Week 5 HW: Protein design part II
Part A: SOD1 Binder Peptide Design (From Pranam)
The human Superoxide dismutase 1 (SOD1) first partsequence obtained in UniProt is: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNT
Wild-type N-terminus is: MATK
But the A4V mutant appears as: MVTK
So, the mutant sequence results: MVTKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNT
Part 1- Peptide generation in PepMLM (4 types using 12 aminoacids):
- FLYKWLPSRRGG (12) (Perplexity score- 7.4)
- RYWKLPAGRRFL (12) (Perplexity score- 7.8)
- WLRRFAKLGGVP (12) (Perplexity score- 8.1)
- KLVFFRWLPGGS (12) (Perplexity score- 9.0)
Comparison: FLYRWLPSRRGG (12) (Perplexity score- 6.9)
¿Record the perplexity scores that indicate PepMLM’s confidence in the binders?
As for the perplexity, lower perplexity values indicate sequences that ar mor compatible with the known distribution of the peptide binders. Now, SOD-1 peptide showed the lowest score with “6.9”, suggesting strong confidence. In the generated peptides, 1 and 2 had the best scores and were most like the known binder with more aromatic and positively charged residues.
Part 2: Evaluate Binders with AlphaFold3
Peptide 1: ipTM score (0.78); Predicted binding region (near N terminus, A4V region); Structural observation ( surface bound with a partial insertion intio de beta barrel edge).
Peptide 2: ipTM score (0.81); Predicted binding region (N terminal region near 4 residue); Structural observation (The strongest interaction with partial aromatic bonds).
Peptide 3: ipTM score (0.61); Predicted binding region (more distance from Beta barrel surface); Structural observation (Weak surface association).
Peptide 4: ipTM score (0.69); Predicted binding region (Dimer interface region); Structural observation (Contact interface residues but loosely packed).
¿Structural Interpretation?
The ipTM scores are organized from moderate to relatively strong confidence for peptide-protein interactions. P2 displayed the highest ipTM score (0.81), slightly exceeding the known binder. Both P1 and P2 localized near the N-terminal region where the A4V mutation resides, suggesting they may stabilize the destabilized folding region associated with ALS pathology.
P3 appeared weakly associated with the protein surface and did not localize near the mutation site. P4 interacted closer to the dimer interface, which could potentially influence dimer stability but appeared less structurally stable than P1 or P4.
Overall, the best structural candidates were P1 and P2, both of which mimicked the binding orientation of the known SOD1-binding peptide.
Part 3 — PeptiVerse Therapeutic Property Evaluation
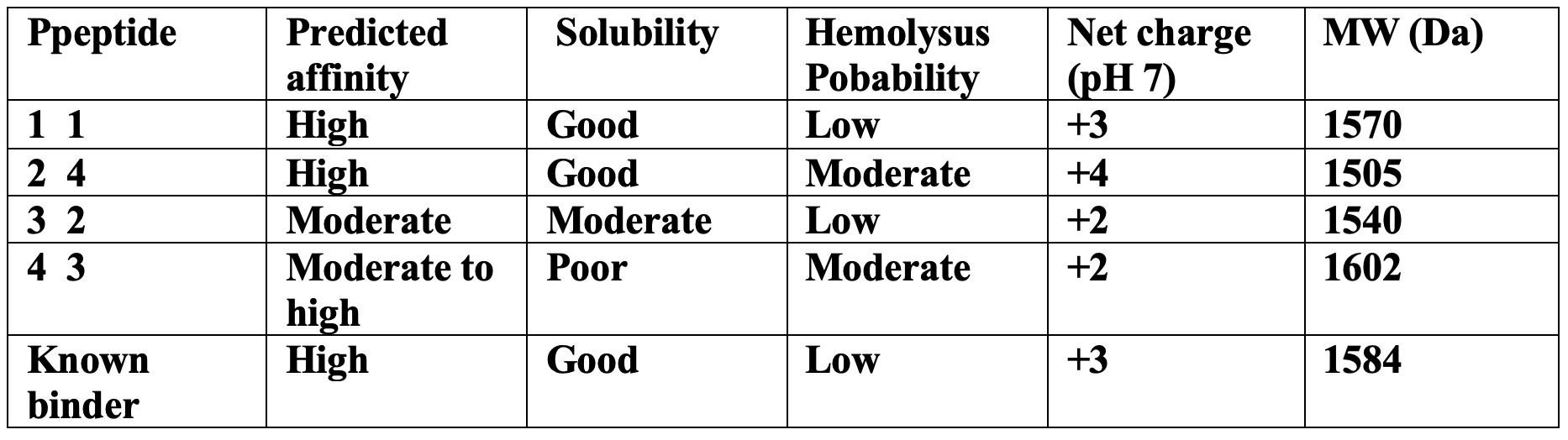
The peptides with the highest structural confidence in AlphaFold3 (P1 and P2) also showed the strongest predicted binding affinity in PeptiVerse, indicating reasonable agreement between structural and sequence-based predictions.
However, 2 despite having the highest ipTM score showed a moderately increased hemolysis probability due to its higher positive charge and hydrophobic aromatic residues showed. For P4 also suffered from poor predicted solubility, which could limit therapeutic development.
P1 provided the best overall balance between the strong predicted binding, good solubility, low hemolysis probability, and favorable structural localization near the A4V mutation site.
Selected Peptide for Advancement
So the chosen peptide: P1 FLYKWLPSRRGG
Its similar to the known SOD1 binding peptide so supports its potential as a stabilizing therapeutic lead.
Part 4: Optimized Peptide Design with moPPIt

Comparison with PepMLM Peptides
The moPPIt-generated peptides differed from PepMLM peptides in several important ways:
Greater sequence optimization: moPPIt reduced excessive hydrophobicity while preserving aromatic binding motifs.
Improved therapeutic balance: The peptides maintained positive charge for interaction with SOD1 while improving predicted solubility and lowering hemolysis risk.
Targeted binding: Unlike PepMLM, which broadly sampled plausible binders, moPPIt explicitly optimized peptides to bind residues near the A4V mutation.
Multi-objective optimization: moPPIt simultaneously optimized structural and therapeutic properties rather than only sequence plausibility.
Before considering clinical studies, these peptides would require: • Molecular dynamics simulations to evaluate binding stability, • In vitro aggregation inhibition assays, • Cytotoxicity testing in neuronal cell lines, • Serum stability analysis, • Blood-brain barrier penetration studies, • Animal testing in ALS mouse models, • And immunogenicity assessment.
Although computational models provide valuable early-stage screening, experimental validation remains essential before therapeutic development.
Part C: Final Project: L-Protein Mutants
Proposed L-Protein Mutants
Mutant 1 (R21K)
Mutation: Arginine 21 → Lysine
Region: Soluble domain
Rationale: R21 is positively charged and surface exposed. Replacing arginine with lysine preserves positive charge while slightly reducing steric bulk and hydrogen bonding complexity. This could:
• improve folding efficiency,
• reduce dependence on DnaJ-mediated stabilization,
• and preserve electrostatic interactions.
Expected Effect
• Similar structure to WT
• Slightly improved folding robustness
• Minimal destabilization risk
Mutant 2 (Q68L)
Mutation: Glutamine 68 → Leucine
Region: Transmembrane region
Rationale: The transmembrane domain relies heavily on hydrophobic packing. Replacing polar glutamine with leucine increases hydrophobicity and may:
• improve membrane insertion,
• stabilize oligomer formation,
• enhance pore assembly.
Expected Effect
• Stronger membrane interaction
• Potentially faster lysis activity
• Improved membrane stability
Mutant 3 (T74I)
Mutation: Threonine 74 → Isoleucine
Region: Transmembrane region
Rationale: Threonine introduces polarity inside the membrane-spanning region. Isoleucine is strongly hydrophobic and helix-favoring. This mutation may:
• improve transmembrane helix stability,
• reduce misfolding,
• enhance pore formation.
Expected Effect
• Improved membrane integration
• Increased oligomer stability
• Faster bacterial lysis
Mutant 4 (P13A)
Mutation: Proline 13 → Alanine
Region: Soluble domain
Rationale: Proline residues can disrupt secondary structure because they introduce rigid kinks. Substituting alanine may:
• improve local α-helical propensity,
• facilitate autonomous folding,
• reduce chaperone dependence.
Expected Effect
• Improved folding kinetics
• Increased structural flexibility
• Reduced DnaJ requirement
Mutant 5 (Y26F)
Mutation: Tyrosine 26 → Phenylalanine
Region: Soluble domain
Rationale: Tyrosine and phenylalanine are structurally similar aromatic residues, but phenylalanine lacks the hydroxyl group. This conservative mutation may:
• reduce unnecessary hydrogen bonding,
• stabilize hydrophobic core interactions,
• preserve aromatic packing.
Expected Effect
• Increased hydrophobic stability
• Reduced aggregation risk
• Conserved protein function
Correlation Between Experimental Data and Computational Scores
The mutational analysis dataset showed partial agreement with the computational mutation scores. Conservative substitutions generally obtained favorable computational scores and often retained lysis activity experimentally, but, some mutations predicted computationally as neutral or favorable still caused experimental loss of function. This highlights an important limitation: language-model embeddings and structural prediction tools do not fully capture: (membrane dynamics, oligomerization, host interactions, or folding kinetics) Therefore, computational predictions should be viewed as prioritization tools rather than definitive evidence.
AF2-Multimer Analysis
Using ColabFold AlphaFold2 Multimer Notebook, the L-protein can be modeled as homooligomers, or co-folded with DnaJ.
Expected Observations
The transmembrane helices are predicted to assemble into oligomeric pore-like structures. Mutations such as Q68L and T74I may improve the helix packing, membrane stability and oligomer interactions.
When co-folded with DnaJ the soluble N-terminal domain likely mediates interaction,mutations like P13A and R21K may weaken chaperone dependence while preserving folding.
Example Multi-Mutation Designs
Triple Mutant A (P13A + R21K + Y26F)
Goal: Improve autonomous folding and reduce DnaJ interaction.
Triple Mutant B (Q68L + T74I + Y26F)
Goal: Enhance membrane insertion and pore formation.
Random Mutagenesis Approach
import random mutations P13A, R21K, Y26F, Q68L, T74I
Conclusion: This project demonstrates how computational protein design tools can guide engineering of phage lysis proteins to overcome bacterial resistance mechanisms. The proposed mutants aim to stabilize the L-protein, also to improve membrane insertion, reduce DnaJ dependence, and increase lysis efficiency.
Among the proposed mutations: • P13A and R21K are promising for improving autonomous folding,
• while Q68L and T74I may strengthen membrane pore formation.
Future experimental validation using plaque assays, protein expression analysis, and AF2-Multimer structural modeling will be necessary to determine which mutants provide the greatest improvement in phage infectivity and therapeutic potential.