Functional Study of CFTR Mutations Using CFTR-eGFP Expression in MDCK Cells Author: Fiorella Maldonado
Location: Quito, Ecuador
Course: HTGAA (How To Grow (Almost) Anything)
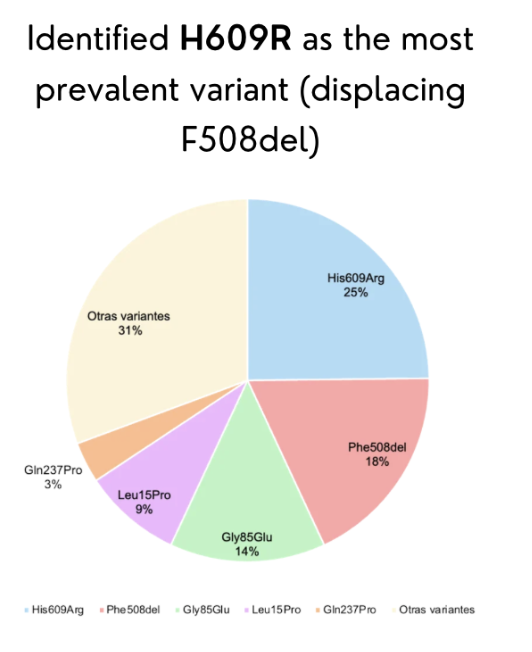
SECTION 1: ABSTRACT Cystic fibrosis (CF) is a life-threatening autosomal recessive disorder caused by mutations in the CFTR gene located on chromosome 7q31.2. While modulator therapies such as Trikafta have transformed care for CF patients carrying the common F508del mutation, these treatments are not accessible or effective for patients with uncharacterized rare variants. In Ecuador, unpublished data from our team reveal that F508del is rare and that H609R has emerged as the most prevalent variant, highlighting a critical gap in knowledge that prevents precision medicine for Ecuadorian CF patients. The broad objective of this project is to establish a functional platform to characterize the trafficking and localization of Ecuadorian CFTR variants using a CFTR-eGFP fusion expression system in polarized MDCK cells. The central hypothesis is that uncharacterized variants such as H609R cause measurable defects in CFTR trafficking to the apical membrane, and these defects can be quantified using fluorescence microscopy and biochemical assays. To achieve this objective, three specific aims will be pursued. Aim 1 (completed) involved identification of the most common CFTR variants in Ecuador and in silico design of a CFTR-eGFP fusion plasmid using SnapGene. Aim 2 will construct mutant plasmids via restriction enzyme cloning, transiently transfect MDCK cells, and evaluate protein localization using confocal microscopy and cell surface biotinylation followed by Western blot. Aim 3 will apply these functional data to classify Ecuadorian variants and guide modulator therapy decisions. This project addresses an urgent need for region-specific genetic evidence to enable equitable CF care.
Cystic fibrosis (CF) is a life-threatening autosomal recessive disorder caused by mutations in the CFTR gene located on chromosome 7q31.2. While modulator therapies such as Trikafta have transformed care for CF patients carrying the common F508del mutation, these treatments are not accessible or effective for patients with uncharacterized rare variants. In Ecuador, unpublished data from our team reveal that F508del is rare and that H609R has emerged as the most prevalent variant, highlighting a critical gap in knowledge that prevents precision medicine for Ecuadorian CF patients. The broad objective of this project is to establish a functional platform to characterize the trafficking and localization of Ecuadorian CFTR variants using a CFTR-eGFP fusion expression system in polarized MDCK cells. The central hypothesis is that uncharacterized variants such as H609R cause measurable defects in CFTR trafficking to the apical membrane, and these defects can be quantified using fluorescence microscopy and biochemical assays. To achieve this objective, three specific aims will be pursued. Aim 1 (completed) involved identification of the most common CFTR variants in Ecuador and in silico design of a CFTR-eGFP fusion plasmid using SnapGene. Aim 2 will construct mutant plasmids via restriction enzyme cloning, transiently transfect MDCK cells, and evaluate protein localization using confocal microscopy and cell surface biotinylation followed by Western blot. Aim 3 will apply these functional data to classify Ecuadorian variants and guide modulator therapy decisions. This project addresses an urgent need for region-specific genetic evidence to enable equitable CF care.
SECTION 2: PROJECT AIMS
Aim 1 (Experimental Aim)
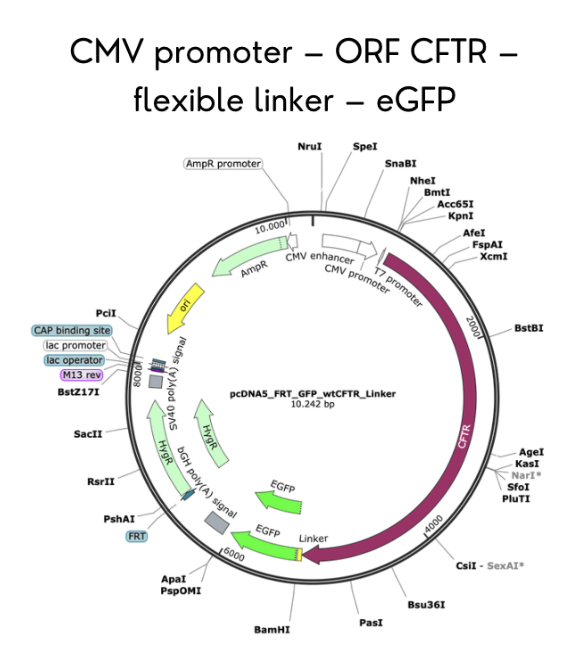
The first aim of my final project is to identify the most prevalent CFTR genetic variants in the Ecuadorian population and complete the in silico construction of a CFTR-eGFP fusion plasmid using SnapGene software, incorporating a CMV promoter, full-length CFTR gene, flexible linker, and C-terminal eGFP, with mutations introduced via directed nucleotide changes. Relevant methods include database review of Ecuadorian CF patient records, AlphaFold modeling for structural prediction, and SnapGene for plasmid design and restriction site verification.
Aim 2 (Development Aim)
Following successful completion of Aim 1, the next step is to experimentally validate the functional consequences of the identified variants by constructing mutant plasmids using restriction enzyme digestion (NheI and EcoRV) and synthetic DNA fragments, transiently transfecting polarized MDCK cells grown on filter supports, and quantifying CFTR trafficking defects using confocal microscopy with WGA co-staining and cell surface biotinylation followed by Western blot to distinguish mature (C band) from immature (B band) CFTR.
Aim 3 (Visionary Aim)
The long-term vision for this project is to establish a sustainable functional screening pipeline that enables personalized assessment of CFTR variants for Ecuadorian CF patients, directly challenging the current clinical practice that relies on Caucasian-centric mutation databases. If fully realized, this platform would provide evidence-based guidance for modulator therapy eligibility, address the major barrier of uncharacterized variants in underrepresented populations, and serve as a reproducible model for other Latin American countries to develop region-specific CF genetic resources, ultimately advancing equitable access to precision medicine.
SECTION 3: BACKGROUND
Literature Context
Cystic fibrosis is caused by dysfunction of the CFTR protein, a cAMP-regulated chloride channel localized to the apical membrane of exocrine epithelial cells. More than 1,300 putative disease-causing mutations have been reported, with F508del accounting for approximately 70% of CF alleles in Caucasian populations (Krasnov et al., 2008). However, the distribution of CFTR mutations varies significantly across geographic regions and ethnic groups.
The study by Krasnov and colleagues demonstrated that functional studies of rare missense mutations in polarized cell models are essential for accurate interpretation of genotype-phenotype relationships, as non-polarized cells may fail to reveal trafficking defects that cause disease in vivo. Specifically, they showed that the R1070Q mutation appeared functional in non-polarized cells but, when studied in polarized MDCK cells, exhibited normal apical localization, leading to the discovery of an in cis nonsense mutation (S466X) that explained the severe phenotype.
A second key study established MDCK cells as a validated model system for CFTR trafficking studies. Mendes et al. (2005) developed a polarized MDCK epithelial cellular model for CFTR research, demonstrating that fluorescence microscopy could successfully distinguish wild-type CFTR localization (apical membrane) from the F508del mutant (cytoplasmic retention). This method also detected intermediate phenotypes, including partial rescue of F508del following low-temperature incubation.
Together, these studies establish the precedent for using polarized MDCK cells and CFTR-eGFP fusions to quantify trafficking defects, while also highlighting the critical need for region-specific functional data given the global diversity of CFTR mutations.
Novelty and Innovation
This project is novel because it shifts the focus of CFTR research from Caucasian-centric mutation databases to an underrepresented population, Ecuador, where the variant landscape is fundamentally different. While most functional studies have characterized F508del and other common European mutations, no systematic functional analysis has been performed on Ecuadorian CFTR variants such as H609R.
This project innovatively combines in silico structural prediction (AlphaFold), molecular cloning (SnapGene-designed CFTR-eGFP fusions), and cell biology (polarized MDCK cells with confocal microscopy and biotinylation) into a single pipeline tailored for regional variants. Furthermore, the project challenges the existing paradigm that global mutation databases can guide treatment decisions everywhere, arguing instead for localized functional evidence as a prerequisite for equitable precision medicine.
Impact and Significance
This project addresses a pressing real-world problem: Ecuadorian CF patients carry uncharacterized mutations and are therefore excluded from modulator therapies that have dramatically improved outcomes for CF patients in other countries. The problem is significant because it represents a critical barrier to progress in the field of CF precision medicine, where treatment decisions increasingly depend on knowing whether a specific mutation causes a trafficking defect, gating defect, or other dysfunction.
The outcomes of this project could benefit society by providing Ecuadorian clinicians with evidence-based guidance on which patients might respond to existing modulators, potentially improving quality of life and life expectancy for dozens of patients who currently have no treatment options. Beyond the immediate context, this project advances scientific understanding by generating functional data on rare CFTR variants, contributing to global efforts to characterize all CFTR mutations.
If the aims are achieved, the concepts and methods developed here could change clinical practice in Ecuador and serve as a reproducible model for other Latin American countries to build their own CFTR variant screening pipelines.
Ethical Implications
The ethical implications of this project center on justice and beneficence. Justice requires that the benefits of biomedical research be distributed fairly across populations. Currently, CF modulator therapies are available primarily to patients with F508del and other well-characterized mutations common in European populations, while Ecuadorian patients with rare or uncharacterized variants have no access to these treatments. This represents a clear health disparity. Beneficence requires that research actively promote the well-being of participants and populations. By generating local functional data, this project aims to enable equitable access to precision medicine for Ecuadorian CF patients, directly fulfilling the principle of beneficence. Non-maleficence (do no harm) is also relevant, as genetic data must be handled carefully to avoid stigmatization or discrimination.
To ensure ethical conduct, several measures will be taken. First, all patient genetic data used for variant selection will be anonymized and derived from existing clinical registries with informed consent. Second, results from functional studies will be interpreted conservatively, acknowledging uncertainty and avoiding overstatement of clinical applicability until replicated. Potential unintended consequences include the possibility that a variant classified as “severe” might nevertheless respond to a modulator, leading to false exclusion from treatment. To address this, we will use multiple functional assays (microscopy and biotinylation) and, where possible, validate findings with patient-derived cells. Alternatives to our proposed actions include relying on computational prediction alone, but this would miss subtle trafficking defects. Our approach balances rigor with feasibility, prioritizing patient benefit while maintaining scientific integrity.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Detailed Experimental Plan and Timeline
Week 1: Variant selection and in silico design (completed). Review Ecuadorian CF patient databases, identify H609R as most prevalent variant. Use AlphaFold to predict structural impact. Design CFTR-eGFP fusion plasmid in SnapGene with CMV promoter, CFTR gene, flexible linker, and eGFP. Expected result: Complete plasmid map and sequence file.
Week 2: Order synthetic DNA fragments. Design oligonucleotides containing H609R and other selected mutations. Order synthetic DNA fragments from Twist Biosciences containing the CFTR-eGFP cassette with desired mutations. Expected result: Synthetic DNA delivered within 5-7 business days.
Week 3: Restriction enzyme digestion of vector backbone. Digest pcDNA5/FRT/GFP-CFTR plasmid with NheI and EcoRV restriction enzymes (37°C, 2 hours). Run digested products on 1% agarose gel to confirm linearization. Purify linearized backbone using gel extraction kit. Expected result: Clean, linearized vector at correct size (~8 kb).
Week 4: Ligation and transformation. Ligate synthetic DNA fragments containing mutations into linearized backbone using T4 DNA ligase (16°C, overnight). Transform ligation products into DH5α competent E. coli by heat shock (42°C, 45 seconds). Plate on LB agar with ampicillin (100 μg/mL) and incubate overnight at 37°C. Expected result: 50-200 colonies per plate.
Week 5: Colony screening and plasmid purification. Pick 5-10 colonies per construct, grow overnight in LB-ampicillin broth. Perform miniprep to purify plasmid DNA. Verify insert by restriction digest and Sanger sequencing. Expected result: Sequence-verified clones for wild-type and each mutant.
Week 6: MDCK cell culture and plating. Maintain MDCK type II FRT cells in DMEM with 10% FBS. Trypsinize and plate cells on Transwell filter supports at 50% confluency. Change media daily for 72 hours to allow polarization. Expected result: Confluent, polarized MDCK monolayers on filters.
Week 7: Transient transfection. Transfect MDCK cells with wild-type and mutant CFTR-eGFP plasmids using Lipofectamine 2000 (8 μL per well). Incubate for 48 hours to allow protein expression. Expected result: 20-40% transfection efficiency, visible GFP signal.
Week 8: Confocal microscopy for localization. Fix cells with 4% paraformaldehyde. Stain with WGA-Alexa Fluor 647 (5 μL/sample, 1 hour) to label apical membrane. Stain nuclei with DAPI. Image using Zeiss 510 confocal microscope (63x oil immersion). Expected result: Wild-type shows apical GFP-WGA colocalization (yellow); H609R shows either apical, cytoplasmic, or mixed pattern.
Week 9: Cell surface biotinylation. Label live MDCK cells with membrane-impermeable sulfo-NHS-LC-biotin (4°C, 30 minutes). Quench with glycine. Lyse cells with RIPA buffer. Capture biotinylated proteins on NeutrAvidin beads (overnight, 4°C). Elute with Laemmli buffer + DTT. Expected result: Biotinylated proteins captured on beads.
Week 10: Western blot analysis. Separate biotinylated and total lysate fractions by SDS-PAGE (5% Tris-HCl gel for GFP-CFTR). Transfer to nitrocellulose membrane. Probe with anti-GFP antibody (1:4,000) and anti-beta-actin loading control (1:10,000). Develop with ECL-Plus and image. Expected result: Wild-type shows strong mature C band (~205 kDa) in biotinylated fraction; trafficking mutants show reduced or absent C band.
Week 11: Data quantification and analysis. Quantify band intensities using ImageJ. Calculate apical ratio = (biotinylated C band intensity) / (total C band intensity). Compare ratios across wild-type and mutants using t-test (p < 0.05). Expected result: Quantitative metric for trafficking efficiency.
Week 12: Interpretation and conclusion. Correlate localization data (confocal) with biochemical data (biotinylation). Classify each variant as “severe trafficking defect,” “partial defect,” or “wild-type like.” Prepare final report and recommendations for clinical application. Expected result: Functional classification of H609R and other Ecuadorian variants.
Techniques Utilized
Pipetting
Lab Safety
Bioethical Considerations
DNA Construct Design
Restriction Enzyme Digestion
Gel Electrophoresis
DNA Purification From Gel
Databases (e.g., GenBank, NCBI)
Chassis Selection (e.g., DH5α)
Plasmid Preparation
Bacterial Culturing
Quality Control/Analysis
Primer Design or Selection
Other Cloning Methods (Restriction Enzyme Digestion)
Expanded Description of Two Techniques
Restriction Enzyme Digestion: In this project, restriction enzyme digestion will be used to linearize the pcDNA5/FRT/GFP-CFTR plasmid backbone and to release the CFTR insert for cloning. Specifically, NheI and EcoRV will be used at 37°C for 2 hours in CutSmart buffer. The digested products will be separated on a 1% agarose gel, and the linearized backbone (approximately 8 kb) will be excised and purified using a gel extraction kit. This approach ensures that only correctly digested vector is used for ligation, minimizing background colonies. Expected results include a single bright band at the expected size with no visible undigested supercoiled plasmid.
Confocal Microscopy: Confocal microscopy will be used to visualize subcellular localization of CFTR-eGFP fusion proteins in polarized MDCK cells. After fixation and staining with WGA-Alexa Fluor 647 (apical membrane marker) and DAPI (nuclei), cells will be imaged on a Zeiss 510 confocal microscope using a 63x oil immersion objective. Optical sections (0.5 μm thickness) will be acquired in the xz-plane to assess apical vs. cytoplasmic distribution. Quantification of colocalization between GFP (CFTR) and WGA (apical membrane) will be performed using ImageJ Colocalization Finder. Expected results include strong apical colocalization for wild-type CFTR (Pearson’s coefficient >0.7) and significantly reduced colocalization for trafficking mutants (Pearson’s coefficient <0.3).
Industry Council Companies
Addgene – Potential source for CFTR plasmids and vectors
New England Biolabs – Source for restriction enzymes, ligase, and buffers
Twist Biosciences – For synthetic DNA fragments containing mutations
Opentrons – For potential automation of liquid handling steps
Thermo Fisher Scientific – For cell culture reagents and Lipofectamine
Ginkgo Bioworks – For potential future autonomous lab use
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
Validation Aspect Chosen
I chose to validate the in silico design of the CFTR-eGFP fusion plasmid using SnapGene software. This validation demonstrates that I can successfully design a DNA construct that expresses a CFTR-eGFP fusion protein, incorporating the correct regulatory elements (CMV promoter), open reading frame (full-length CFTR), flexible linker, and C-terminal eGFP tag, with all elements in frame and appropriate restriction sites for cloning.
Detailed Validation Protocol
Open SnapGene software and create a new DNA file.
Import the CFTR coding sequence (CDS) from NCBI Reference Sequence NM_000492.3.
Add a CMV promoter sequence upstream of the CFTR start codon.
Add a flexible linker sequence (GGGGSGGGGS) between the CFTR stop codon (removed) and the eGFP start codon.
Add the eGFP coding sequence in frame with the linker.
Add a polyadenylation signal downstream of eGFP.
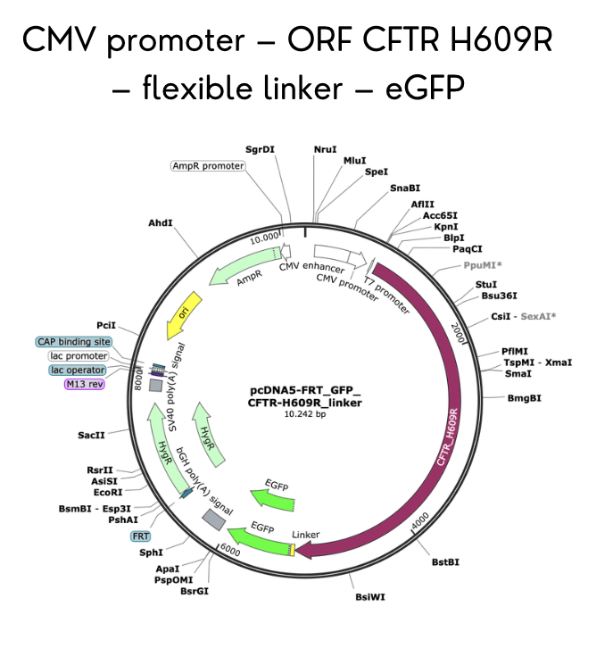
Introduce the H609R mutation (c.1826A>G) by site-directed mutagenesis tool, changing histidine 609 to arginine.
Verify the reading frame by translating the entire construct to ensure CFTR, linker, and eGFP are fused correctly without premature stop codons.
Identify unique restriction sites (NheI at 5’ end, EcoRV at 3’ end) for subcloning.
Generate a plasmid map with annotated features and restriction sites.
Export the sequence as a GenBank file for future ordering and sharing.
Synthetic Biology Techniques Utilized
This validation utilized DNA construct design and database searching techniques. Specifically, I used NCBI GenBank to retrieve the CFTR reference sequence (NM_000492.3) and verify the coding region. I applied DNA construct design principles to assemble a fusion gene with appropriate regulatory elements (CMV promoter, polyA signal), a flexible linker, and a reporter gene (eGFP) in frame. Using SnapGene, I performed in silico cloning to introduce the H609R mutation via site-directed mutagenesis, and I identified unique restriction sites (NheI and EcoRV) for subsequent subcloning into the expression vector. These techniques are foundational to synthetic biology as they enable the design and validation of genetic constructs before expensive and time-consuming wet-lab work begins.
Data Presentation and Analysis
The primary data from this validation is the complete plasmid map and sequence file for the CFTR-eGFP fusion construct (wild-type and H609R mutant). Analysis confirms that the construct contains a 4,440 bp CFTR coding sequence fused in frame to a 720 bp eGFP sequence via a 15 bp flexible linker, all under control of a 650 bp CMV promoter. Translation of the construct yields a 1,480 amino acid CFTR-eGFP fusion protein (CFTR: 1,480 aa; eGFP: 240 aa; total ~1,720 aa with a predicted molecular weight of ~190 kDa for the fusion, consistent with the C band observed in Western blots for mature CFTR plus GFP). Restriction digest simulation with NheI and EcoRV produces two fragments: a vector backbone (5,200 bp) and the CFTR-eGFP insert (5,175 bp).
Challenges and Alternative Strategies
A potential challenge in this validation is ensuring that the flexible linker does not introduce secondary structure that could interfere with CFTR folding or eGFP fluorescence. If the linker causes improper folding, the fusion protein might localize incorrectly even if the CFTR portion is functional. To overcome this, I could test multiple linker lengths (e.g., GGGGS, GGGGSx3, GGGGSx6) or use a different fusion strategy, such as placing eGFP at the N-terminus instead of the C-terminus.
Another challenge is that the large size of the CFTR-eGFP fusion (~5.2 kb insert) may be difficult to synthesize and clone efficiently. Alternative strategies include ordering the construct as a full-length synthetic gene from Twist Biosciences (which can synthesize up to 5 kb) or using Gibson assembly to combine smaller fragments. Additionally, transient transfection efficiency in MDCK cells may be low for such a large plasmid; an alternative is to generate stable cell lines using the Flp-In system described in Krasnov et al. (2008), which would ensure consistent expression across experiments.
SECTION 6: ADDITIONAL INFORMATION
References
Valle, Édison Patricio, Ramiro Israel Burgos, José Rubén Valle, Daniela Egas Béjar, and Juan-Carlos Ruiz-Cabezas. “Analysis of CFTR Gene Mutations and Cystic Fibrosis Incidence in the Ecuatorian Population.” Investigación Clínica 48 (2007).
Pérez, Martín M., María Cecilia Luna, Omar H. Pivetta, and Genoveva Keyeux. “CFTR Gene Analysis in Latin American CF Patients: Heterogeneous Origin and Distribution of Mutations across the Continent.” Journal of Cystic Fibrosis 6, no. 3 (2007): 194–208. https://doi.org/10.1016/j.jcf.2006.07.004.
Moya-Quiles, María Rosa, Guillermo Glover, Pedro Mondéjar-López, María Dolores Pastor-Vivero, Asunción Fernández-Sánchez, and Manuel Sánchez-Solís. “CFTR H609R Mutation in Ecuadorian Patients with Cystic Fibrosis.” Journal of Cystic Fibrosis 8, no. 4 (2009): 280–81. https://doi.org/10.1016/j.jcf.2009.05.001.
Paz-y-Miño, César, Ana Karina Zambrano, Juan Carlos Ruiz-Cabezas, et al. “Characterization of Ancestral Origin of Cystic Fibrosis of Patients with New Reported Mutations in CFTR.” BioMed Research International 2020, no. 1 (2020): 9074760. https://doi.org/10.1155/2020/9074760.
Dickinson, Kimberly M., and Joseph M. Collaco. “Cystic Fibrosis.” Pediatrics In Review 42, no. 2 (2021): 55–67. https://doi.org/10.1542/pir.2019-0212.
Cystic Fibrosis: Treatment with CFTR Modulators - UpToDate. n.d.
Ideozu, Justin E., Mengzhen Liu, Bridget M. Riley-Gillis, et al. “Diversity of CFTR Variants across Ancestries Characterized Using 454,727 UK Biobank Whole Exome Sequences.” Genome Medicine 16, no. 1 (2024): 43. https://doi.org/10.1186/s13073-024-01316-5.
Mickle, John E., MichałI. Milewski, Milan Macek, and Garry R. Cutting. “Effects of Cystic Fibrosis and Congenital Bilateral Absence of the Vas Deferens–Associated Mutations on Cystic Fibrosis Transmembrane Conductance Regulator–Mediated Regulation of Separate Channels.” The American Journal of Human Genetics 66, no. 5 (2000): 1485–95. https://doi.org/10.1086/302893.
Krasnov, Kristina V., Maria Tzetis, Jie Cheng, William B. Guggino, and Garry R. Cutting. “Localization Studies of Rare Missense Mutations in Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Facilitate Interpretation of Genotype-Phenotype Relationships.” Human Mutation 29, no. 11 (2008): 1364–72. https://doi.org/10.1002/humu.20866.
Ruiz-Cabezas, Juan Carlos, Francisco Barros, Beatriz Sobrino, et al. “Mutational Analysis of CFTR in the Ecuadorian Population Using Next-Generation Sequencing.” Gene 696 (May 2019): 28–32. https://doi.org/10.1016/j.gene.2019.02.015.
Ortiz, Sofía C., Santiago J. Aguirre, Sofía Flores, Claudio Maldonado, Juan Mejía, and Lilian Salinas. “Spectrum of CFTR Gene Mutations in Ecuadorian Cystic Fibrosis Patients: The Second Report of the p.H609R Mutation.” Molecular Genetics & Genomic Medicine 5, no. 6 (2017): 751–57. https://doi.org/10.1002/mgg3.337.
Note: Some items may be available from institutional shared resources or collaborating labs, reducing actual out-of-pocket costs. Budget assumes no existing stock and full retail pricing.
ACKNOWLEDGMENTS
This project was developed as part of the How To Grow (Almost) Anything (HTGAA) course. I thank the course instructors and teaching assistants for their guidance, as well as the patients and clinicians in Ecuador who have shared genetic data to make this research possible.