Week 4 HW: Protein design part I
Part A. Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Reasoning: 1 Dalton = 1 g/mol. So 100 Daltons = 100 g/mol. 500 g of meat is mostly water, protein, fat, etc. Assuming meat is ~20% protein by mass (typical for muscle), that gives 100 g of protein. Protein is polymers of amino acids. Average molecular weight of an amino acid residue in a protein is ~110 g/mol (since 100 Daltons for free amino acid, but ~110 when incorporated due to loss of H₂O). Moles of residues = 100 g / 110 g/mol ≈ 0.91 mol. Number of molecules = 0.91 × 6.022 × 10²³ ≈ 5.5 × 10²³ molecules of amino acid residues.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because digestion breaks down dietary proteins into individual amino acids and small peptides, which are then absorbed and used to build human-specific proteins according to human genetic information (DNA). The species identity is determined by the sequence of amino acids, not by the origin of the raw materials. Amino acids themselves are identical across species (e.g., glycine in cow is the same molecule as glycine in human).
- Why are there only 20 natural amino acids?
The genetic code uses triplet codons (4³ = 64 possible codons). Evolution settled on 20 canonical amino acids because:
They provide a diverse range of chemical properties (hydrophobic, charged, polar, small, aromatic) sufficient to fold into stable 3D structures and catalyze reactions. More than 20 would increase translation errors and metabolic cost; fewer would limit functional diversity. The aminoacyl-tRNA synthetase system co-evolved to precisely match these 20 with tRNA molecules.
- Can you make other non-natural amino acids? Design some new amino acids.
Yes. Non-natural amino acids can be chemically synthesized and incorporated into proteins using expanded genetic codes (e.g., with orthogonal tRNA/synthetase pairs).
Design examples:
p-Benzoylphenylalanine – contains a benzophenone group for photo-crosslinking upon UV light. Azidohomoalanine – contains an azide group for click chemistry. 4-Fluorotryptophan – fluorine substitution to probe protein dynamics via ¹⁹F NMR. Naphthylalanine – larger aromatic side chain for enhanced π-stacking.
- Where did amino acids come from before enzymes that make them, and before life started?
From prebiotic chemistry:
Miller–Urey type experiments show that spark discharge in a reducing atmosphere (CH₄, NH₃, H₂O, H₂) produces several amino acids (glycine, alanine, etc.). Meteorites (e.g., Murchison meteorite) contain dozens of amino acids, including some non-biological ones. Hydrothermal vents could have synthesized amino acids from HCN, CO, and H₂S under high pressure/temperature. Formose reaction and Strecker synthesis (aldehyde + NH₃ + HCN → aminonitrile → amino acid) are plausible prebiotic routes. Thus, amino acids predate enzymes; enzymes later evolved to optimize their production.
- Why are most molecular helices right-handed?
This is a deep biophysical and chemical question. Possible reasons:
Chirality of building blocks: L-amino acids and D-sugars bias helix handedness through steric interactions between side chains and the backbone. Lowest energy conformation: For polypeptides, right-handed α-helix has less steric clash between side chains and the backbone carbonyl oxygen compared to left-handed. Evolutionary selection: Once early proteins fixed right-handed helices, all further evolution built on that scaffold. In abiotic systems (e.g., quartz, certain liquid crystals), right-handedness can arise from subtle energy differences due to parity violation in weak nuclear forces, though this effect is tiny.
- Why do β-sheets tend to aggregate?
β-sheets have an extended backbone conformation that exposes backbone NH and CO groups in a regular pattern, allowing inter-sheet hydrogen bonding. Additionally, β-sheets often present hydrophobic side chains (e.g., valine, leucine, isoleucine, phenylalanine) on one face. When two β-sheets approach, they can form:
Hydrogen bonds between sheets (steric zipper). Hydrophobic collapse of exposed nonpolar surfaces. This combination makes β-sheets prone to self-association into larger fibrils.
- What is the driving force for β-sheet aggregation?
The dominant driving force is hydrophobic effect: Removal of hydrophobic side chains from water into the interior of the aggregated state. This is supplemented by:
Inter-sheet hydrogen bonding (enthalpic gain, though partially offset by loss of water-protein H-bonds). van der Waals packing of side chains in the dry interface. Experiments show that mutations replacing hydrophobic residues with charged or polar ones reduce aggregation, confirming hydrophobicity as the key driver.
- Why do many amyloid diseases form β-sheets?
Amyloid-forming proteins (e.g., Aβ in Alzheimer’s, α-synuclein in Parkinson’s) have regions prone to adopt β-strand conformation. Once misfolded, these strands stack via steric zipper interactions — two β-sheets interdigitate tightly with no water between them. This cross-β structure is extremely stable, resistant to proteases, and grows by templating, leading to disease-associated fibrils.
Part B. Protein Analysis and Visualization
- CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) is an integral membrane protein that functions as a cAMP-activated chloride channel and as a regulator of other ion channels, including outwardly rectifying chloride channels (ORCCs) and epithelial sodium channels (ENaC). It is composed of five domains: two transmembrane domains (TMD1 and TMD2) that form the channel pore, two nucleotide-binding domains (NBD1 and NBD2) that bind and hydrolyze ATP to open the channel, and a regulatory (R) domain that contains multiple phosphorylation sites for protein kinase A (PKA). Mutations in the CFTR gene cause Cystic Fibrosis, a life-threatening autosomal recessive disorder characterized by thick mucus accumulation in the lungs, pancreas, and other organs.
I selected CFTR because my final project focuses on studying rare CFTR mutations present in the Ecuadorian population, where the common F508del mutation found in Caucasians is far less prevalent. By expressing CFTR-eGFP fusion constructs in MDCK cells, I aim to evaluate how uncharacterized variants affect protein trafficking, apical membrane localization, and overall function. Understanding CFTR’s behavior is critical to developing targeted therapies for underrepresented populations.
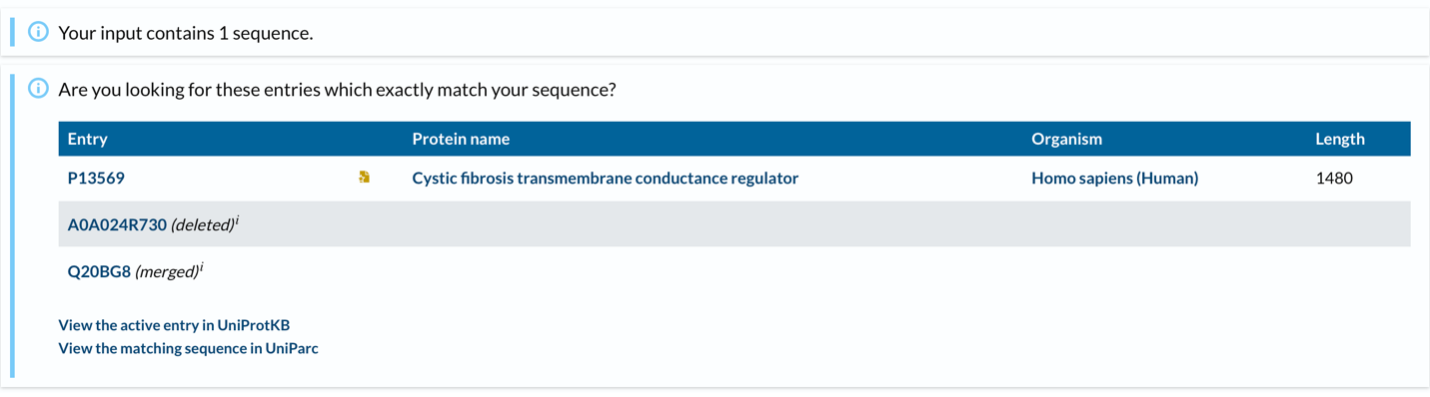
- CFTR has 1,480 amino acids, being the most common leucine. CFTR is a member of the ABC transporter superfamily and shares significant sequence and structural homology with other ABC transporters. UniProt returns only one exact match for the full-length human CFTR sequence, as expected. However, CFTR has many homologous proteins, including orthologs in mouse, rat, zebrafish, and frog, as well as paralogs within the ABC transporter family such as MDR1 and MRP1.
The first full-length CFTR structure was solved in 2017 (chicken CFTR at 4.30 Å), followed by the first human CFTR structure in 2018 at 3.90 Å. No. The best full-length CFTR structure has a resolution of 3.90 Å, which is worse than the 2.70 Å threshold for “good quality.” However, individual domains like NBD1 have been solved at high resolution (1.87 Å). Yes. CFTR structures contain ATP, magnesium ions (Mg²⁺), cholesterol, and CFTR modulator drugs such as Lumacaftor (VX-809). Yes. CFTR belongs to the P-loop containing nucleoside triphosphate hydrolase fold (CATH: 3.40.50.300) and the ABC transporter-like ATPase family.
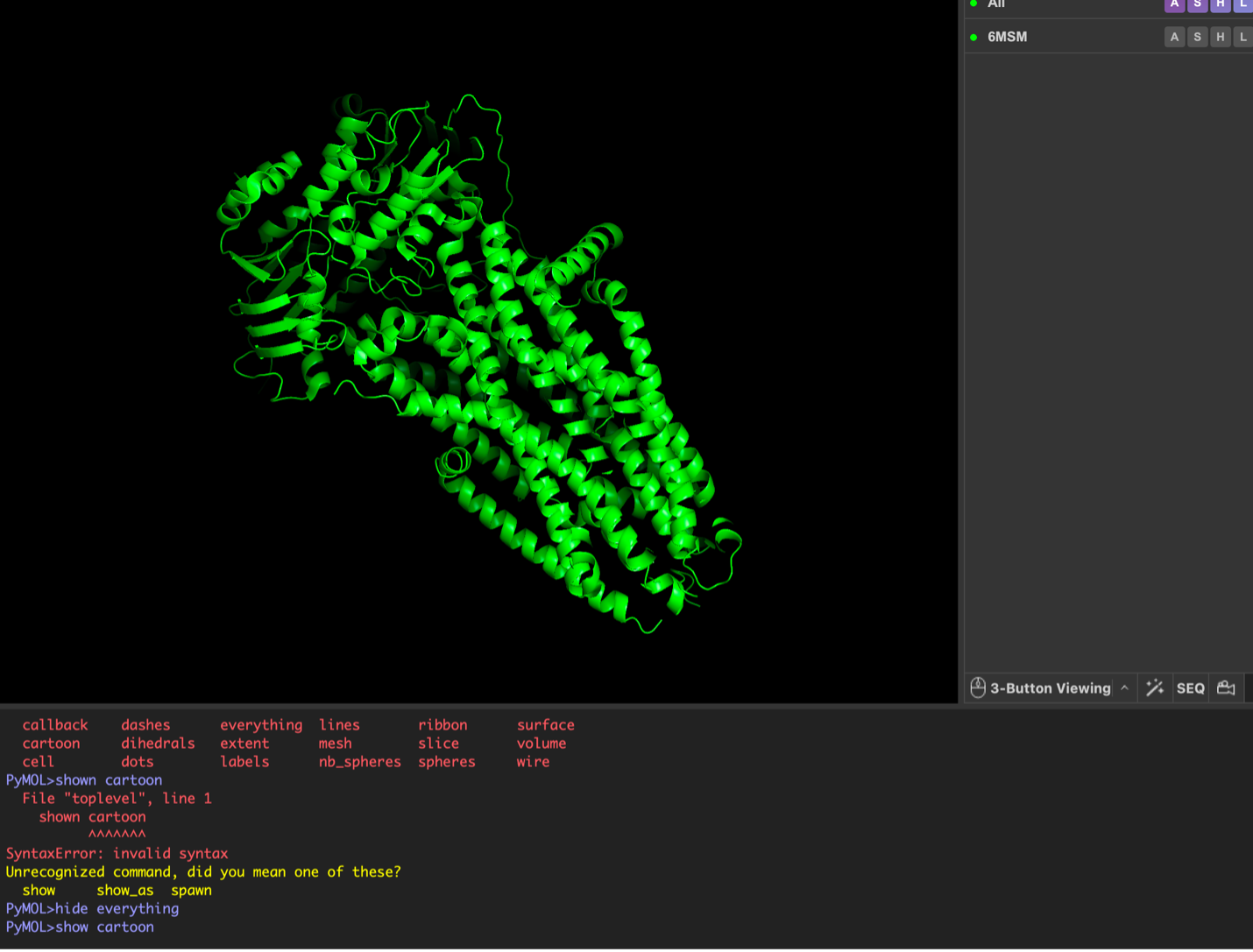
images from Pymol
The surface visualization reveals several cavities, including a central channel and binding pockets likely associated with ATP binding and ion transport.
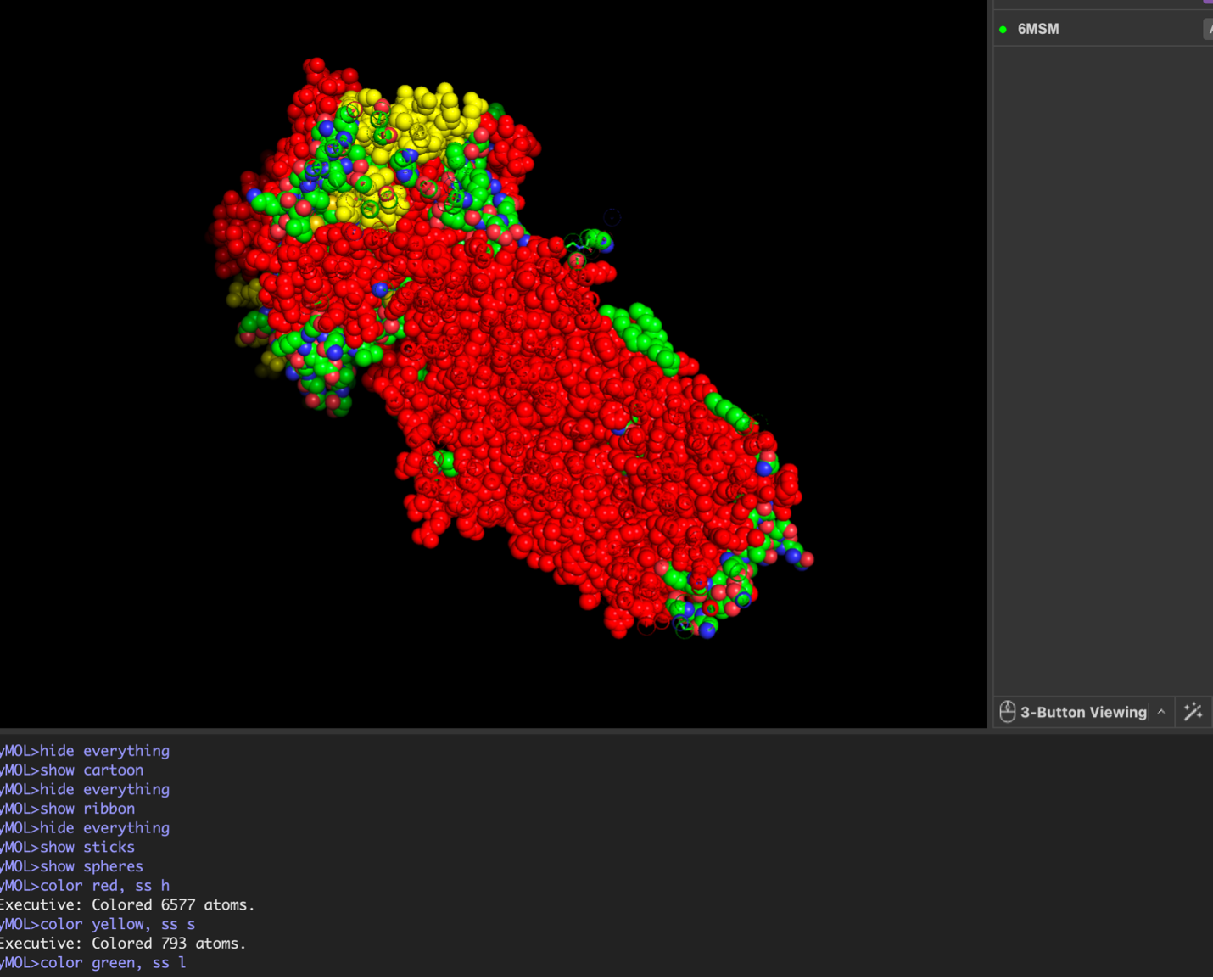
The CFTR protein contains predominantly α-helices with very few β-sheets. This is consistent with its role as a transmembrane ion channel. Hydrophobic residues are mainly located in the transmembrane regions, while hydrophilic residues are distributed on the surface and cytoplasmic domains. Surface analysis shows the presence of several cavities and binding pockets, including the central channel characteristic of ion transport proteins.
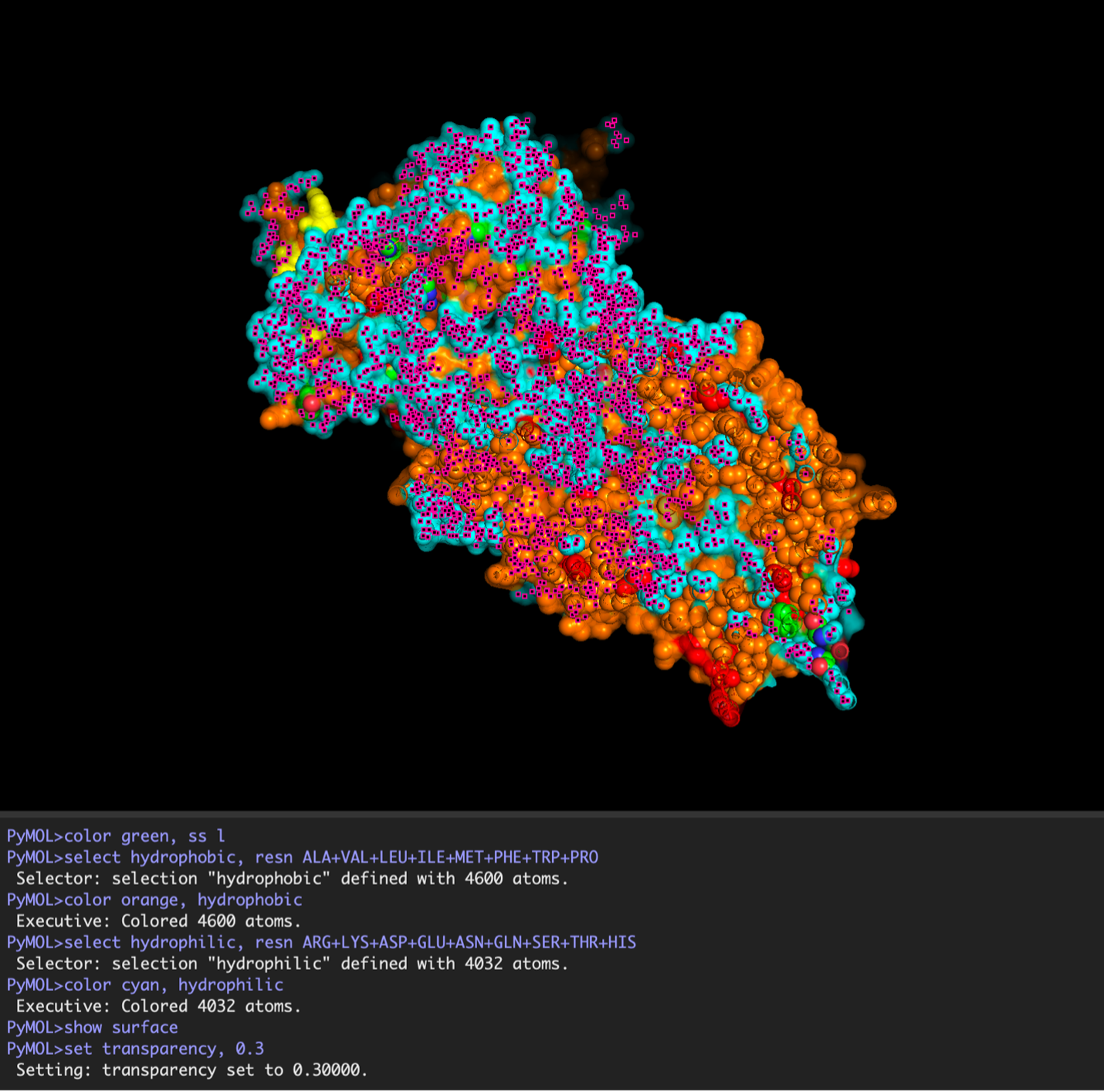
 Hydrophobic residues are mainly located in the transmembrane regions, while hydrophilic residues are found on the outer surface.
Hydrophobic residues are mainly located in the transmembrane regions, while hydrophilic residues are found on the outer surface.
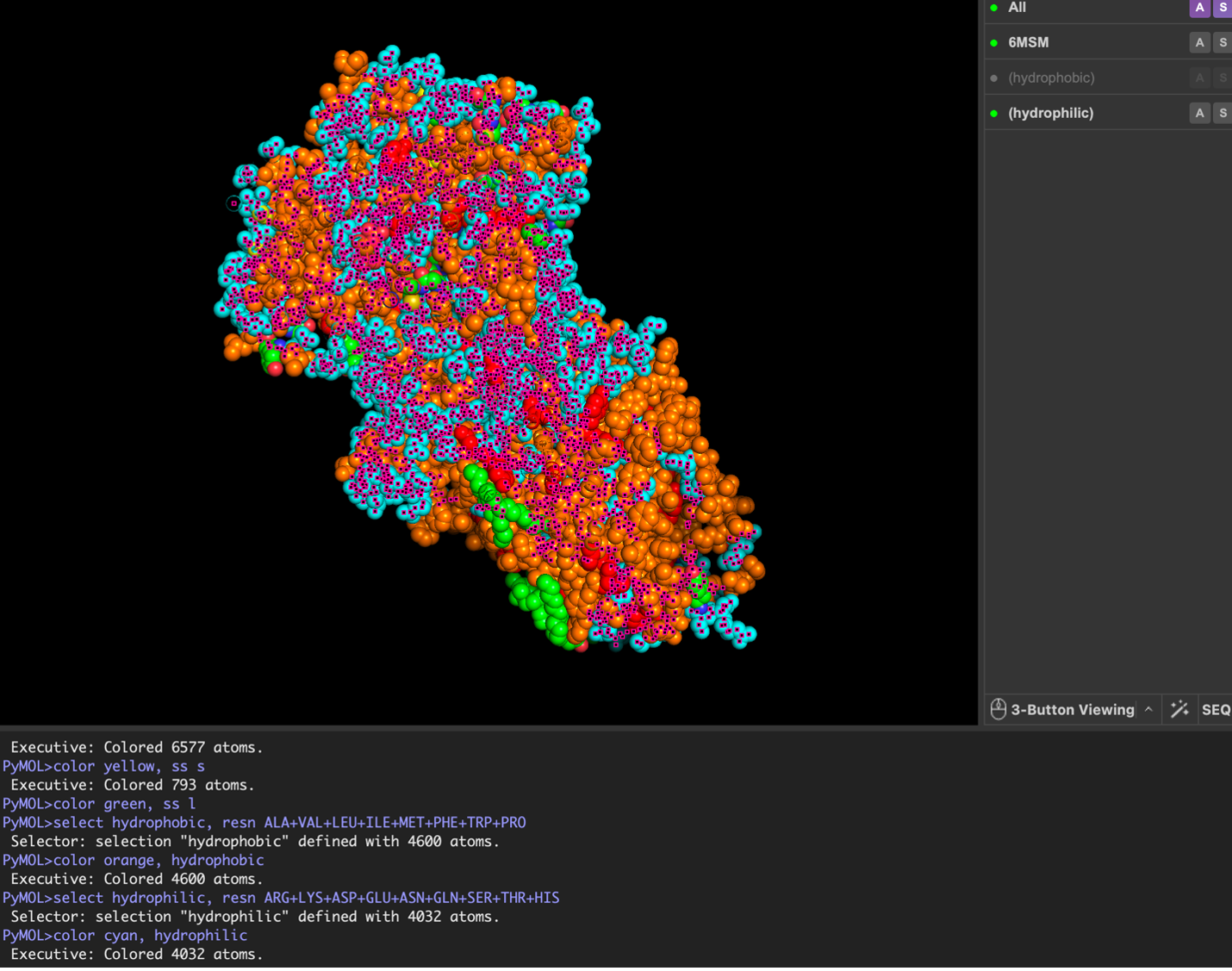
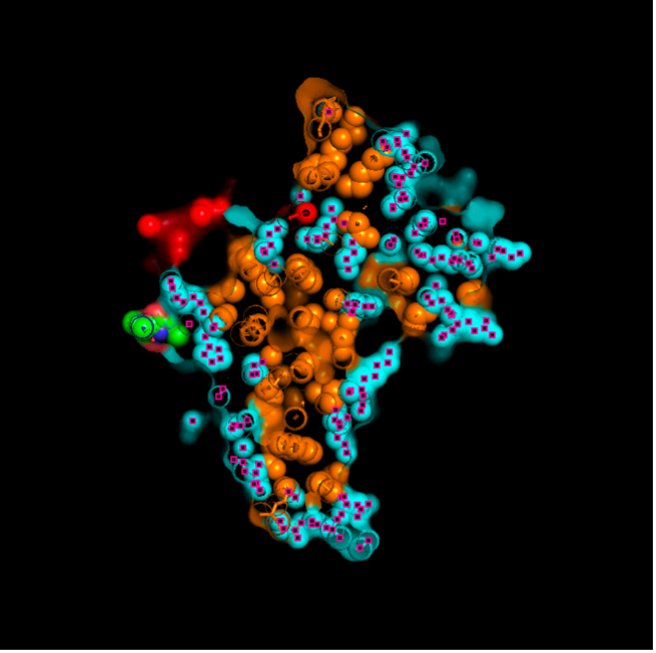
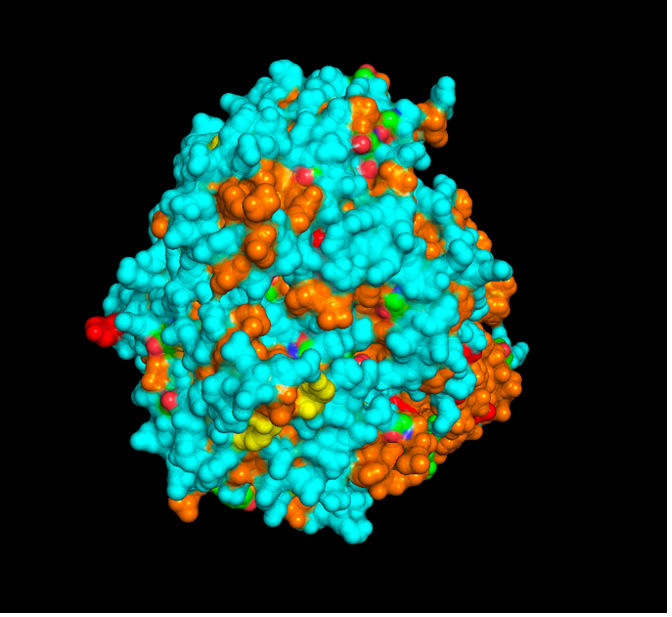
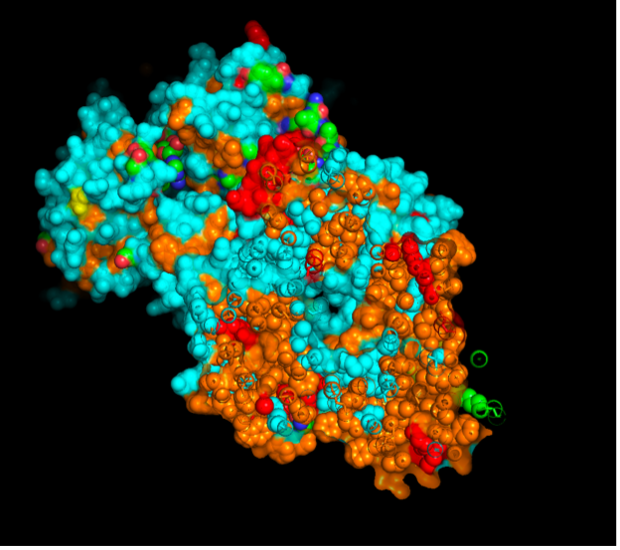
 Surface visualization reveals several cavities and a central channel consistent with binding pockets.
Surface visualization reveals several cavities and a central channel consistent with binding pockets.
C1. Protein Language Modeling
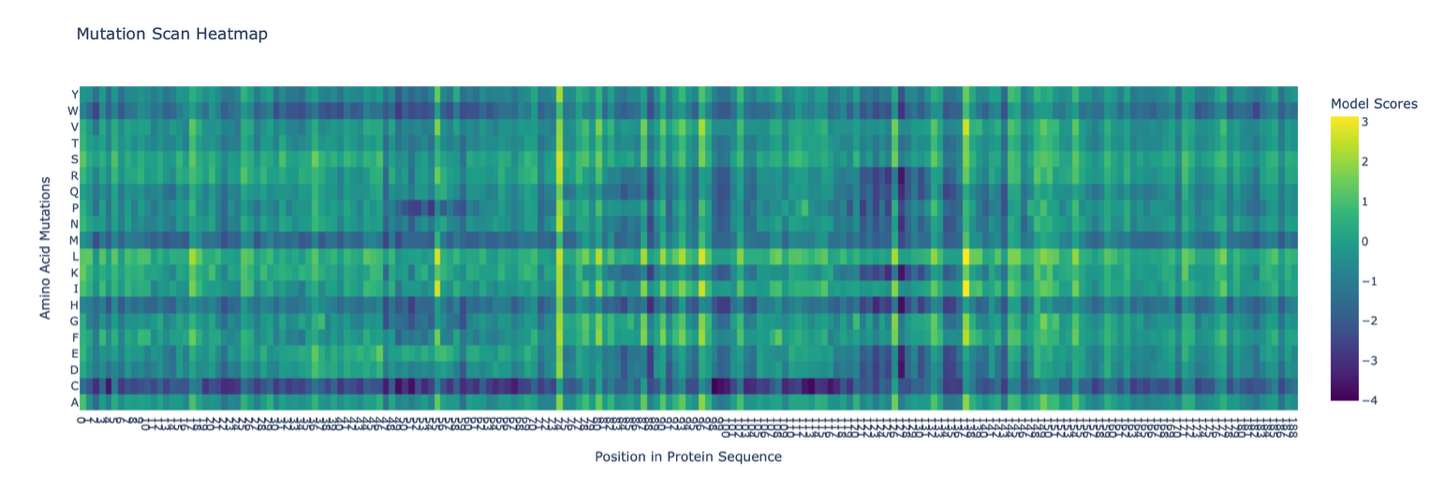
- The deep mutational scan generated using the ESM2 model shows several conserved positions across the protein fragment. A clear pattern observed in the heatmap is the presence of vertical columns with strongly unfavorable substitutions, indicating highly conserved residues. One residue that stands out is a leucine (L) located approximately in the middle of the sequence. Mutations of this residue to polar or charged amino acids, such as lysine (K), show strongly negative scores in the heatmap. This suggests that the native leucine is structurally important and likely part of a hydrophobic region. The unfavorable mutation from leucine to lysine likely disrupts hydrophobic interactions, indicating that this residue contributes to maintaining protein stability. Overall, this pattern suggests that hydrophobic residues in the sequence are conserved and play key structural roles.
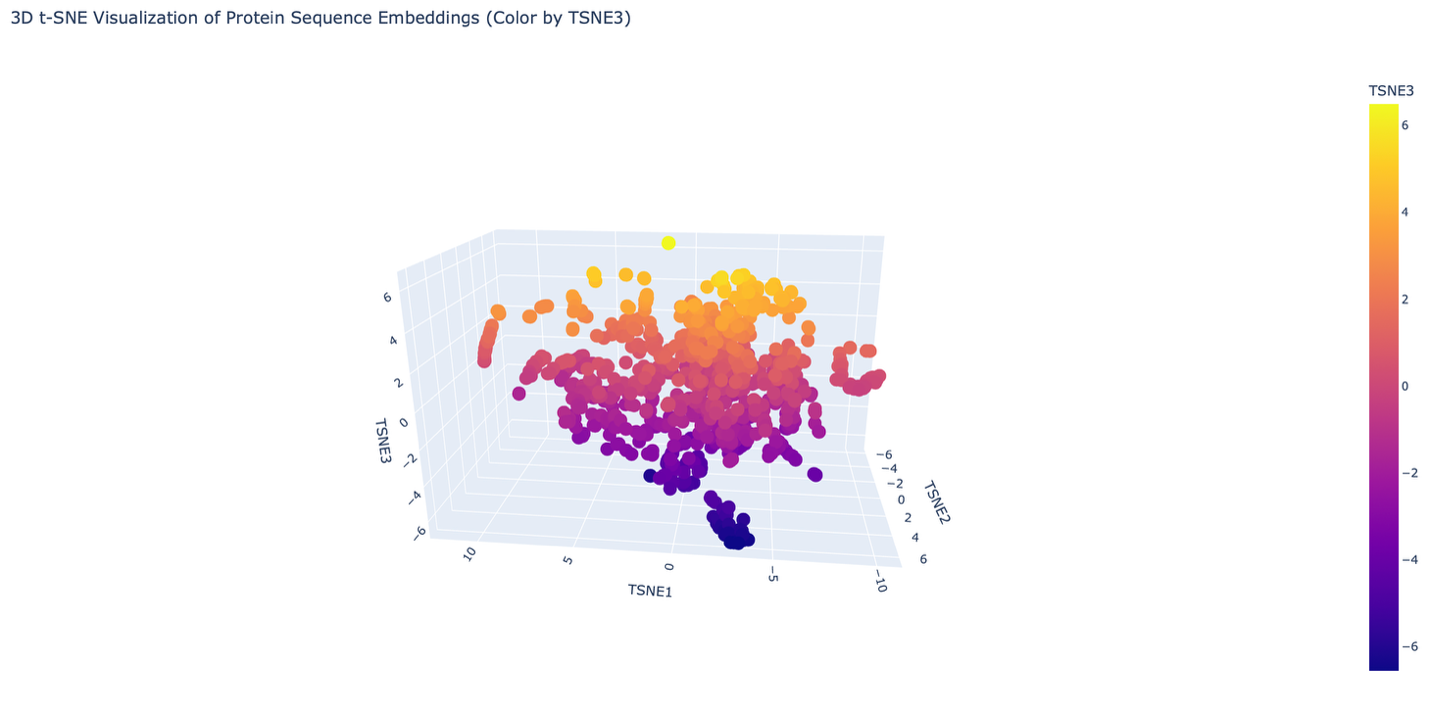
- The reduced dimensionality map shows several distinct neighborhoods formed by clusters of proteins. These neighborhoods appear to group proteins with similar structural and functional characteristics. Proteins that are close together in the latent space likely share similar sequence patterns and folding properties. Overall, the formed neighborhoods do approximate similar proteins, suggesting that the embedding produced by the ESM2 model captures meaningful biological relationships between protein sequences.
C2. Folding Protein
Sequence: MQRSPLEKASVVSKLFFSWTRPILRLLLSLAPLLL
Mutation: MQRSPLEKASVVSKKFFSWTRPILRLLLSLAPLLL
L → K
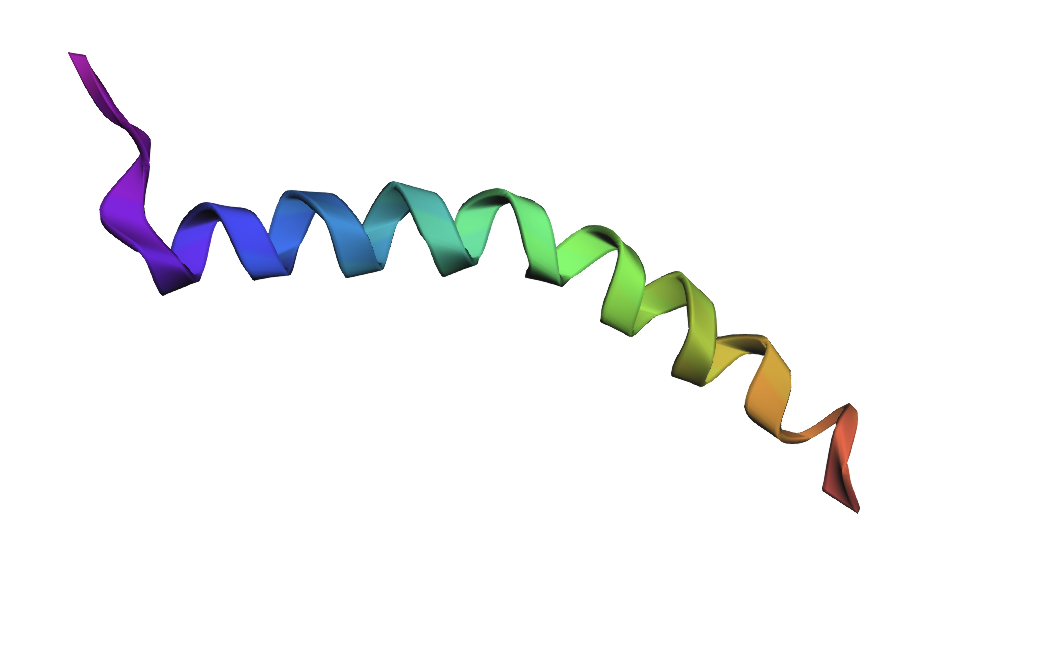
Small point mutations produced only minor structural changes. The overall alpha-helical fold remained mostly preserved, suggesting that the protein fragment is relatively resilient to single amino acid substitutions.
- Larger mutation: MQRSPLEKASVVSKDDDKKKNNNQQQLSAPLLL

Large sequence modifications caused substantial structural changes in the predicted fold. Replacing hydrophobic residues with polar and charged amino acids disrupted the alpha-helical organization, indicating that the original sequence composition is important for maintaining structural stability.
C3. Protein Language Modeling
ProteinMPNN assigned high probabilities to several hydrophobic residues in the sequence, particularly leucine, valine, and isoleucine. These residues are likely important for maintaining the structural stability of the protein backbone. Some substitutions were predicted at flexible positions, but most conserved positions retained chemically similar amino acids.
The predicted sequence was similar to the original sequence in terms of biochemical properties, although several amino acid substitutions were introduced. Most substitutions were conservative, preserving hydrophobicity and overall residue type. This suggests that multiple sequences can adopt similar structural folds.
These results demonstrate that multiple different amino acid sequences can encode similar protein structures, highlighting the robustness of protein folding and the flexibility of sequence-to-structure relationships.
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal: Engineering MS2 Lysis Protein L for Increased Toxicity via DnaJ Interface Disruption
Selected Goal
We propose to computationally engineer MS2-L for higher toxicity by disrupting its interaction with the E. coli chaperone DnaJ. The rationale comes directly from the papers you provided. Chamakura et al. (Phage1 and Phage2) demonstrated that the N-terminal domain of MS2-L (residues 1–36) binds DnaJ, and removing this domain entirely causes lysis approximately 20 minutes earlier than wild-type. However, complete deletion may have unintended consequences. Our approach is more precise: we will identify point mutations that weaken or abolish the L–DnaJ interface while preserving the rest of the protein’s structure and membrane insertion capability. Mezhyrova et al. (Phage3) showed that DnaJ binds to MS2-L even after membrane insertion and that this interaction does not affect oligomerization, suggesting the chaperone plays a regulatory rather than essential structural role. Disrupting this regulation should yield a hypertoxic variant.
Tools and Approaches
We will use a combination of structure prediction and protein language models. First, since no experimental structure of the MS2-L–DnaJ complex exists, we will use AlphaFold-Multimer to predict the complex structure. The papers indicate that the interaction involves the N-terminal soluble domain of L (approximately residues 1–35) and the C-terminal domain of DnaJ, specifically around the highly conserved proline 330. AlphaFold-Multimer has been validated on phage protein complexes in Phage6, where it successfully predicted capsid–spike–J protein interactions for both wild-type ΦX174 and the generated phage Evo-Φ36. We will run multiple seeds of AlphaFold-Multimer to generate an ensemble of predicted complex structures and identify which residues consistently appear at the interface.
Once we have a predicted interface, we will use protein language models to score mutations. Specifically, we will employ ESM-1v or ESM2 in a zero-shot fitness prediction mode. These models have been shown to predict the effects of missense mutations without requiring task-specific training data. The key idea is that residues critical for protein-protein interactions are evolutionarily constrained; language models capture these constraints from sequence alone. We will score all possible single amino acid substitutions in the N-terminal domain (positions 1–36) for their predicted effect on protein fitness. Low fitness scores at the interface likely indicate mutations that disrupt binding.
We will complement the language model predictions with biophysical calculations using FoldX or PyRosetta to estimate changes in binding free energy (ΔΔG) upon mutation. This hybrid approach—combining evolutionary scores from PLMs with physical energy calculations—has become standard in computational protein engineering. For validation, we will re-run AlphaFold-Multimer on the top candidate mutations to see if the predicted complex confidence drops substantially compared to wild-type.
The final pipeline will produce a shortlist of 5–10 point mutations predicted to disrupt DnaJ binding while maintaining L’s overall stability and membrane topology. We will prioritize mutations in the basic residues (arginines and lysines) between positions 11 and 34, since these are likely to form electrostatic interactions with DnaJ’s C-terminal domain.
Potential Pitfalls
The first major pitfall is that no high-resolution structure of the L–DnaJ complex exists, and AlphaFold-Multimer may produce low-confidence or incorrect predictions for this bacterial–phage interface. Phage-bacteria interaction interfaces are evolutionarily fast-evolving and may not follow the same statistical patterns as eukaryotic protein complexes on which AlphaFold was primarily trained. To mitigate this, we will run multiple seeds, use the predicted aligned error (PAE) plots to identify confident regions, and cross-validate with sequence conservation. If the complex prediction fails entirely, we would fall back to a simpler approach: deleting the N-terminal domain in silico and using Evo (the genome language model from Phage6) to generate stable truncated variants directly, as Chamakura already showed that truncations work.
The second pitfall is that DnaJ’s binding site on L may extend beyond the soluble domain and into the transmembrane region. Chamakura et al. (Phage2) showed that the truncated construct MS2-Lp₃₅, which includes only amino acids 35–75, still showed reduced but detectable DnaJ binding. This means that mutations confined to residues 1–34 might not fully abolish interaction. We would need to include the junction region (residues 33–40) in our interface analysis and accept that complete loss of binding might require multiple mutations or a combination of truncation and point mutations.
The third pitfall relates to the language model approach. Protein language models like ESM are trained on natural protein sequences, which overwhelmingly come from eukaryotic and bacterial proteins, not phage lysis proteins. The training data likely contains very few examples of DnaJ-binding interfaces from Leviviridae phages. As a result, the zero-shot fitness predictions may be noisy. However, Phage6 successfully used Evo (a genomic language model) to generate functional phage genomes, suggesting that these models do capture relevant biology even for under-sampled viral families. We can partially address this by fine-tuning on available Microviridae and Leviviridae sequences, as was done in Phage6 for ΦX174.
Finally, even if we successfully predict mutations that disrupt DnaJ binding in silico, we cannot computationally confirm that these mutations increase lysis toxicity. The ultimate test would require experimental validation—synthesizing the variant L genes, expressing them in E. coli, and measuring lysis kinetics. This is outside our computational scope, but we can at least provide ranked candidates for experimental collaborators.