicted solubility and lowering hemolysis risk.
Targeted binding: Unlike PepMLM, which broadly sampled plausible binders, moPPIt explicitly optimized peptides to bind residues near the A4V mutation.
Multi-objective optimization: moPPIt simultaneously optimized structural and therapeutic properties rather than only sequence plausibility.
Before considering clinical studies, these peptides would require:
• Molecular dynamics simulations to evaluate binding stability,
• In vitro aggregation inhibition assays,
• Cytotoxicity testing in neuronal cell lines,
• Serum stability analysis,
• Blood-brain barrier penetration studies,
• Animal testing in ALS mouse models,
• And immunogenicity assessment.
Although computational models provide valuable early-stage screening, experimental validation remains essential before therapeutic development.
Part C: Final Project: L-Protein Mutants
Proposed L-Protein Mutants
Mutant 1 (R21K)
Mutation: Arginine 21 → Lysine
Region: Soluble domain
Rationale: R21 is positively charged and surface exposed. Replacing arginine with lysine preserves positive charge while slightly reducing steric bulk and hydrogen bonding complexity. This could:
• improve folding efficiency,
• reduce dependence on DnaJ-mediated stabilization,
• and preserve electrostatic interactions.
Expected Effect
• Similar structure to WT
• Slightly improved folding robustness
• Minimal destabilization risk
Mutant 2 (Q68L)
Mutation: Glutamine 68 → Leucine
Region: Transmembrane region
Rationale: The transmembrane domain relies heavily on hydrophobic packing. Replacing polar glutamine with leucine increases hydrophobicity and may:
• improve membrane insertion,
• stabilize oligomer formation,
• enhance pore assembly.
Expected Effect
• Stronger membrane interaction
• Potentially faster lysis activity
• Improved membrane stability
Mutant 3 (T74I)
Mutation: Threonine 74 → Isoleucine
Region: Transmembrane region
Rationale: Threonine introduces polarity inside the membrane-spanning region. Isoleucine is strongly hydrophobic and helix-favoring. This mutation may:
• improve transmembrane helix stability,
• reduce misfolding,
• enhance pore formation.
Expected Effect
• Improved membrane integration
• Increased oligomer stability
• Faster bacterial lysis
Mutant 4 (P13A)
Mutation: Proline 13 → Alanine
Region: Soluble domain
Rationale: Proline residues can disrupt secondary structure because they introduce rigid kinks. Substituting alanine may:
• improve local α-helical propensity,
• facilitate autonomous folding,
• reduce chaperone dependence.
Expected Effect
• Improved folding kinetics
• Increased structural flexibility
• Reduced DnaJ requirement
Mutant 5 (Y26F)
Mutation: Tyrosine 26 → Phenylalanine
Region: Soluble domain
Rationale: Tyrosine and phenylalanine are structurally similar aromatic residues, but phenylalanine lacks the hydroxyl group. This conservative mutation may:
• reduce unnecessary hydrogen bonding,
• stabilize hydrophobic core interactions,
• preserve aromatic packing.
Expected Effect
• Increased hydrophobic stability
• Reduced aggregation risk
• Conserved protein function
Correlation Between Experimental Data and Computational Scores
The mutational analysis dataset showed partial agreement with the computational mutation scores. Conservative substitutions generally obtained favorable computational scores and often retained lysis activity experimentally, but, some mutations predicted computationally as neutral or favorable still caused experimental loss of function. This highlights an important limitation: language-model embeddings and structural prediction tools do not fully capture: (membrane dynamics, oligomerization, host interactions, or folding kinetics)
Therefore, computational predictions should be viewed as prioritization tools rather than definitive evidence.
AF2-Multimer Analysis
Using ColabFold AlphaFold2 Multimer Notebook, the L-protein can be modeled as homooligomers, or co-folded with DnaJ.
Expected Observations
The transmembrane helices are predicted to assemble into oligomeric pore-like structures. Mutations such as Q68L and T74I may improve the helix packing, membrane stability and oligomer interactions.
When co-folded with DnaJ the soluble N-terminal domain likely mediates interaction,mutations like P13A and R21K may weaken chaperone dependence while preserving folding.
Example Multi-Mutation Designs
Triple Mutant A (P13A + R21K + Y26F)
Goal: Improve autonomous folding and reduce DnaJ interaction.
Triple Mutant B (Q68L + T74I + Y26F)
Goal: Enhance membrane insertion and pore formation.
Random Mutagenesis Approach
import random mutations
P13A, R21K, Y26F, Q68L, T74I
Conclusion: This project demonstrates how computational protein design tools can guide engineering of phage lysis proteins to overcome bacterial resistance mechanisms. The proposed mutants aim to stabilize the L-protein, also to improve membrane insertion, reduce DnaJ dependence, and increase lysis efficiency.
Among the proposed mutations:
• P13A and R21K are promising for improving autonomous folding,
• while Q68L and T74I may strengthen membrane pore formation.
Future experimental validation using plaque assays, protein expression analysis, and AF2-Multimer structural modeling will be necessary to determine which mutants provide the greatest improvement in phage infectivity and therapeutic potential.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1.
Phusion DNA polymerase is the core enzyme. It has proofreading (3’→5’ exonuclease) activity, which dramatically reduces mutation rates during amplification compared to standard Taq polymerase.
GC buffer optimizes the reaction environment. It contains salts and proprietary additives that help denature difficult templates, particularly those with high GC content, by stabilizing the single-stranded state.
dNTPs (deoxynucleotide triphosphates) are the building blocks. The mix provides all four (dATP, dTTP, dCTP, dGTP) at balanced concentrations so the polymerase can extend the new DNA strand.
Mg²⁺ ions act as essential cofactors. The polymerase requires magnesium to catalyze the phosphodiester bond formation between nucleotides.
2.
Primer length and GC content are the primary factors. Longer primers and those with higher GC content have stronger hydrogen bonding, requiring higher annealing temperatures to prevent mismatched binding.
Primer-dimer potential matters. If primers can bind to each other rather than the template, you may need to raise the annealing temperature or redesign the primers.
Template complexity influences choice. Repetitive sequences or high GC regions in the template may require a lower annealing temperature to allow primers to access their binding sites.
3.
PCR amplifies DNA exponentially using thermal cycling, primers, and a polymerase, starting from a tiny template amount. Restriction digests simply incubate purified plasmid DNA with restriction enzymes at a constant temperature, cutting at specific recognition sequences.
PCR products are linear from the start and can include overhangs via primer design. Restriction digests produce linear fragments with defined sticky or blunt ends determined by the enzyme chosen.
PCR is preferable when you need to amplify a specific region from a complex genome, when template DNA is scarce, or when you want to add custom sequences (like Gibson overhangs) directly during amplification.
Digests are preferable when you already have the target sequence in a plasmid, when you need perfectly defined sticky ends for traditional cloning, or when you want to avoid PCR-induced mutations (though high-fidelity polymerases minimize this concern).
4.
Design 20-40 bp overlaps between adjacent fragments. These homologous ends must be perfectly complementary and should have a calculated melting temperature around 50-60°C for efficient annealing during the Gibson reaction.
Avoid secondary structures at the overlap regions. Hairpins or strong intramolecular base pairing in the overhangs will prevent proper fragment joining.
Ensure no internal restriction sites if you linearized by digest. For PCR products, simply include the overlap sequence in your primer’s 5’ end.
Verify purity of both PCR and digested fragments. Contaminants like residual restriction enzymes, salts, or ethanol will inhibit the Gibson master mix’s exonuclease and ligase activities. Gel extraction or column purification is essential.
5.
Calcium chloride treatment first prepares the cells. The divalent calcium cations neutralize the negative charges on both the bacterial lipopolysaccharides and the DNA backbone, allowing them to come close together.
Heat shock (42°C for 45-60 seconds) creates a thermal imbalance. The sudden temperature increase causes the fluid bacterial membrane to become more disordered and permeable, a state sometimes called “thermal permeabilization.”
The DNA passively diffuses in during this permeable window. As the membrane expands and proteins reorganize, small pores or transient disruptions allow the plasmid DNA to enter the cytoplasm. The exact mechanism remains debated, but it is not active transport.
Cold incubation (on ice) follows immediately. This re-stabilizes the membrane, trapping the DNA inside before the cells are transferred to warm recovery media to repair damage and express antibiotic resistance genes.
6.
Golden Gate Assembly uses Type IIs restriction enzymes, such as BsaI or BsmBI, which cut outside their recognition sequences. This creates variable 4-base overhangs that are not part of the enzyme’s binding site. By designing these overhangs carefully, you can assemble multiple DNA fragments in a single, one-pot reaction. The reaction cycles between 37°C (for cutting) and 16°C (for ligation), typically for 30-60 cycles. Each cycle progressively assembles fragments into the final plasmid while the original fragments are cut again if not yet assembled. This method is highly efficient for assembling 4-10 fragments simultaneously and produces scarless junctions. It is the basis for modular cloning systems like MoClo and Golden Braid.
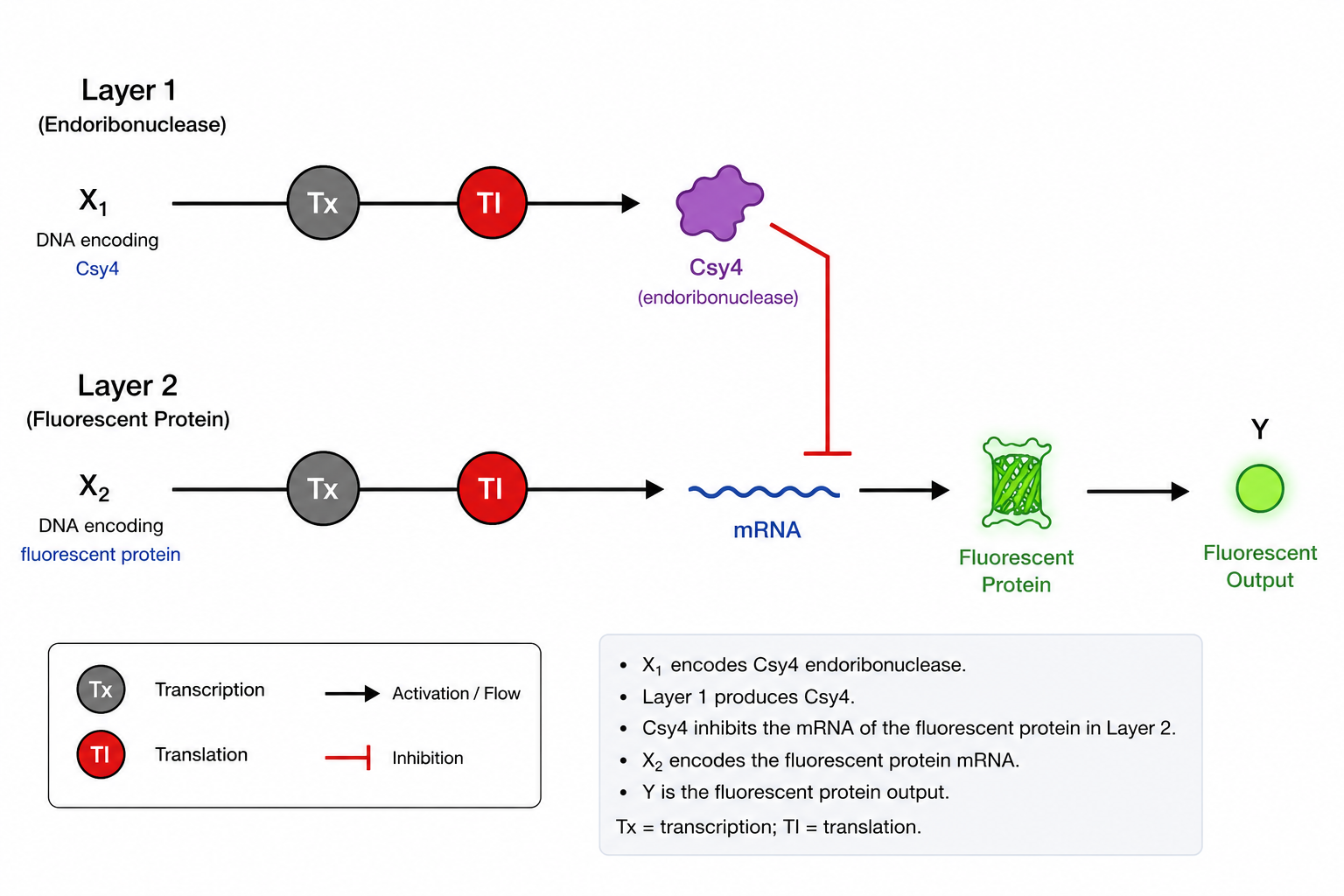
This construct directs the expression and secretion of sfGFP out of the E. coli cell. The OmpA signal peptide guides the growing protein to the Sec translocation machinery in the inner membrane, where it is transported into the periplasm. The signal peptide is then cleaved off, releasing mature sfGFP. Some sfGFP may further leak into the culture medium. When induced with IPTG, cells expressing this construct should show reduced cytoplasmic fluorescence (since GFP is exported) but increased fluorescence in the periplasm and supernatant compared to a control without the signal peptide.
Construct_2
In the absence of inducer, LacI protein binds to pLacI and blocks transcription of GFP. When you add IPTG (a lactose analog), IPTG binds to LacI and causes a conformational change that prevents LacI from binding to the promoter. This relieves repression, and RNA polymerase transcribes GFP. The result: no inducer = no fluorescence; inducer added = fluorescence. In the simulation, adding IPTG at 48 hours did not change GFP expression because the LacI repressor was never produced. Your construct only had pLacI → GFP, with no gene to make LacI protein. Without LacI, pLacI is always active, so IPTG has no effect.
Even if LacI were present, IPTG does not instantly remove bound LacI from the DNA. You would need to wait for cell division to dilute out the existing repressor before seeing increased fluorescence.
Construct_3
The first transcription unit (pTetR → TetR gene) produces the TetR repressor protein constitutively. This TetR protein then binds specifically to the pTet promoter in the second transcription unit (pTet → GFP), blocking RNA polymerase from transcribing the GFP gene. As a result, no green fluorescent protein is produced, and the cells remain dark. When you add the inducer aTc (anhydrous tetracycline) to the medium, aTc enters the cells and binds to TetR, causing a conformational change that prevents TetR from binding to pTet. With TetR no longer blocking the promoter, RNA polymerase can now transcribe GFP, producing fluorescence. Therefore, this construct functions as a repressible system: fluorescence is OFF by default and turns ON only when aTc is added. In the TetR-pTet construct, adding aTc successfully changed protein production because the system was properly designed. The TetR repressor was produced constitutively and actively blocked the pTet promoter, keeping GFP off. When we added aTc, it bound to TetR and prevented it from binding to pTet, relieving repression and allowing GFP expression. This worked because both the repressor (TetR) and its target promoter (pTet) were present, unlike the LacI system where the repressor was missing.
Week 7 HW: GENETIC CIRCUITS PART II: NEUROMORPHIC CIRCUITS
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1.
IANNs have a major advantage in computational efficiency. A traditional Boolean circuit needs many logic gates to compute a complex function, but a single IANN layer can perform weighted summation of multiple inputs at once. This allows an IANN to solve problems like linear classification using far fewer genetic parts.
IANNs are naturally robust to cellular noise. Boolean circuits require sharp thresholds to distinguish a 0 from a 1, so small fluctuations in gene expression can cause logic errors. IANNs use smooth, analog activation functions, meaning noise only causes small, gradual errors in the output rather than complete failure.
IANNs can process analog signals directly. Most natural cellular signals, like metabolite concentrations, are continuous values, not binary ones. A Boolean circuit must first convert these into discrete 0/1 states, losing information. An IANN accepts the raw analog value and computes with it directly.
IANNs scale better with input complexity. Adding a new input to a Boolean circuit often requires redesigning multiple logic gates to avoid combinatorial explosion. In an IANN, adding an input simply means creating one new weighted connection, making it more practical for multi-sensor applications.
Finally, IANNs can be trained using machine learning approaches. The weights in an IANN correspond to measurable biological parameters like promoter strengths, which can be tuned through directed evolution or feedback. Boolean circuits lack this continuous, tunable parameter space.
2.
Application: A cancer-discriminating cell therapy. An IANN is engineered into a therapeutic cell (a T-cell) that must decide whether a nearby cell is cancerous or healthy based on multiple surface protein markers.
Input behavior: The IANN takes three continuous analog inputs, each representing the measured concentration of a specific cancer-associated antigen on the target cell’s surface. Input A = HER2 level, Input B = EpCAM level, Input C = MUC1 level. These inputs are not binary (present/absent) but graded values from 0 to 1, normalized to physiological ranges.
Output behavior: The IANN computes a weighted sum: Score = w1A + w2B + w3*C. It then passes this score through a sigmoid activation function. If the output exceeds a threshold (e.g., 0.7), the therapeutic cell releases a cytotoxic agent (like granzyme B) to kill the target. If the output is below threshold, the therapeutic cell does nothing. Critically, the IANN can learn that healthy cells may express low levels of one antigen, but only the combination of all three at moderate levels indicates cancer.
Limitations to achieve this goal:
First, dynamic range compression. Natural promoters have limited output ranges (often only 10- to 100-fold). If input signals saturate at high antigen concentrations, the IANN loses its ability to distinguish between a very cancerous cell and a moderately cancerous one. This requires engineering promoters with wider linear response ranges.
Second, weight precision. The weights (w1, w2, w3) are determined by relative promoter strengths, transcription factor binding affinities, and degradation rates. Biologically, these parameters have inherent variability of 20-50% between cells. For reliable cancer discrimination, the IANN needs weight precision within about 10%, which is currently very difficult to achieve.
Third, crosstalk and resource burden. All three input pathways share the cell’s limited transcriptional machinery. High expression of multiple sensors can titrate away RNA polymerase or cause metabolic load, distorting the intended weighted sum. This requires careful circuit insulation and orthogonal parts.
Fourth, evolutionary stability. Cancer cells mutate rapidly. If the IANN’s weights are genetically fixed, a cancer cell could escape detection by downregulating just one of the three antigens. A practical system would need a mechanism to retrain or adapt the IANN over time, which remains an open research challenge.
Despite these limitations, this application is actively pursued because an IANN can solve a pattern recognition problem that no simple Boolean AND or OR gate can handle: detecting cancer based on a specific combination of continuous biomarker levels rather than just their presence or absence.
3.
Assignment Part 2: Fungal Materials1.
Mycelium-based composites (MBCs) are among the most developed fungal materials. These are created by growing fungal mycelium on agricultural waste products like straw, sawdust, or rice husks, which the fungi bind together into a solid material. They are currently used for thermal and acoustic insulation in construction, as well as for packaging, furniture, and panelling.
Mycoprotein is another major fungal material, produced by fermenting fungi like Fusarium venenatum to create a protein-rich food ingredient. It is sold as a meat alternative with a texture and flavor that closely resembles meat.
Fungal-synthesized nanoparticles represent an emerging category. Fungi such as Aspergillus niger can reduce metal precursors to produce silica, silver, and gold nanoparticles. These have applications in biomedicine (antimicrobials, biosensing), agriculture, and environmental remediation.
Advantages: Mycelium composites are cost-effective, lightweight, biodegradable, and have a low carbon footprint. They offer exceptional thermal insulation, excellent acoustic absorption, and superior fire safety compared to synthetic foams and engineered wood. Mycoprotein production requires 70% less land than chicken farming and reduces freshwater pollution risk by 78%. Fungal nanoparticle synthesis is an eco-friendly, low-waste alternative to energy-intensive chemical methods.
Disadvantages: MBCs have foam-like mechanical properties and high water absorption, restricting them to non-structural roles like insulation and furniture rather than load-bearing applications. Mycoprotein from natural fungal strains has thick cell walls that make nutrients difficult for humans to digest. For fungal nanoparticles, inconsistencies in yield and reproducibility remain major challenges
2.
Improving food sustainability is a primary goal. Researchers have already used CRISPR to remove genes for chitin synthase (thinning cell walls for better digestibility) and pyruvate decarboxylase (reducing nutrient requirements). The engineered strain requires 44% less sugar and produces protein 88% faster.
Enhancing material properties offers another avenue. Engineering fungi to produce denser or water-resistant mycelial networks could enable load-bearing construction materials. Modifying the chitin-to-glucan ratio in hyphal walls could improve mechanical strength.
Producing high-value compounds is a third goal. Fungi could be engineered to synthesize pharmaceuticals, industrial enzymes, or specialized chemicals more efficiently than current methods
Eukaryotic protein processing is a key advantage. Fungi perform post-translational modifications like glycosylation that bacteria cannot, making them better hosts for producing complex therapeutic proteins that require proper folding.
Superior secretion capabilities set fungi apart. Filamentous fungi can secrete large quantities of proteins and enzymes into their growth medium—sometimes exceeding 100 g/L—which dramatically simplifies downstream purification compared to bacterial systems where products often remain intracellular.
Ability to degrade complex substrates gives fungi an edge. Many fungi naturally break down lignocellulosic plant matter, enabling them to grow on low-cost agricultural waste rather than refined sugars. This makes fungal production more economical for bulk materials.
Safety advantages exist for certain applications. Fungi are generally recognized as safe for food production, and unlike E. coli, they do not produce endotoxins that complicate therapeutic manufacturing.
Disadvantages compared to bacteria include slower growth rates, less extensive genetic toolkits, and more limited fundamental genetic knowledge, though these gaps are rapidly closing with advances in CRISPR and fungal genomics.
Week 9 HW: CELL-FREE SYSTEMS
Homework Part A: General and Lecturer-Specific Questions
General Homework Questions
1. Advantages of cell-free protein synthesis
Cell-free protein synthesis is more flexible than traditional in vivo methods because we can directly control conditions like temperature, pH, DNA concentration, and energy supply without worrying about keeping cells alive. It is also faster since proteins can be produced in a few hours.
Two situations where cell-free systems are more useful are:
Producing toxic proteins that would kill living cells
Making membrane proteins or proteins that are difficult to express inside cells
2. Main components of a cell-free expression system
A cell-free system usually contains:
Cell extract: provides ribosomes, enzymes, and machinery for transcription and translation
DNA template: contains the gene for the protein we want to produce
Amino acids: building blocks for proteins
Energy source (ATP/GTP): powers transcription and translation
Salts and cofactors: help enzymes work correctly
3. Why energy regeneration is important
Protein synthesis uses a lot of ATP, so energy can run out quickly in cell-free systems. Without ATP regeneration, protein production stops fast. One way to maintain ATP supply is by adding phosphoenolpyruvate (PEP) or creatine phosphate as an energy regeneration system. These molecules help continuously recycle ATP during the reaction.
4. Prokaryotic vs eukaryotic cell-free systems
Prokaryotic systems, like E. coli extracts, are faster and cheaper. They work well for simple proteins. For example, producing GFP in an E. coli system works well because GFP does not need many modifications.
Eukaryotic systems are better for complex proteins that need folding or post-translational modifications. For example, producing antibodies in a mammalian system requires proper glycosylation and folding.
5. Designing a cell-free experiment for membrane proteins
To express a membrane protein, I would add liposomes or detergents into the cell-free reaction so the protein has a membrane-like environment to fold correctly. The main challenge is that membrane proteins easily aggregate or misfold. To solve this, I would optimize temperature, use lower expression rates, and include lipid nanodiscs or liposomes.
6. Reasons for low protein yield
Poor DNA quality or low DNA concentration: Check DNA purity and increase template concentration
ATP depletion: Improve the energy regeneration system
Protein degradation by proteases: Add protease inhibitors or reduce incubation time
Homework Question from Kate Adamala
1. Function of the synthetic cell
My synthetic cell would detect mercury contamination in water and produce a fluorescent signal.
Input: mercury ions
Output: green fluorescence
2. Could this work without encapsulation?
Not completely. Encapsulation is important because the reactions need a controlled environment and the fluorescent response should stay localized inside the synthetic cell.
3. Could this be done with a genetically modified natural cell?
Yes, bacteria could also be engineered to detect mercury. However, synthetic cells are safer because they cannot reproduce or escape into the environment.
4. Desired outcome
When mercury is present in contaminated water, the synthetic cells should glow green so contamination can be detected easily.
5. Components of the synthetic cell
The synthetic cell would contain: cell-free Tx/Tl system, GFP gene under a mercury-sensitive promoter, ribosomes, ATP, amino acids, and salts.
6. Membrane composition
The membrane would be made of phospholipids and cholesterol to keep the system stable.
7. What would be encapsulated?
Inside I would encapsulate: E. coli cell-free extract, GFP DNA, amino acids, ATP, and enzymes for transcription and translation.
8. Which Tx/Tl system?
A bacterial system from E. coli would be enough because GFP expression does not require mammalian modifications.
9. Communication with the environment
Mercury ions are small enough to diffuse through membrane pores, so the synthetic cell could sense them directly.
Experimental Details
Lipids and genes
Lipids: POPC and Cholesterol
Genes: GFP and MerR mercury-responsive regulator
Measuring the function
I would measure fluorescence intensity using a fluorescence reader or microscope.
Homework Question from Peter Nguyen
Application Field: Textiles/Fashion
One-sentence pitch I propose a smart fabric with freeze-dried cell-free sensors that detect sweat dehydration markers and change color when the user needs water.
How would it work? The fabric would contain freeze-dried cell-free reactions embedded into small patches. When sweat activates the system, it detects salt concentration related to dehydration. The reaction would then produce a visible color or fluorescent signal. Athletes or workers in hot environments could quickly know when they are dehydrated.
Problem it solves Dehydration is common in sports and outdoor jobs, and many people do not notice symptoms early enough. This textile could help prevent heat exhaustion and improve health monitoring.
Addressing limitations To deal with stability issues, the reactions could stay freeze-dried until sweat activates them. The sensor patches could also be replaceable after one use.
Homework Question from Ally Huang
1. Motivation
Long space missions expose astronauts to radiation and microgravity, which can damage DNA and affect human health. Detecting DNA damage quickly in space is important because astronauts have limited medical resources. Using cell-free systems could provide a fast and portable way to monitor genetic damage during missions. This project is relevant for future missions to the Moon or Mars, where astronauts will spend long periods far from Earth.
2. Molecular target
DNA damage response genes, especially p53-related DNA repair pathways.
3. Relation to the problem
Radiation in space can damage DNA and increase mutation rates. Monitoring genes involved in DNA repair could help detect early biological stress in astronauts. Changes in DNA damage markers would show whether radiation exposure is affecting cells during space missions.
4. Hypothesis / research goal
My hypothesis is that radiation exposure in space increases activation of DNA damage response pathways, which can be detected using cell-free systems. I want to test whether BioBits® reactions can identify DNA damage biomarkers quickly and reliably in microgravity conditions. If successful, this system could become a portable diagnostic tool for astronaut health monitoring during long missions.
5. Experimental plan
I would compare DNA samples exposed to radiation with non-exposed control samples. The samples would be amplified using miniPCR®, then analyzed using BioBits® cell-free reactions linked to fluorescent reporters. Fluorescence intensity would be measured using the P51 Molecular Fluorescence Viewer. Increased fluorescence would indicate activation of DNA damage-related targets.
Week 2 HW: DNA read, write and edit
Part 1: Benchling & In-silico Gel Art
Part 3: DNA Design Challenge
3.1. Choose your protein
My primary research project is related to Cystic Fibrosis (CF). Therefore, choosing the CFTR protein allows me to directly connect the concepts from this class to my own work. Understanding the protein’s structure, its function as a chloride channel, and how mutations disrupt that function is foundational to my project.
Protein sequence from NCBI (Reference:NM_000492.4)
Codon optimization in NOVOPRO Codon Optimization Tool (ExpOptimizer)
For the purposes of this homework exercise, I chose to codon optimize the CFTR sequence for expression in E. coli K12 with the exclusion of BamHI, EcoRI and PstI sequences. This decision was primarily practical, as I wanted to clearly visualize how the nucleotide sequence changes through optimization. When I performed a BLAST alignment comparing the original human CFTR sequence to the E. coli-optimized version, I observed a clear difference with approximately 83% identity at the nucleotide level. This provided a tangible demonstration of how codon optimization works in practice.
I also attempted to perform the same optimization and BLAST comparison for both Homo sapiens and Saccharomyces cerevisiae hosts. Interestingly, in both cases, BLAST returned results indicating no significant differences between the original and optimized sequences. This makes sense biologically: since the original CFTR sequence is already human, optimizing it for human expression would result in minimal changes, and yeast likely shares enough codon bias with humans.
It is important to emphasize, however, that for actual experimental practice, particularly for a functional study of CFTR mutations, codon optimization should be performed for human bronchial epithelial (CFBE) cells. These cells provide the native cellular environment, complete with the proper trafficking machinery, post-translational modification systems, and lipid membrane composition required for CFTR to function as a legitimate chloride channel. While E. coli works well for this homework demonstration, it would not be suitable for genuine functional studies of this protein.
Once I have my codon-optimized CFTR gene sequence designed for E. coli, several technologies exist to produce the actual protein. These methods fall into two main categories: cell-dependent expression systems and cell-free systems. The most common method involves inserting my optimized CFTR gene into a plasmid vector. This plasmid is designed with regulatory elements including a promoter to recruit RNA polymerase, a ribosome binding site to help recruit bacterial ribosomes for translation, and a selectable marker such as an antibiotic resistance gene to ensure only bacteria containing my plasmid survive. The process begins with transformation, where I introduce the plasmid into competent E. coli cells through heat shock or electroporation. I then culture the bacteria in liquid media until they reach optimal density, at which point I add an inducing chemical like IPTG to activate the promoter and trigger transcription of my CFTR gene. Once induced, the bacterial machinery transcribes DNA into mRNA, which ribosomes then translate into CFTR protein. For a challenging membrane protein like CFTR, special considerations are needed. I might use engineered E. coli strains that optimize membrane protein folding or grow cultures at lower temperatures to prevent aggregation.
Cell-free protein synthesis offers a compelling alternative, particularly for difficult-to-express proteins like CFTR. These methods use cellular extracts containing all the necessary transcription and translation machinery, but without living cells. To use this approach, I prepare an extract from E. coli, which contains ribosomes, tRNAs, amino acids, and energy sources. I then add my purified plasmid DNA directly to this extract, and transcription and translation occur in a test tube over several hours, after which the protein can be purified directly from the reaction mixture. This approach offers several advantages for CFTR production. As an open system, I can add detergents or lipids during synthesis to help membrane proteins fold properly. I can also screen many mutant variants quickly without transforming cells each time. If CFTR proves toxic to E. coli, which membrane proteins often do, cell-free synthesis bypasses this entirely. Additionally, I can easily incorporate modified amino acids for structural studies.
If I could choose any DNA to sequence, I would want to sequence the CFTR gene from individuals diagnosed with or suspected of having Cystic Fibrosis. This choice is directly connected to my main project and interest in CF research. Sequencing the CFTR gene from patient samples would allow me to identify which specific mutation or mutations an individual carries. This is critically important because Cystic Fibrosis is caused by over 2,000 different known mutations in the CFTR gene, and these mutations can affect the protein in different ways. Some mutations, like F508del, cause the protein to misfold and never reach the cell surface. Others, like G551D, allow the protein to reach the surface but prevent the channel from opening properly. Still other mutations result in premature stop codons or splicing errors that produce no functional protein at all. By sequencing each patient’s CFTR gene, I could determine their specific genotype, which directly informs their prognosis and guides treatment decisions. This is particularly relevant today because of the development of mutation-specific therapies with modulators. These drugs aren’t effective in every mutations, highlighting why knowing the exact DNA sequence is essential for personalized medicine. Beyond individual patient care, sequencing CFTR genes from large populations would contribute to our understanding of how different variants correlate with disease severity, how new variants arise, local variants and potentially identify rare mutations that might respond to existing or future therapies. For someone working on CF, having this sequence information is the foundation upon which everything else is built.
For sequencing the CFTR gene, I would employ a combination approach using whole exome sequencing followed by targeted CFTR gene sequencing, both performed on the Illumina platform. This dual strategy is particularly valuable for clinical scenarios involving suspected Cystic Fibrosis or other disorders with overlapping symptoms. Illumina sequencing is classified as second-generation sequencing, also called next-generation sequencing or NGS. Unlike first-generation Sanger sequencing, which reads one DNA fragment at a time, Illumina’s massively parallel approach sequences millions of fragments simultaneously, making it highly efficient for clinical diagnostics. I have selected Illumina because it offers accuracy with very low error rates, making it ideal for detecting disease-causing mutations where precision is paramount. Additionally, Illumina produces shorter reads which provides the high accuracy needed for clinical decision-making. This is particularly important for CFTR sequencing, as knowing the exact mutation determines which targeted therapies a patient may receive.
The input for this sequencing approach would be genomic DNA isolated from a patient blood sample. The essential steps for library preparation would begin with DNA isolation and quality assessment, where I extract genomic DNA from the patient sample and verify its purity and integrity using UV spectrophotometry and fluorometric assays. Next, for whole exome sequencing, I would use hybridization-based probes to capture all exonic regions, while for targeted CFTR sequencing, a custom enrichment panel can specifically capture the CFTR gene regions including both exons and introns. The captured DNA is then fragmented into small pieces suitable for Illumina sequencing. Following fragmentation, Illumina-specific adapters are ligated to both ends of the fragments, as these adapters are complementary to the oligos on the flow cell and enable cluster generation. PCR amplification is then performed to increase the quantity of the library, which is especially important when working with limited starting material. Finally, the prepared libraries are quantified to ensure optimal loading concentration onto the sequencer.
The essential steps of Illumina sequencing technology begin with cluster generation through bridge amplification. The library is loaded onto a flow cell where fragments hybridize to complementary oligos on the surface. Through bridge amplification, each fragment forms a clonal cluster of identical molecules, which amplifies the fluorescent signal for detection. The core technology that decodes the bases is called sequencing by synthesis. This process uses fluorescently labeled nucleotides with reversible terminators. In each cycle, DNA polymerase adds a single complementary nucleotide to each growing strand. Unbound nucleotides are washed away, and a laser excites the fluorescent labels. A camera captures images of the flow cell, recording which base was incorporated in each cluster based on the emission wavelength. The fluorescent dye and terminator are then cleaved, allowing the next cycle to begin. This cyclic reversible termination process repeats for the desired number of cycles to achieve a specific read length. The images are processed by Illumina’s DRAGEN Bio-IT Platform or similar software, which converts fluorescence intensities into base calls with associated quality scores.
The output of this sequencing approach includes several file types and reports. Raw FASTQ files contain the sequence reads and quality scores for each base call. Aligned BAM files show the reads aligned to the human reference genome, allowing visualization of the CFTR region. Variant call format files, or VCF files, list all identified variants including single nucleotide variants, and small insertions and deletions. For CFTR specifically, this includes critical variants that affect splicing. Finally, a clinical report provides a curated interpretation of identified variants, classifying them as pathogenic, likely pathogenic, variants of uncertain significance, or benign based on established guidelines. This comprehensive approach would enable accurate diagnosis of CFTR-related disorders, guide mutation-specific therapy decisions, and potentially identify novel variants for further study.
5.2 DNA Write
For DNA synthesis, I would like to synthesize an mRNA construct encoding a fully functional, wild-type CFTR protein, designed for use as an mRNA replacement therapy. This approach would operate independently of current modulator therapies and could benefit patients regardless of their specific variant type, including those with nonsense or frameshift mutations that do not respond to existing drugs.
The concept involves delivering synthetic CFTR mRNA to airway epithelial cells, where it would be translated directly into functional CFTR protein by the cell’s own ribosomes. This bypasses the need to correct the patient’s mutated endogenous gene. However, this strategy would ideally require co-delivery of RNA interference molecules such as siRNA or shRNA to silence the expression of the endogenous mutated CFTR transcript. This would prevent potential dominant-negative interactions or production of toxic truncated protein fragments from the patient’s own gene.
I would use the silicon-based high-throughput DNA synthesis platform developed by Twist Bioscience. This technology represents the current industry standard for commercial gene synthesis and is ideally suited for producing the approximately 4,500 base pair CFTR coding sequence I require.
Twist’s synthesis method is classified as second-generation high-throughput DNA synthesis and relies on phosphoramidite chemistry miniaturized and parallelized on a silicon microarray chip. Unlike conventional column-based synthesizers that produce one sequence at a time, Twist’s platform synthesizes thousands to millions of oligonucleotides simultaneously on a single silicon wafer. I have chosen this technology because it offers the optimal balance of throughput, accuracy, and cost-effectiveness for my CFTR project. Additionally, all synthesized products are sequence-verified by next-generation sequencing, ensuring the accuracy critical for downstream therapeutic applications.
The essential steps begin with sequence design and optimization, where my codon-optimized CFTR coding sequence is analyzed for complexity factors like high GC content and repeats. A silicon wafer is then functionalized with a chemical linker to enable oligonucleotide attachment. The core process is parallel oligonucleotide synthesis using phosphoramidite chemistry, where each synthesis cycle involves four reactions: deblocking, coupling, capping, and oxidation. These cycles occur across thousands of features on the chip with stepwise coupling efficiency. Once synthesis is complete, the oligonucleotides are chemically cleaved and eluted from the silicon surface. For my full-length CFTR gene, these oligonucleotides undergo amplification and assembly through PCR-based methods and Gibson Assembly to join fragments into the complete 4.4 kb coding sequence. Finally, rigorous quality control and sequence verification using next-generation sequencing confirms accuracy before delivery.
Regarding limitations, speed remains a factor with standard turnaround of approximately 10 business days. Accuracy is excellent at the per-base level, but cumulative error rates for sequences of several thousand base pairs range from 0.3% to 1.4%, which is why NGS verification is essential. For scalability, the platform excels at producing many sequences in parallel but has inherent limits on individual sequence length. Twist’s Gene Fragments are offered up to 5 kb, which accommodates my CFTR coding sequence but would require additional assembly steps for larger constructs.
5.3 DNA Edit
For DNA editing, I would want to edit the CFTR gene directly in patient-derived cells to permanently correct disease-causing mutations at the source. Unlike my previously described mRNA replacement therapy, which provides a temporary workaround, gene editing offers the potential for a one-time, permanent cure by repairing the endogenous gene and restoring normal CFTR expression under its native regulatory control. This approach would be particularly valuable because it maintains the cell’s natural mechanisms for regulating CFTR expression, rather than relying on exogenous delivery.
The technology I would use to perform these edits is CRISPR-Cas9 for initial exploration, but for therapeutic application I would preferentially employ base editing or prime editing depending on the specific mutation. These represent second-generation CRISPR technologies that offer greater precision and safety for clinical applications.
CRISPR-Cas9 is classified as a programmable nuclease system derived from bacterial adaptive immune systems. The essential steps begin with designing a single guide RNA (sgRNA) complementary to the target region adjacent to a protospacer adjacent motif (PAM) sequence. To overcome the limitation of CRISPR-cas9 with the errors caused by the double strand breake, I would use base editing, a more precise technology that does not require this type of brakes. Base editors fuse a catalytically impaired Cas protein (nickase Cas9) to a deaminase enzyme. For example, for correcting G551D, a point mutation where glycine is replaced by aspartate, I would use an adenine base editor (ABE) that converts an A•T base pair to G•C. The essential steps involve designing an sgRNA that positions the Cas9 nickase to expose the target base within the editing window, typically 4-8 nucleotides. The deaminase chemically converts the target adenine to inosine, which is read as guanine during replication.
In terms of preparation, the input components would include a base editor mRNA or protein and the corresponding sgRNA. These components would be delivered to patient-derived bronchial epithelial cells or induced pluripotent stem cells (iPSCs) for ex vivo editing, with the goal of eventually moving to in vivo delivery via lipid nanoparticles or AAV vectors optimized for lung targeting.
The limitations of these methods must be carefully considered. Base editing offers higher efficiency and fewer double-strand breaks, but is limited to transition mutations (A→G or C→T) and cannot correct insertions or deletions. It would be necessary to consider off-target mutations.