Week 5 HW: Protein Design Part II
SOD1 Binder Peptide Design
PepMLM Peptide Perplexity Scores
| Binder | Pseudo Perplexity |
|---|---|
| KLYYPAALRHKE | 20.698044578085693 |
| WRVPAVAAAWKK | 7.38320080665346 |
| WLYYVVALALWX | 17.40309350786337 |
| WLYGATGAEHKK | 11.275864207596417 |
| FLYRWLPSRRGG* | 20.63523127283615 |
*Known SOD1-binding peptide added for comparison
Evaluating Binders with AlphaFold3
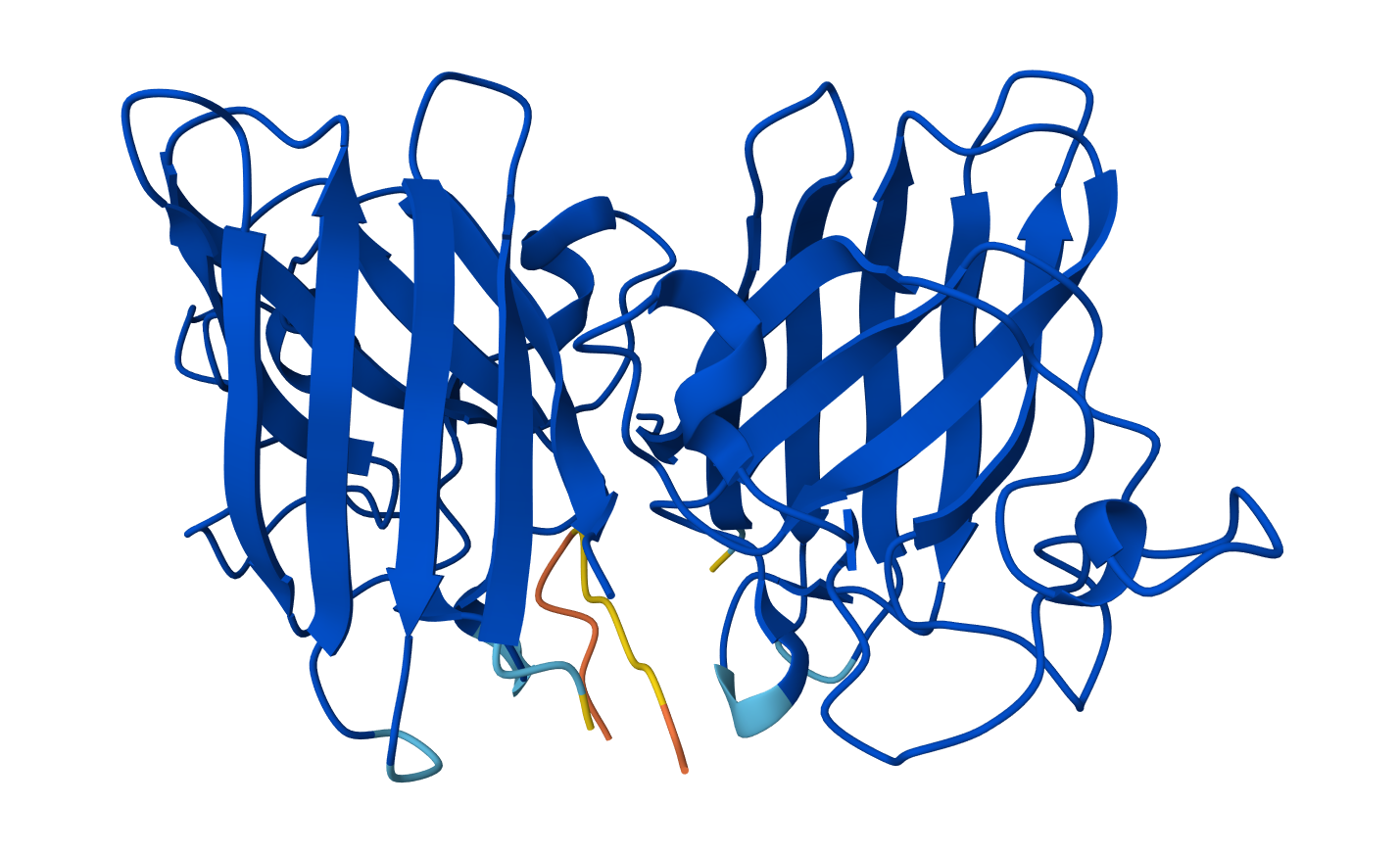
Binder: KLYYPAALRHKE
ipTM score: 0.87
This peptide appears to bind near the N-terminus where A4V sits. The peptide engages with the β-barrel region of the SOD1 protein and is surface-bound.
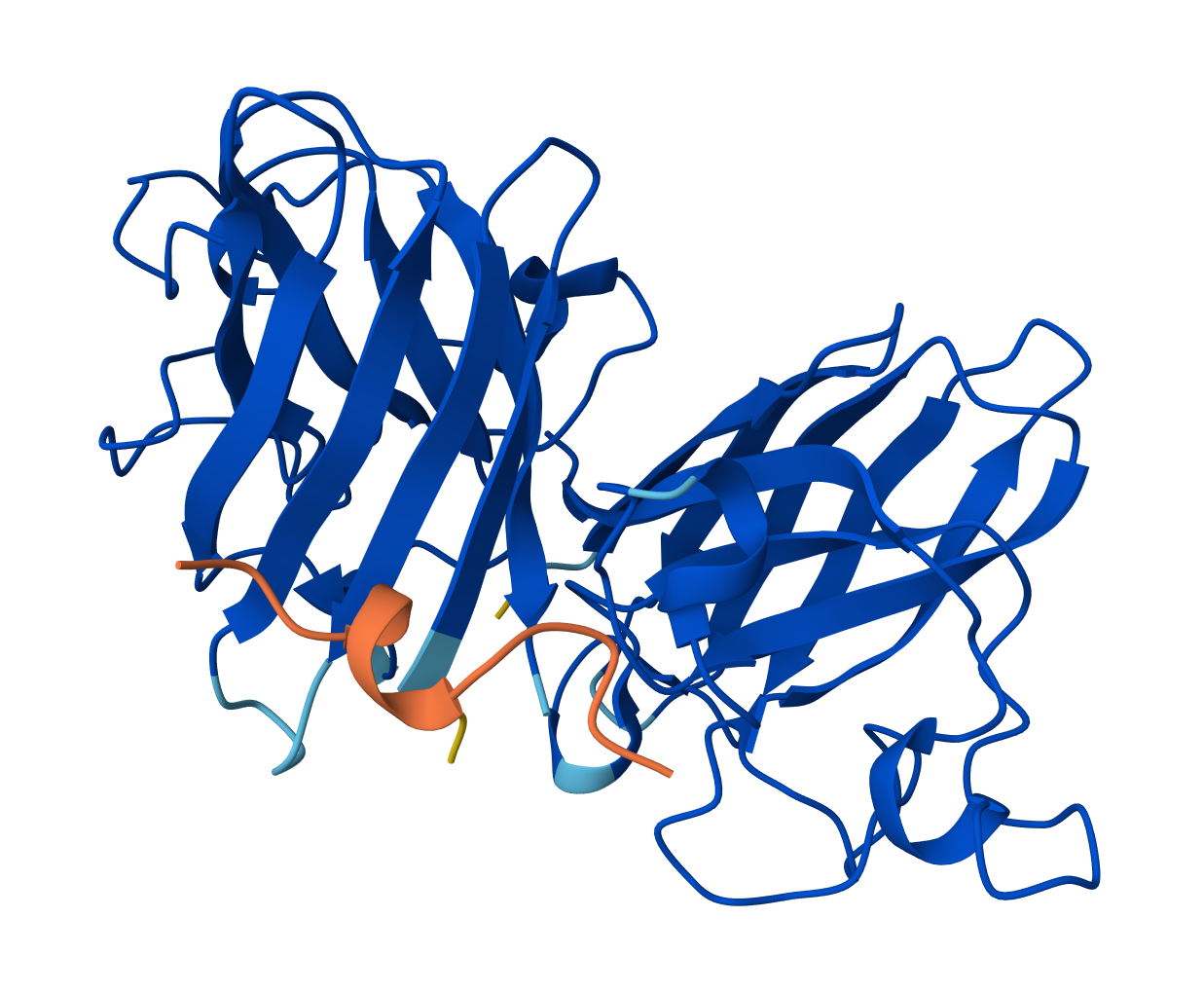
Binder: WRVPAVAAAWKK
ipTM score: 0.75
This peptide appears to bind near the N-terminus where A4V sits. The peptide engages with the dimer interface and it appears to be partially buried.
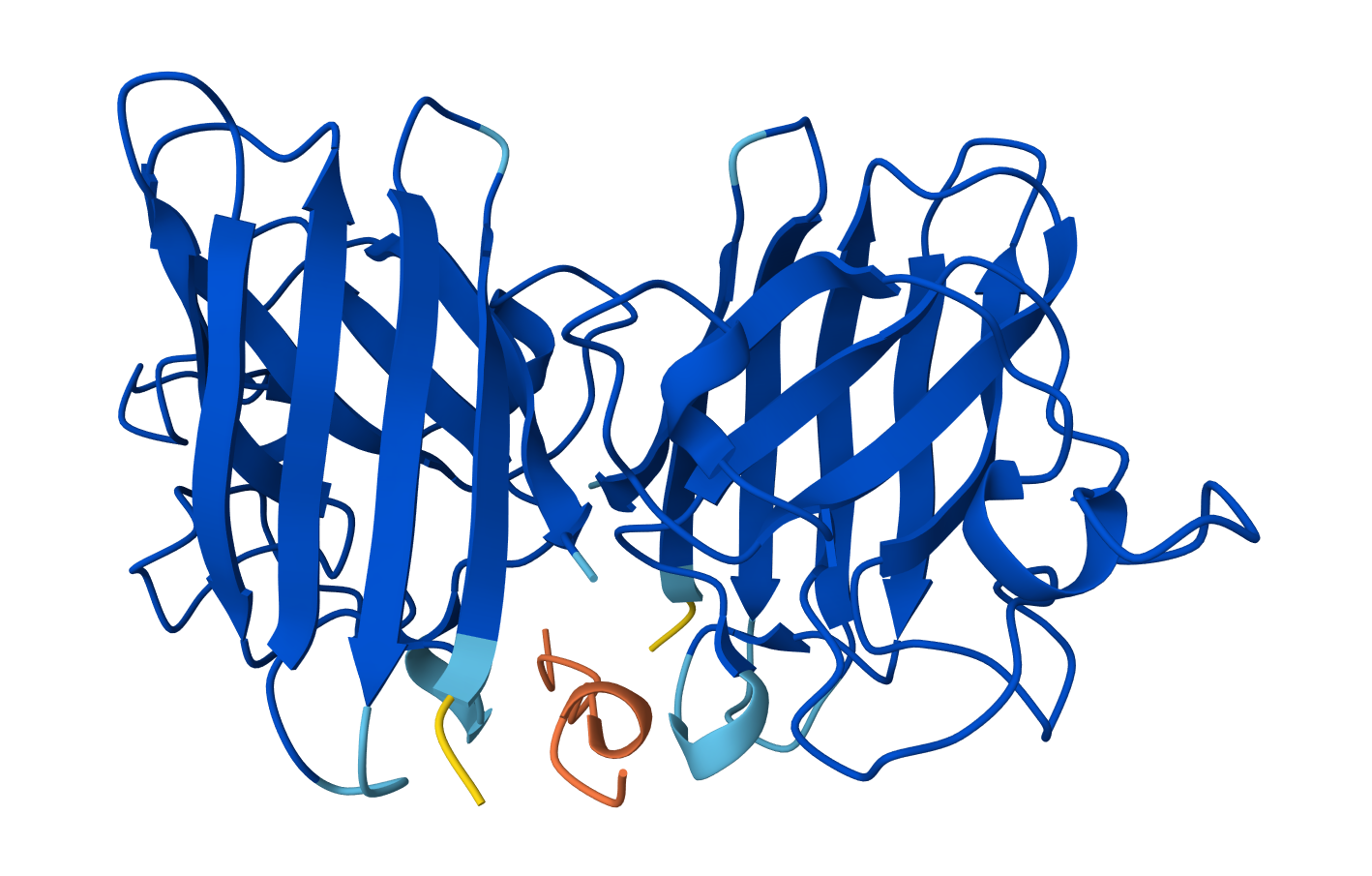
Binder: WLYYVVALALWX
ipTM score: 0.81
This peptide does not appear to bind near the N-terminus where A4V sits. The peptide engages with the β-barrel region of the SOD1 protein and is surface-bound.
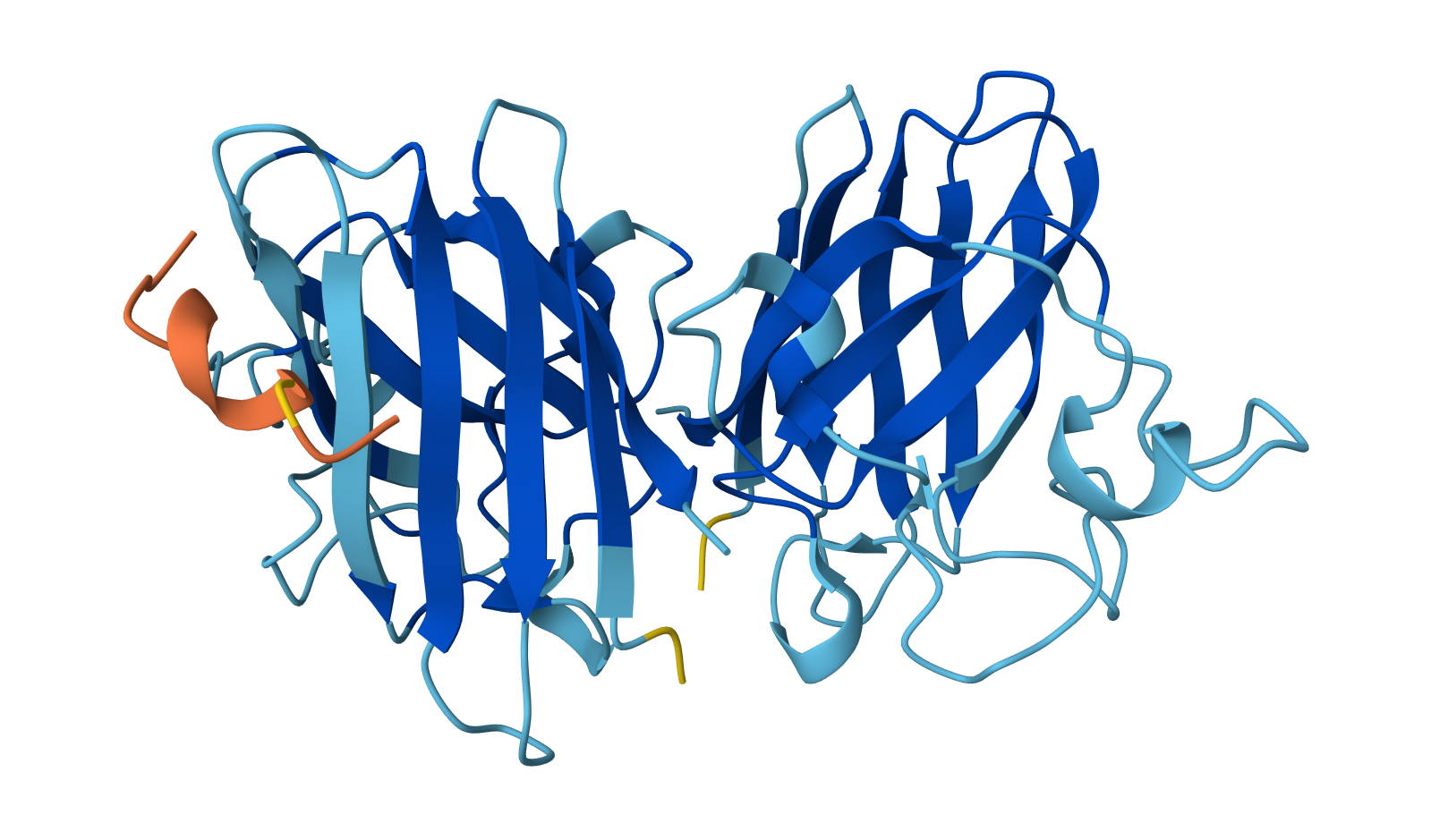
Binder: WLYGATGAEHKK
ipTM score: 0.78
This peptide does not appear to bind near the N-terminus where A4V sits. The peptide engages with the β-barrel region of the SOD1 protein and is surface-bound.
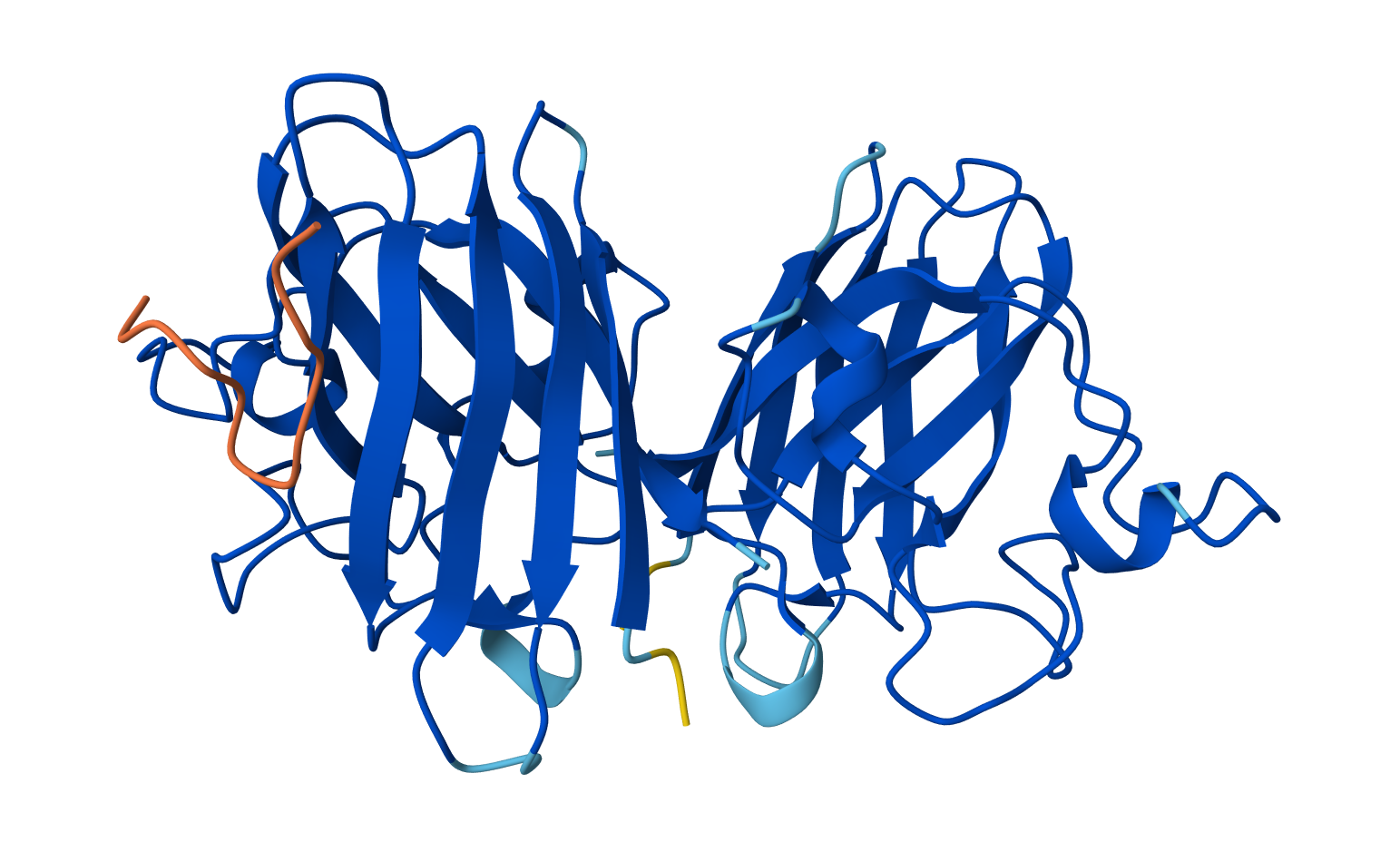
Binder: FLYRWLPSRRGG
ipTM score: 0.89
This peptide is a known SOD1-binding peptide that was added for comparison. It appears to bind near the N-terminus where A4V sits. The peptide engages with the dimer interface and it appears to be partially buried.
Across the five peptides, the known binder has the highest reported ipTIM = 0.89. The four PepMLM-generated peptides show ipTM values of 0.87 (KLYYPAALRHKE), 0.81 (WLYYVVALALWX), 0.78 (WLYGATGAEHKK), and 0.75 (WRVPAVAAAWKK). This means the designed peptides show moderately strong predicted interactions, but none match or exceed the known binder. The closest is KLYYPAALRHKE at 0.87, which is slightly below the reference.
Evaluating Properties of Generated Peptides in the PeptiVerse
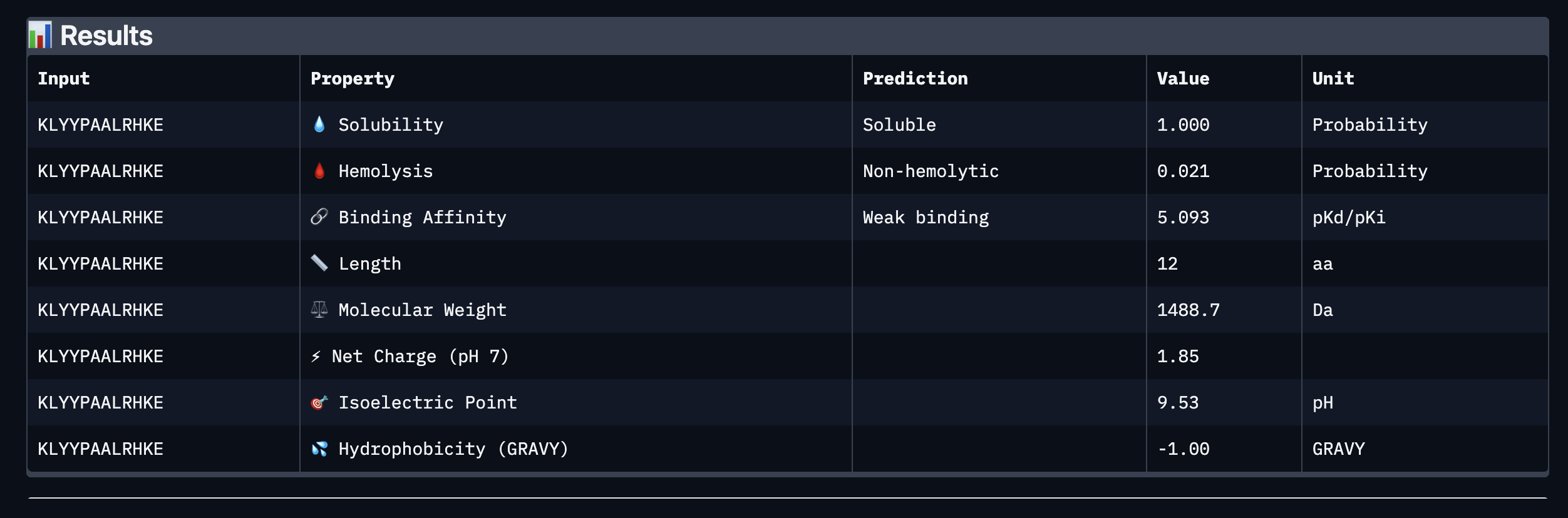
Binder: KLYYPAALRHKE
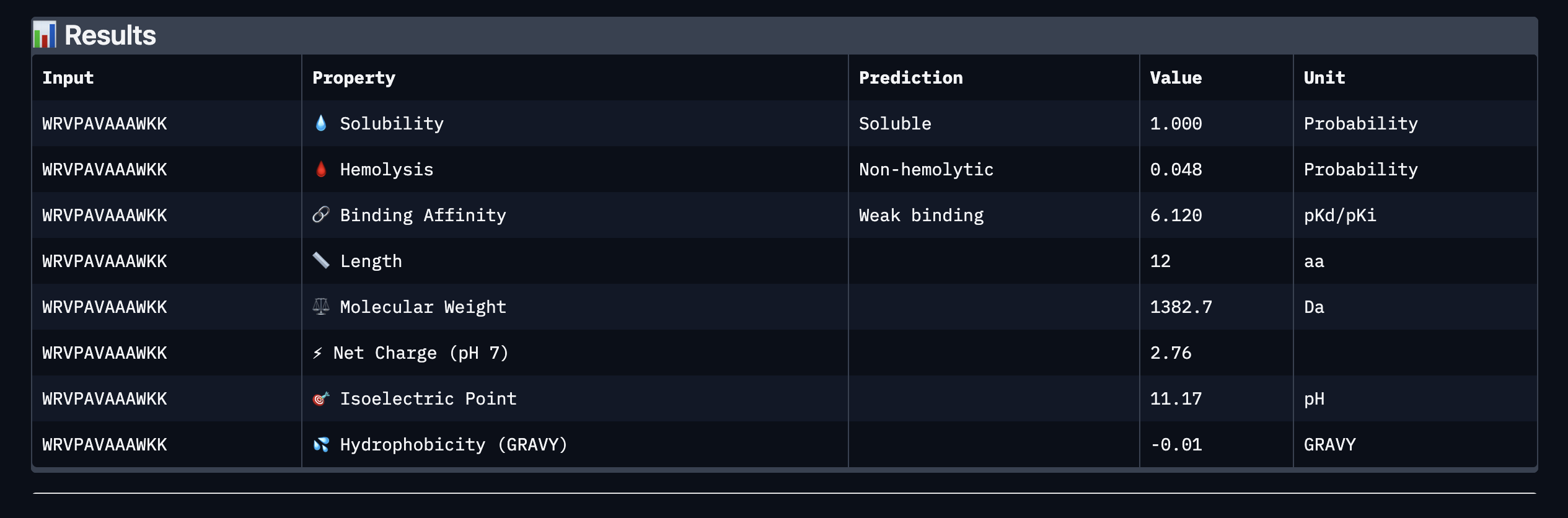
Binder: WRVPAVAAAWKK
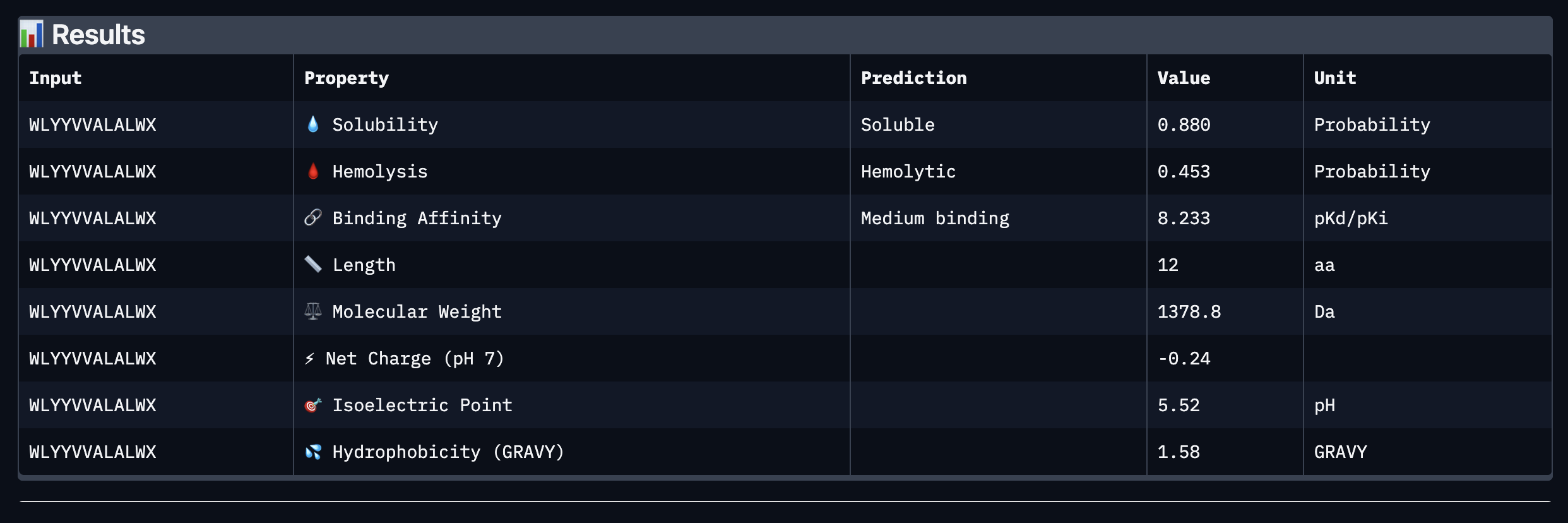
Binder: WLYYVVALALWX
Binder: WLYGATGAEHKK
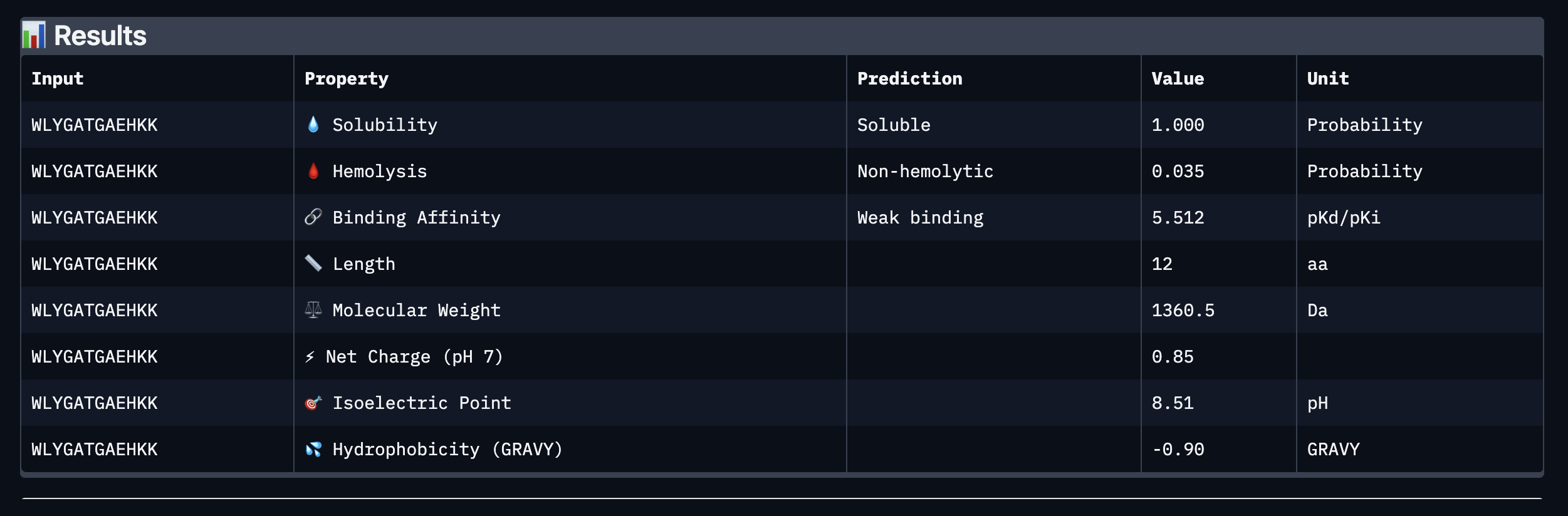
Comparing the AlphaFold3 structural predictions to the PeptiVerse therapeutic property predictions reveals that higher ipTM does not always correspond to stronger predicted binding affinity. KLYYPAALRHKE has the highest ipTM among the designed peptides (0.87) yet the weakest PeptiVerse binding affinity (pKd 5.093), while WLYYVVALALWX has a lower ipTM (0.81) but the strongest predicted affinity (pKd 8.233, medium binding). Notably, WLYYVVALALWX, the only peptide with medium binding, is also the only one flagged as hemolytic (0.453 probability) and has the highest hydrophobicity (GRAVY 1.58) and lowest solubility (0.880). This suggest that there is a trade-off between binding strength and therapeutic safety. Of the four PepMLM-generated peptides, I would advance KLYYPAALRHKE as it best balances predicted binding and therapeutic properties. It has the highest structural confidence from AlphaFold3, binds near the A4V site on SOD1, and has excellent solubility (1.000), the lowest hemolysis risk (0.021), and favorable hydrophilicity (GRAVY −1.00).
Generating Optimized Peptides with moPPIt
| Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| NKENFPKKKCKW | 0.9695186857134104 | 0.75 | 7.140074253082275 | 0.6160803437232971 |
| KPCGRGKRDAEH | 0.9709703326225281 | 0.8333333134651184 | 7.246584892272949 | 0.00822020135819912 |
| EQRKTDGCLLKI | 0.9666469506919384 | 0.75 | 6.097865581512451 | 0.8978230953216553 |
| KQKVCETYFRKN | 0.9696119148284197 | 0.8333333134651184 | 7.353872299194336 | 0.907169759273529 |
The moPPit generated peptides differ from the PepMLM peptides in several ways. The moPPit peptides show more consistent, moderate binding affinities (6.1–7.4) with uniformly high non-hemolytic probabilities (~0.97), while the PepMLM peptides had a wider spread. Additionally, moPPit provides motif scores indicating how well each peptide matches known binding motifs, with KQKVCETYFRKN (0.907) and EQRKTDGCLLKI (0.898) scoring highest. Before advancing any of these peptides to clinical studies, one would need to validate predictions experimentally through in vitro binding assays, cell-based hemolysis and cytotoxicity testing, solubility measurements, pharmacokinetic profiling (half-life, bioavailability), selectivity screening for off-target interactions, and ultimately in vivo efficacy and toxicology studies in animal models.
BRD4 Drug Discovery Platform Tutorial (Boltz Lab)
(View Full Screen)
L-Protein Mutants
Mutagenesis
Does the experimental data correlate with the scores from the ESM2 model?
The experimental data seems to correlate with the notebook scores somewhat, but not perfectly. In some cases, the predictions match the experimental results pretty well. For example, mutations at the start methionine had very negative LLR scores, and experimentally those mutations completely got rid of protein production and lysis. There were also some mutations like P13L, S15A, A45P, and I46F where the notebook suggested the mutation would be tolerated, and the experimental data showed that those mutations still worked.
At the same time, there were definitely some cases where the notebook scores did not match the experimental results. Some mutations had high or favorable LLR scores but still lost function in the experimental data. This happened at positions like C29, Y39, and K50. In some of those cases, the protein still seemed to be made, but it no longer caused lysis. That makes me think the language model is better at predicting whether a mutation is generally tolerated in the sequence than whether the protein will still do its exact job.
So overall, I would say the embeddings do capture useful information about the protein, but they do not fully predict function. The scores are helpful as a guide, but the experimental data matters more when deciding which mutations are actually good candidates.
Five proposed mutations
I chose these mutations by looking for positions that either had a positive or near-neutral LLR score, showed a positive effect in the experimental data, or seemed to tolerate substitutions well.
Transmembrane region:
A45L: I picked A45L because position 45 seems pretty tolerant to mutation. The LLR score for leucine here is strongly positive, and experimentally A45P still worked, which means this position can handle substitutions. Since leucine is also a very common hydrophobic amino acid in transmembrane regions, I think this mutation has a good chance of working.
I46F: I chose I46F because it was already shown experimentally to keep both lysis activity and protein production. It is also a conservative hydrophobic substitution, which makes sense in the transmembrane region.
Soluble region:
P13L: I chose P13L because both the notebook score and the experimental data support it. It had a slightly positive LLR score, and experimentally it gave lysis = 1 and protein = 1, so this seems like a strong choice.
R18G: I chose R18G because it worked experimentally even though the LLR score was somewhat negative. Since both R18G and R18I were functional, that suggests this position is more flexible than the model predicted.
R30L: I chose R30L because it was functional in the experimental data and had a nearly neutral LLR score. Also, position 30 tolerated another mutation (R30Q) as well, so this looks like another flexible site where substitution is possible without losing function.
Overall, I picked these mutations by combining the notebook predictions with the experimental data, but I relied more on the experimental results whenever the two did not match. That seemed like the best way to choose mutations that actually have a chance of working.