Week 4 HW: Protein Design Part I
Conceptual Questions
1. Why are there only 20 natural amino acids?
- There aren’t only 20 amino acids. There are just 20 that biology standardized early on in evolution. Proteins are built using translation. Once that system had evolved changing it was difficult because every protein in every organism depended on it. That creates evolutionary lock-in often referred to as a “frozen standard.” The current amino acids were selected due to their component atoms, functional groups, biosynthetic cost, use in a protein core or on the surface, solubility and stability. There are reasons for the selection of every amino acid.
2. Where did amino acids come from before enzymes that make them, and before life started?
- Abiotic chemistry on early Earth. Amino acids are chemically natural products when carbon, nitrogen, hydrogen, oxygen, and energy mix. Meteorites can also contain amino acids, therefore, some could have come to Earth from space. Geochemical environments like hydrothermal vents, mineral surfaces, metal ions, heat gradients, and pH differences can drive reactions that form amino acids from simpler molecules. Before enzymes chemistry did the job.
3. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- A helix made from D-amino acids will form a left-handed α-helix.
4. Can you discover additional helices in proteins?
- Yes there are algorithms that can scan protein structures and assign different helices based on hydrogen-bond patterns and geometry. Proteins contain more than just the regular α-helix. There are also rarer helices such as 3₁₀ helices, π helices, polyproline helices, and collagen triple helices. With computational design or mutation experiments, you can often convert loops or disordered regions into helices.
5. Why are most molecular helices right-handed?
- Most molecular helices are right-handed because the building blocks of life are chiral molecules, and biology chose one handedness early on. Once that choice locked in, the geometry of bonding and steric constraints naturally favor right-handed helices for those particular molecular configurations. A right-handed α-helix lets hydrogen bonds line up cleanly while avoiding atomic collisions. A left-handed α-helix is theoretically possible but energetically unfavorable with L-amino acids.
6. Why do β-sheets tend to aggregate?
A β-sheet is a protein secondary structure where the backbone is stretched out into strands that sit next to each other, stabilized by hydrogen bonds between the backbone carbonyl and amide groups. The hydrogen-bond donors and acceptors often remain partially unsatisfied at the sheet edges. When another β-strand comes nearby, it can complete those hydrogen bonds. So strands stack. Then stacks stack. Then you get fibrils.
What is the driving force for β-sheet aggregation?
- β-sheet aggregation is driven by the combination of unsatisfied backbone hydrogen bonds seeking partners, hydrophobic interactions between sheet faces, favorable side-chain packing, and nucleation-dependent polymerization that lowers free energy as aggregates grow.
7. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- 6 × 10²³ amino acid molecules. Meat is usually about 20% protein by mass (varies by cut, fat content, species). So 500g of meat would contain about 100g of protein. We are given that on average an amino acid is about 100 Daltons (100 g/mol per amino acid).
- 100 g ÷ (100 g/mol per amino acid) = 1 mole of amino acids
- 1 mole = 6.022 × 10²³ molecules (Avogadro’s number)
- Therefore, you are ingesting 6 × 10²³ amino acid molecules
8. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- When you eat beef or fish your digestive system breaks them down biochemically. Proteins are broken into amino acids, fats into fatty acids, carbohydrates into simple sugars. By the time nutrients cross your intestinal wall, there is no “cow” left just raw chemical building blocks.
9. Why do many amyloid diseases form β-sheets?
Amyloid diseases happen when protein folding goes wrong. A β-sheet is essentially the most efficient way for a polypeptide backbone to hydrogen bond with itself when the protein is unfolded or partially misfolded. That creates a highly stabilized, repetitive lattice.
Can you use amyloid β-sheets as materials?
- Yes, amyloid β-sheet structures can be used as materials. Amyloid fibrils have remarkable mechanical properties. Their stiffness rivals silk and even steel on a weight basis. They self-assemble spontaneously under mild conditions (water, room temperature). They’re nanoscale fibers with predictable dimensions. People are exploring amyloid-based materials for biomaterials and scaffolds for tissue engineering, nanowires and conductive materials, drug delivery systems, adhesives and coatings, and biodegradable plastics or films made from peptide assemblies.
Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
Luciferase (Luz)💡
- For my protein I chose the fungal luciferase protein (Luz). I have selected this protein because it is responsible for the light producing reaction in the fungal bioluminescence pathway (FBP).
Identify the amino acid sequence of your protein.
Luciferase (Luz) 💡 Info:
The length of the protein is 271 amino acids. The most common amino acid is: Leucine (L), which appears 24 times. There are 250 protein sequence homologs for this luciferase protein. This protein would be a part of the luciferase protein family.
Identify the structure page of your protein in RCSB
- An RCSB structure entry could not be found for fungal luciferase. So instead I chose to do the bacterial luciferase (Lux) for this step.
Bacterial Luciferase (Lux) 💡 RCSB Info:
The structure was solved in 1996. It is a good quality structure the resolution is 1.50 Å. The structure was determined in the absence of substrates. Bacterial luciferase (Lux) belongs to the alkanal monooxygenase family structure classification.
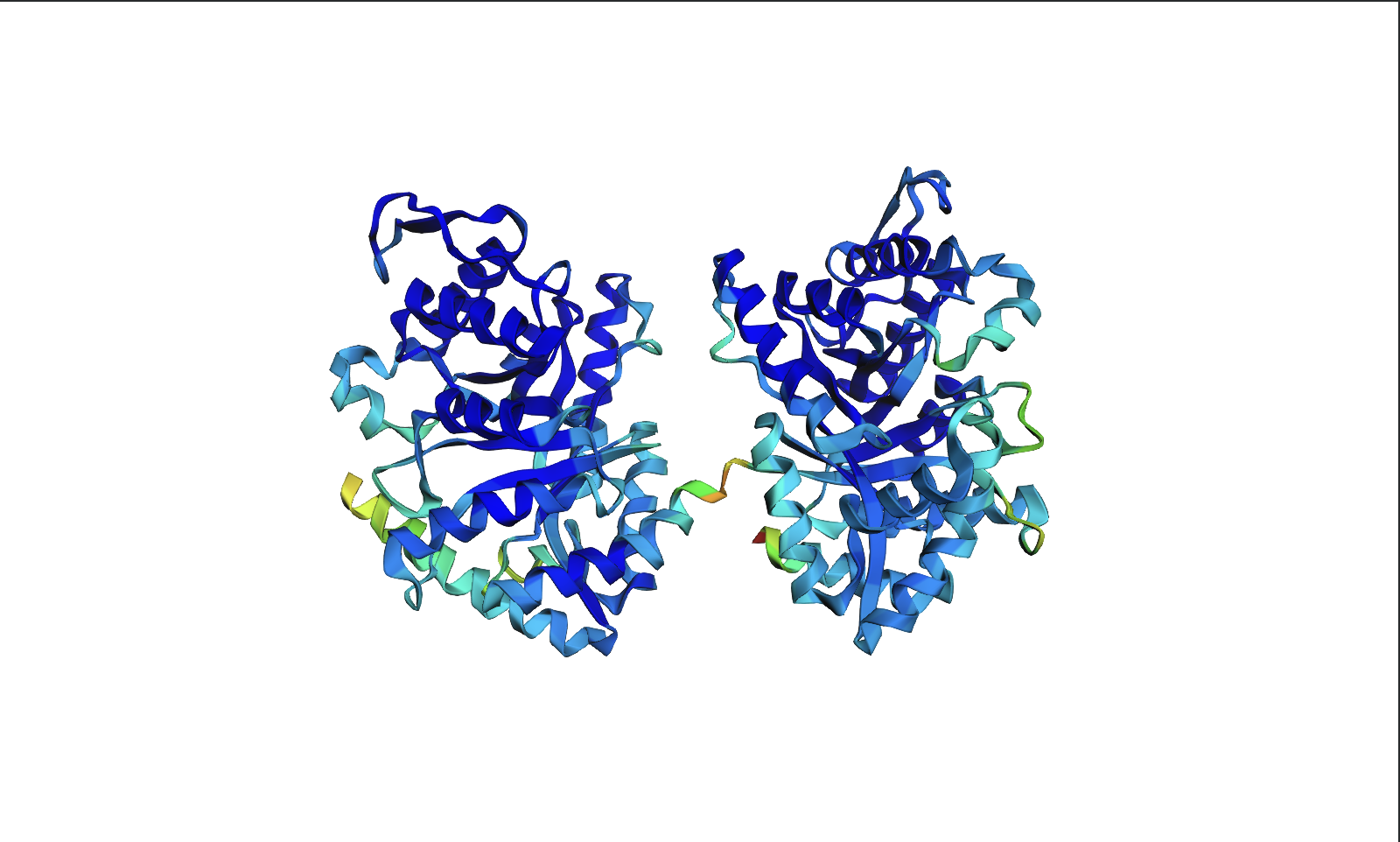
Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon:
Ribbon:

Ball and Stick:

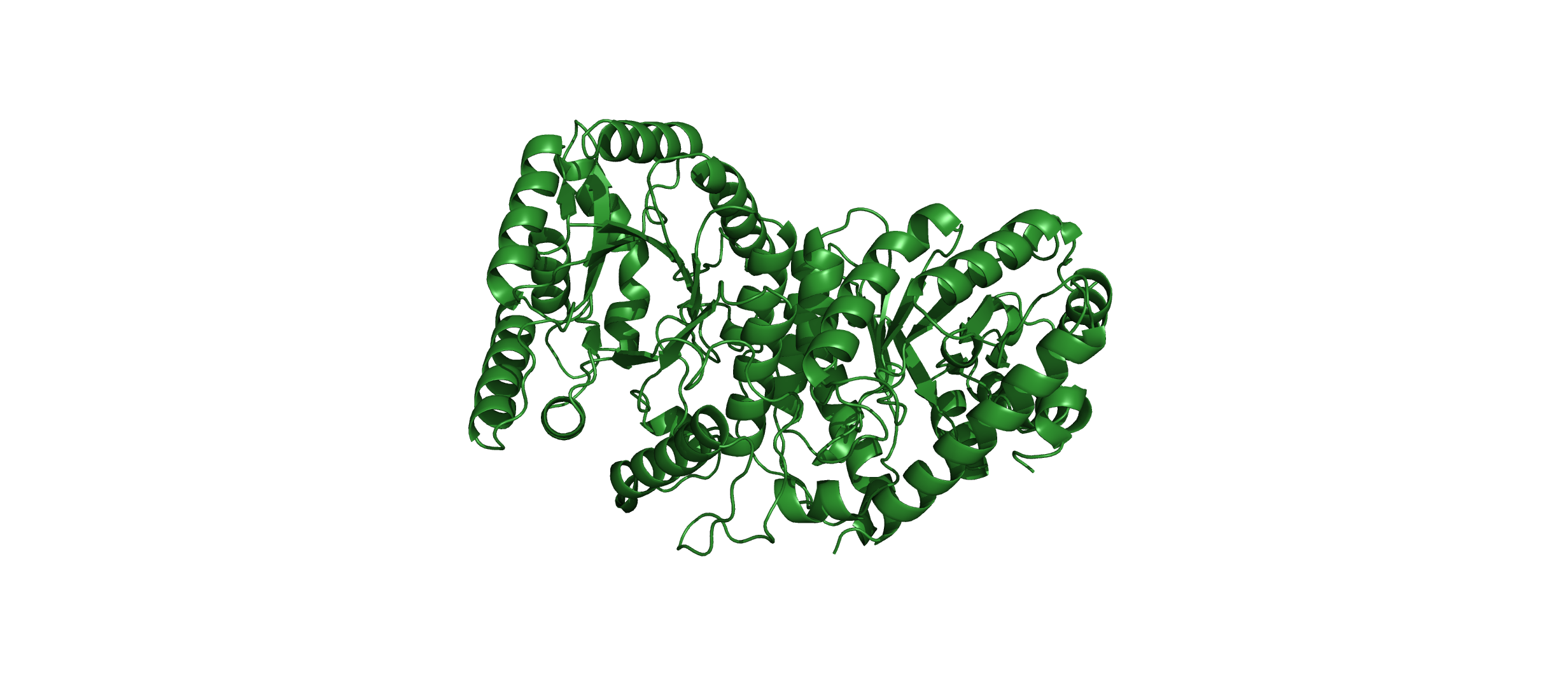
Color the protein by secondary structure. Does it have more helices or sheets?
- The protein has more helices than sheets. I counted roughly 9 sheets and 24 helices.
Secondary structure:
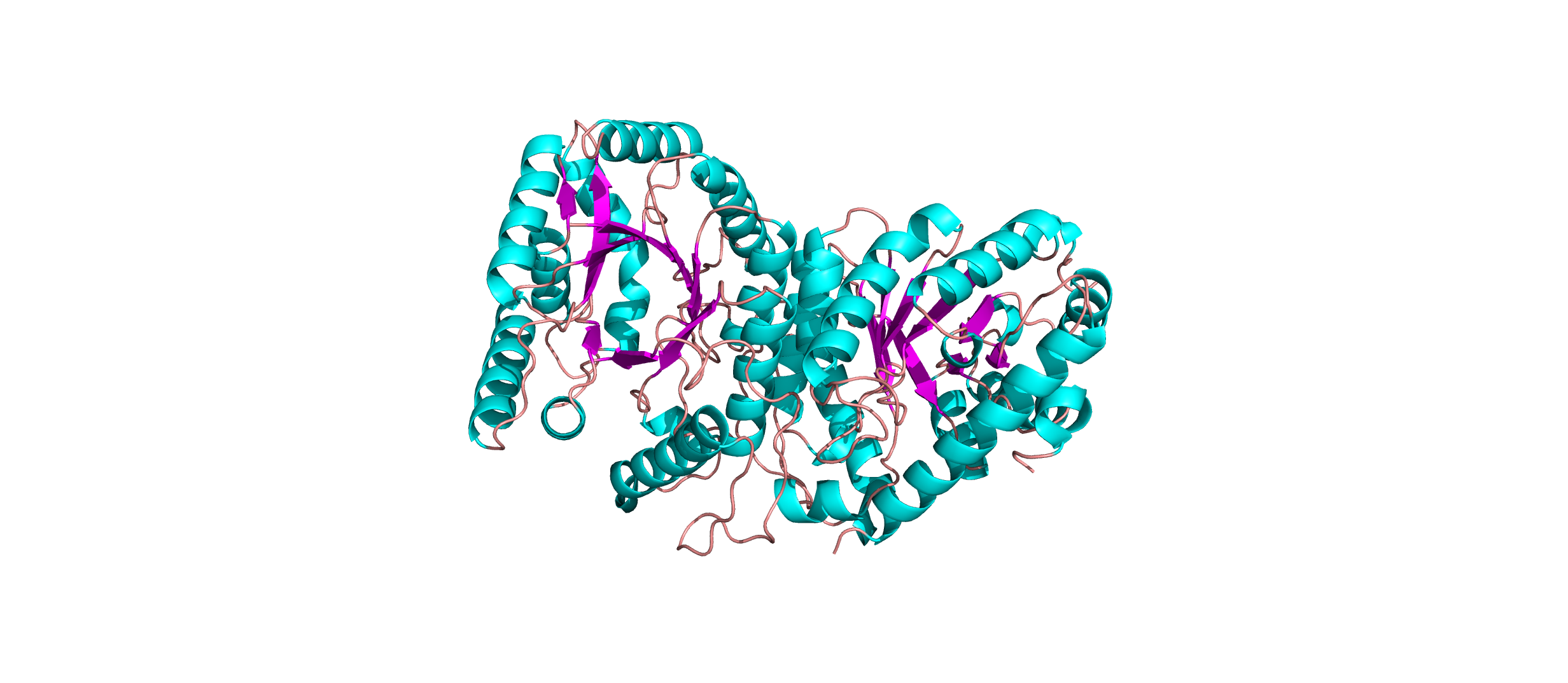
- The protein has more helices than sheets. I counted roughly 9 sheets and 24 helices.
Secondary structure:
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- The hydrophobic residues (yellow) cluster predominantly in the interior of the helical bundles and sheet cores, while the hydrophilic/charged residues (green, blue, red) decorate the surface and loop regions. That pattern tells us the protein has a well formed hydrophobic core. Also, the charged residues are surface-biased. The blue (positively charged) and red (negatively charged) patches tend to sit on solvent-exposed faces and flexible loops.
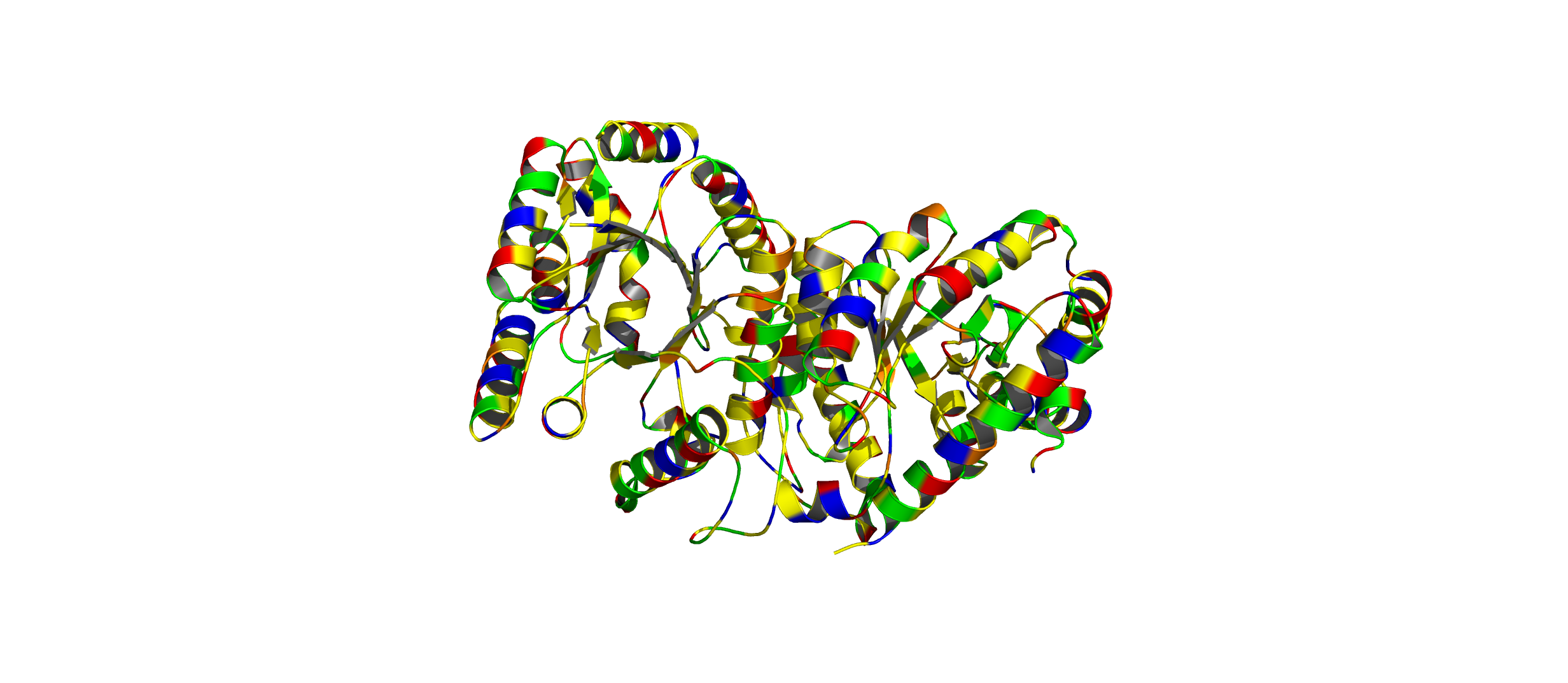
- The hydrophobic residues (yellow) cluster predominantly in the interior of the helical bundles and sheet cores, while the hydrophilic/charged residues (green, blue, red) decorate the surface and loop regions. That pattern tells us the protein has a well formed hydrophobic core. Also, the charged residues are surface-biased. The blue (positively charged) and red (negatively charged) patches tend to sit on solvent-exposed faces and flexible loops.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
- Yes there are holes.
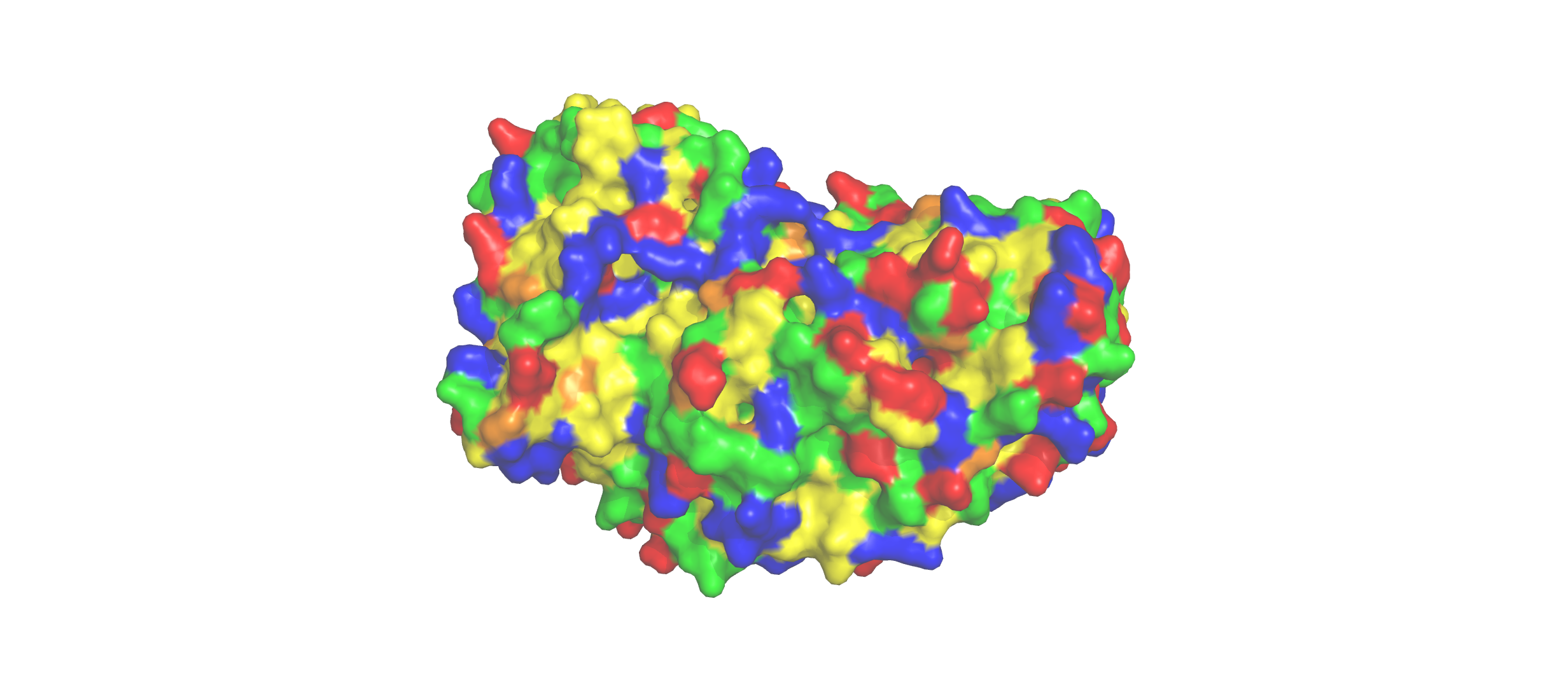
- Yes there are holes.
Using ML-Based Protein Design Tools
Protein Language Modelling
Deep Mutational Scans
- Position 30 (Leucine) shows strongly negative scores for most substitutions. This indicatces that most mutations at this site are disfavored. Particularly proline and glycine, indicating this residue is highly conserved. This is consistent with the position being structurally or functionally critical. Proline would disrupt backbone geometry, and glycine would introduce excessive flexibility.
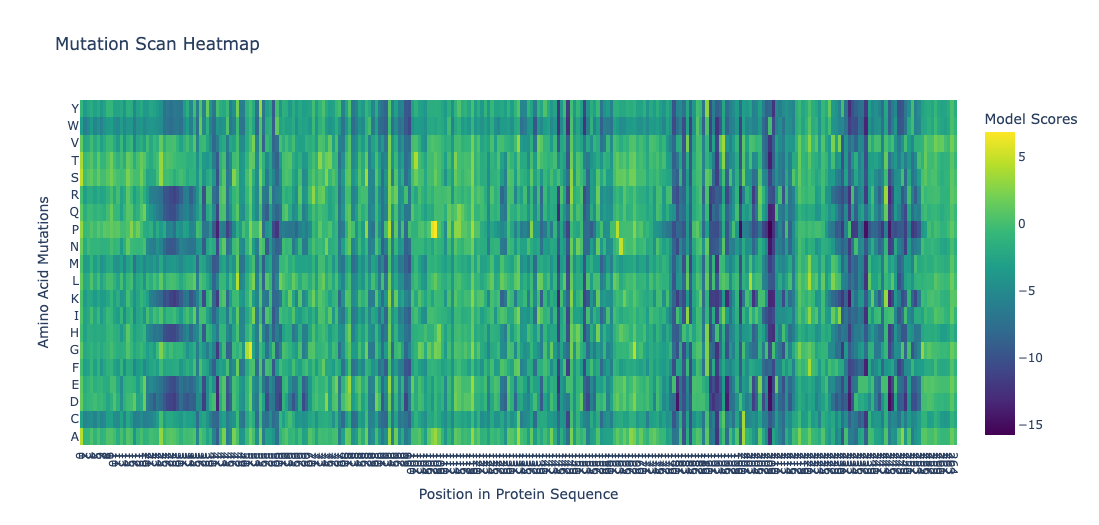
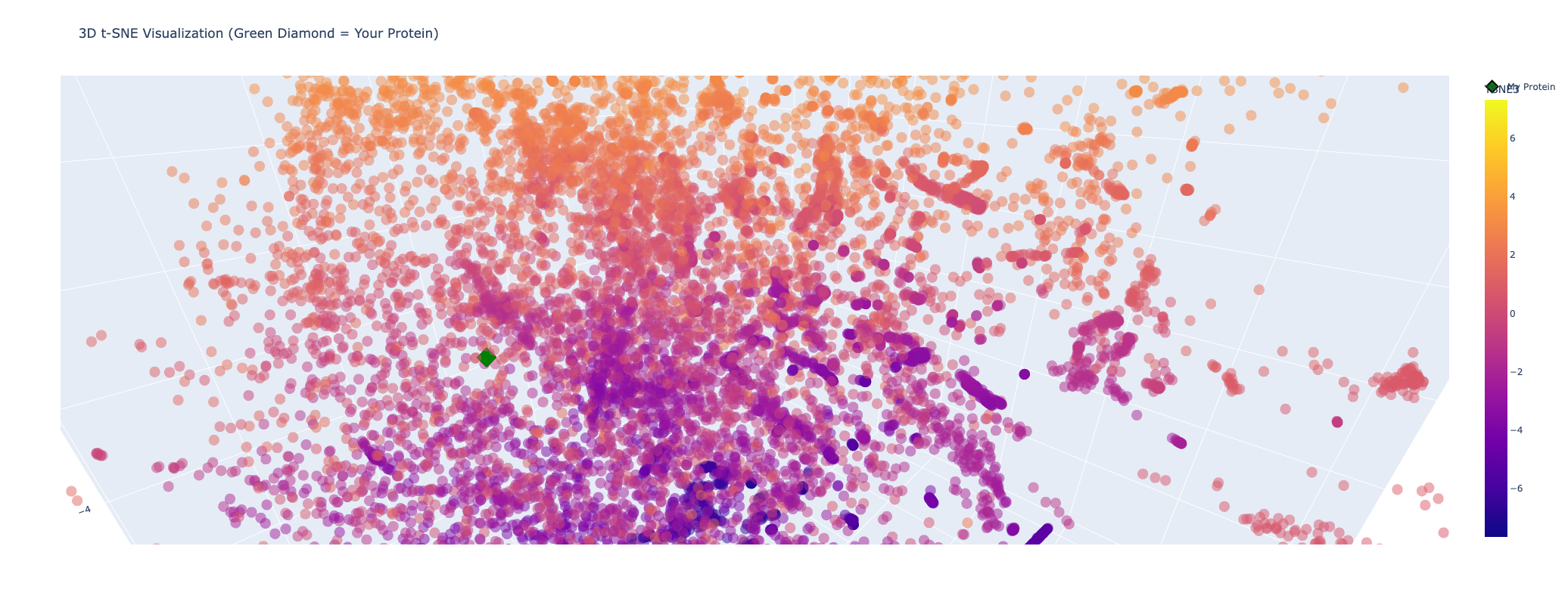
Latent Space Analysis
The latent space does approximates similar proteins. It appears to group them both by evolutionary origin (e.g., similar bacterial species) and biological function (e.g., repressors), demonstrating that the ESM2 embeddings capture meaningful biological features.
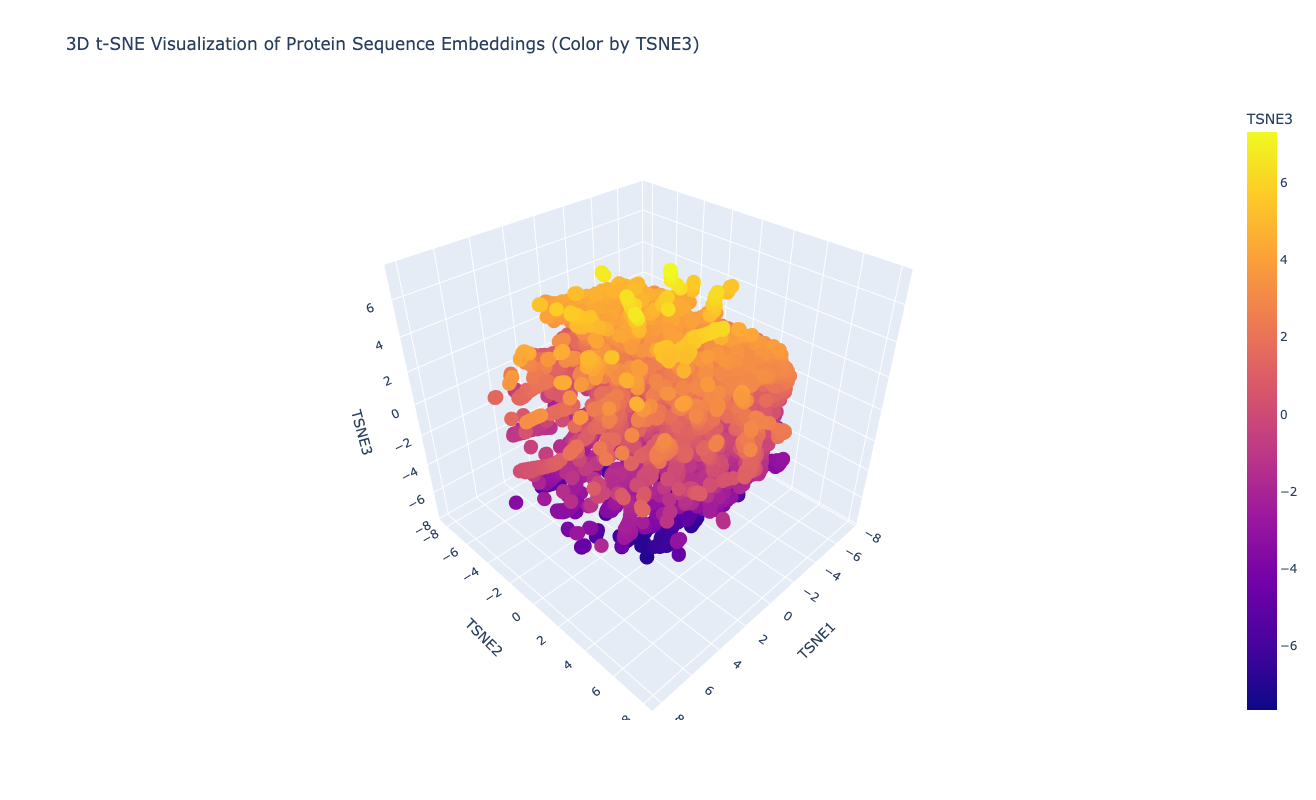
My protein (Luciferase) is positioned within a cluster of structurally related enzymes in the latent space. Its closest neighbors share similar structure classifications, indicating a shared evolutionary origin and structural fold, specifically relating to ATP-dependent ligation and metabolic signaling. This is interesting because luciferases are typically considered to be oxidoreductases. This could mean that the structure of fungal luciferase resembles ATP-dependent ligation and metabolic signaling enzymes more than classic oxidoreductases. Or it could also be possible that fungal luciferase might share an evolutionary ancestor with those enzyme families despite having diverged in function.
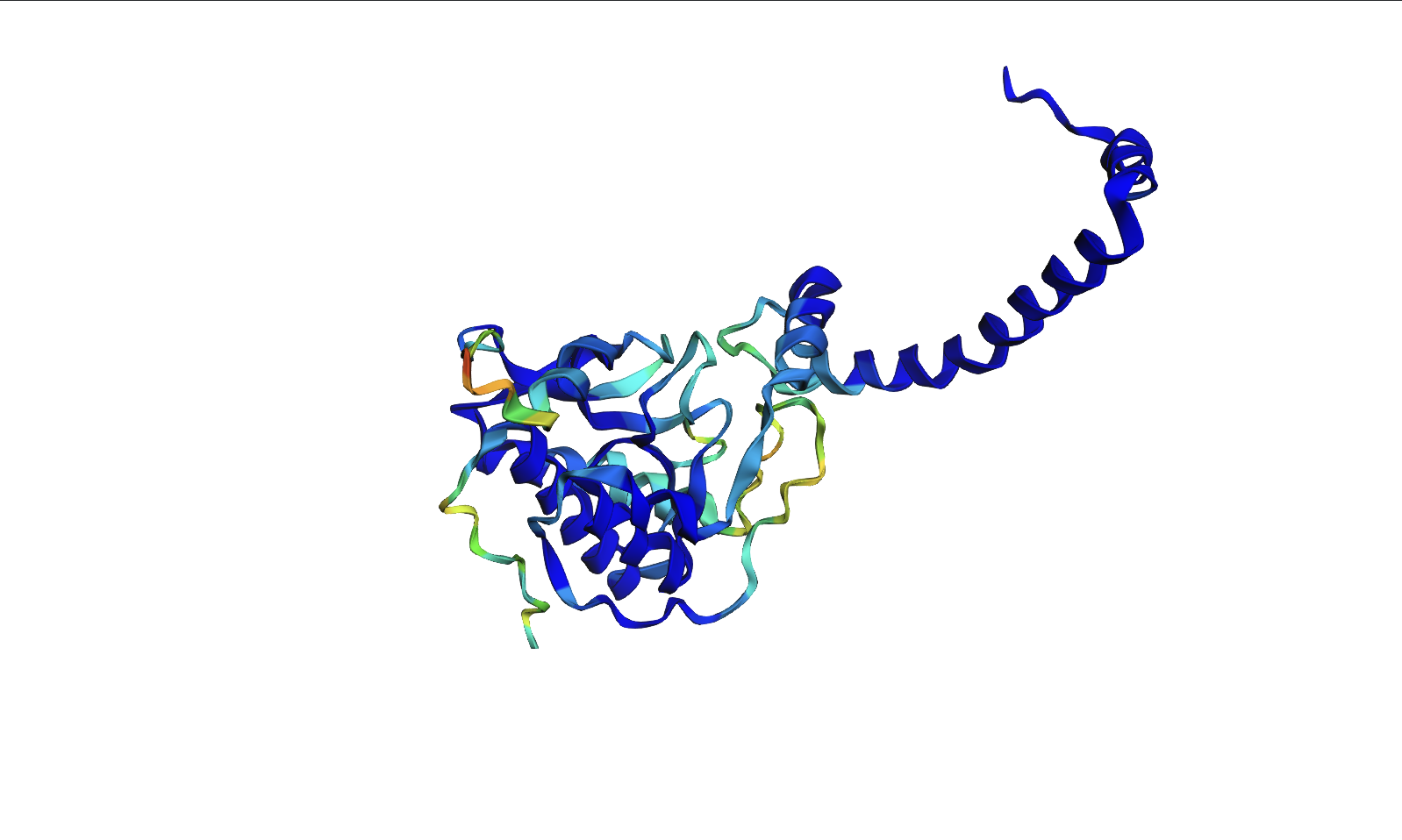
Protein Folding
- The ESMFold predicted coordinates match the original structure. The protein structure is resilient to mutations. I attempted several single base mutations and even larger segments with no impact on the protein structure. It seems that you would have to make significant changes to the sequence to alter the structure of the protein. I did notice that the confidence started to decrease in certain areas however after various mutations.
Protein Generation
- The ProteinMPNN amino acid heatmap shows that the model is highly confident at structurally constrained positions. Comparing the predicted sequence to the original, many positions share the same or chemically similar residues, indicating that ProteinMPNN recovers the biophysical constraints imposed by the backbone structure. There is a stretch of X residues in the predicted sequence that appears to correspond to a low confidence region in the heatmap, likely due to missing or poorly resolved coordinates in the input structure. Overall, the predicted sequence is not a reconstruction of the original but rather an alternative solution. A sequence that folds into a similar 3D structure, diverging at positions where the backbone tolerates variation.
Original:
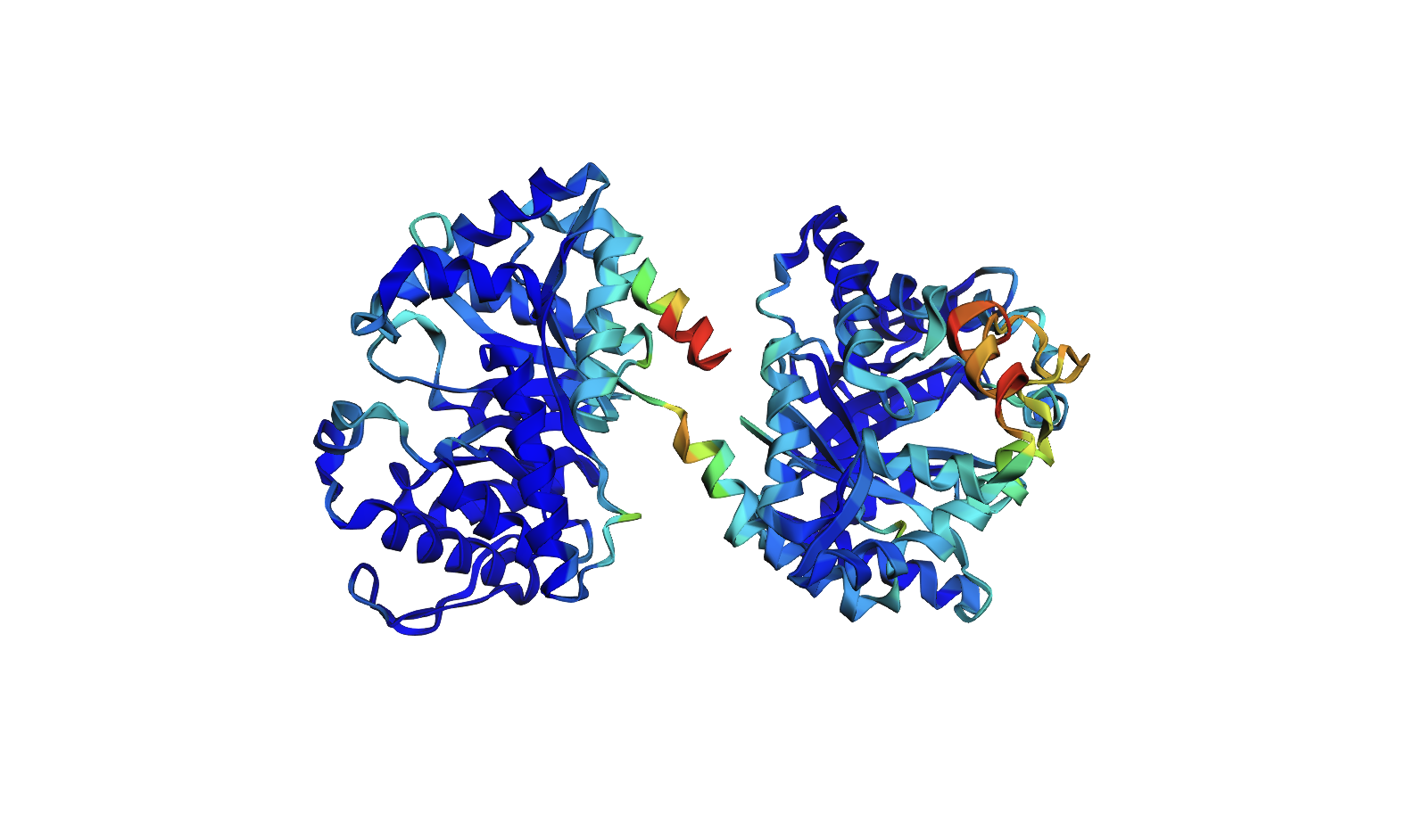
Predicted: