Individual Final Project

Section 1: Abstract
Engineering Brighter Autonomous Bioluminescence
Autonomous bioluminescence offers a major advantage over conventional bioluminescent systems because it eliminates the need for repeated addition of external luciferin. In plants, the fungal bioluminescence pathway is especially attractive because it uses caffeic acid, a metabolite that already exists in plant metabolism. However, current glowing plants are still limited by pathway flux, enzyme efficiency, and overall light output. The goal of this project is to engineer a brighter autonomous bioluminescence system by combining improved fungal bioluminescence enzymes with upstream metabolic enhancements that increase precursor availability. My hypothesis is that replacing the original pathway enzymes with brighter variants, including truncated nnLuz v4, nnH3H v2, and mcitHispS, and then increasing caffeic acid supply through added metabolic modules such as BnC3’H1 and TAL/HpaB/HpaC, will produce stronger self-sustained light emission than earlier fungal pathway designs. The specific aims are to first validate improved core pathway components, then compare enhancer strategies in modular construct designs, and finally identify the best architecture for future transient expression in Nicotiana benthamiana and stable transformation in Nicotiana tabacum. Methods include modular DNA design in Benchling, synthesis of gene cassettes, hierarchical DNA assembly, sequence verification, and comparative testing of optimized enzyme combinations. For the current class stage, an initial simplified test will evaluate whether nnLuz v4 truncated and nnH3H v2 can generate light in an inducible expression system, providing an early validation step before full plant pathway assembly.
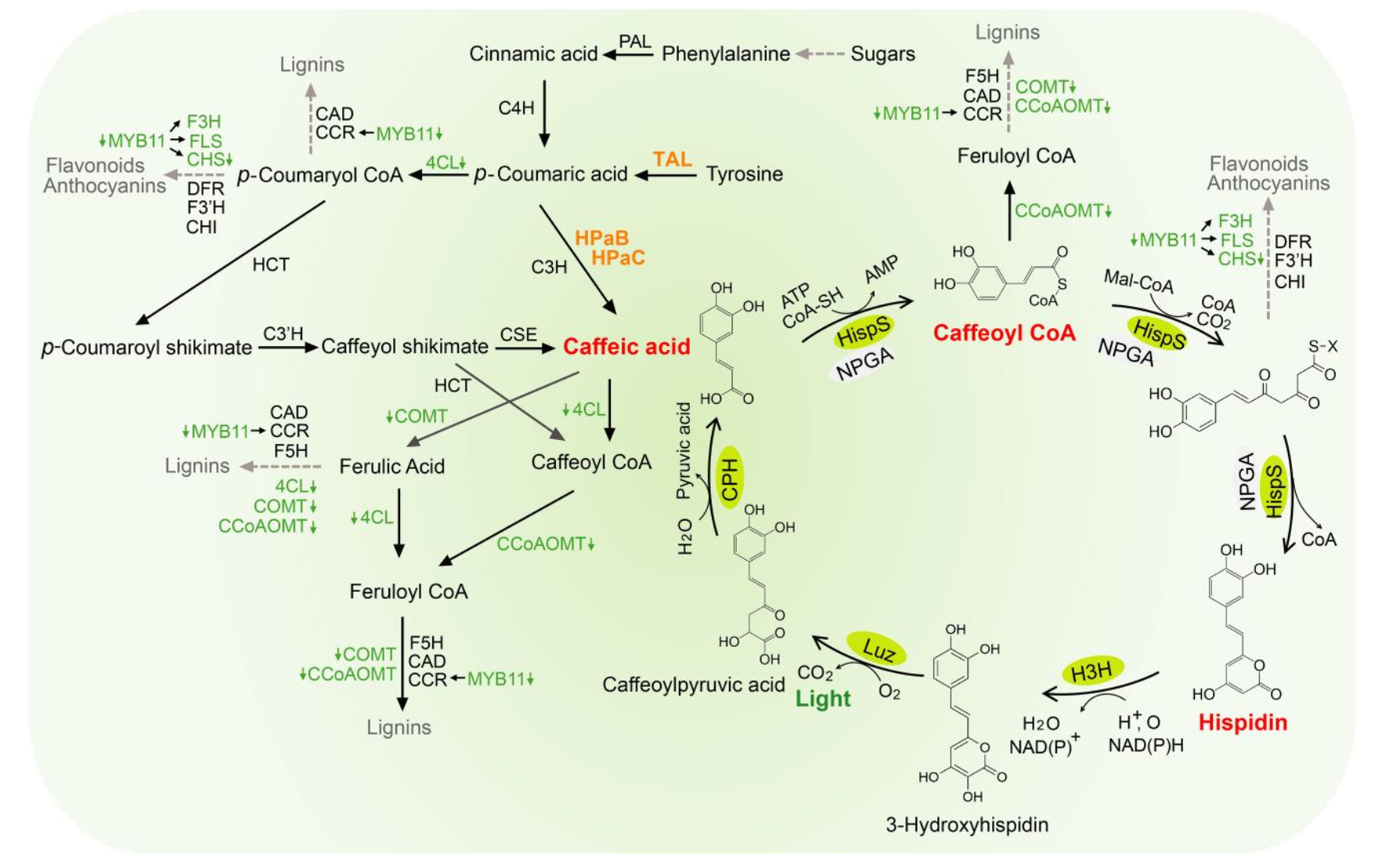
Figure 1. Bioluminescence Pathway.
Section 2: Project Aims
Aim 1: Experimental Aim
The first aim of my final project is to validate that the optimized fungal bioluminescence enzymes nnLuz v4 truncated and nnH3H v2 produce measurable light when co-expressed in E. coli, by utilizing a pET-28a(+) expression plasmid (BamHI/HindIII cloning sites), BL21(DE3) competent E. coli cells, IPTG induction, and exogenous hispidin supplementation, with luminescence quantified in 96-well black plates on a plate reader. Because neither the v4 mutation set nor the ΔN20 truncation has previously been tested in combination, this experiment provides a fast, low-cost functional readout of whether the two independently-validated improvements stack and create a brighter enzyme pair before committing to full pathway assembly in plants. Tools and resources include Benchling for construct design and sequence verification (referenced against Shakhova et al. 2024, Nature Methods and Patent CN116732084A), Twist Bioscience for synthesis of the codon-optimized gene cassette, remote Opentrons automation for liquid handling, and standard molecular biology reagents (kanamycin selection, LB media, NEB OneTaq for verification PCR). A detailed step-by-step protocol for this aim is provided in the Experimental Design section.
Aim 2: Development Aim
The second aim of my final project is to assemble and compare modular versions of an improved autonomous bioluminescence pathway for plant expression by combining an upgraded fungal core pathway with different upstream precursor boosting modules. Following a successful Aim 1, I plan to build and test pathway versions containing the brighter core enzymes (mcitHispS, NpgA, nnLuz_v4 truncated, nnCPH, and nnH3H_v2) together with caffeic acid precursor enhancers, including BnC3’H1 and the bacterial TAL/HpaB/HpaC module, to determine which combination produces the strongest autonomous signal. These constructs will be assembled into pCAMBIA1300 and delivered via Agrobacterium tumefaciens (GV3101), first evaluated in Nicotiana benthamiana through transient agroinfiltration before selecting the best performing design for stable transformation in Nicotiana tabacum. This stage extends the project from an enzyme pair validation in E. coli into a complete, self-sustaining plant bioluminescence system, while systematically testing whether boosting upstream caffeic acid availability further amplifies the gains achieved through core enzyme optimization. Once a winning architecture is established, future work can layer in additional improvements such as a Bioluminescence Resonance Energy Transfer (BRET) module for emission tuning and miRNA-based silencing strategies to fine tune pathway flux.
Aim 3: Visionary Aim
The third aim of my final project is to develop a much brighter and more robust autonomous bioluminescence platform that could enable new classes of living reporters, biosensors, and potentially useful light producing plants. If fully realized, this project could push plant bioluminescence beyond novelty and toward practical applications by creating systems that are easier to detect, quantify, and deploy without external substrate addition. More broadly, a sufficiently bright autonomous system could expand the use of bioluminescence for noninvasive monitoring of biological processes and help establish living light producing systems as useful tools in synthetic biology, agriculture, and environmental sensing.
Section 3: Background
Background and Literature Context
The fungal bioluminescence pathway (FBP) was first fully characterized in Neonothopanus nambi by Kotlobay et al. (2018, PNAS), who identified the core enzymes hispidin synthase (HispS), hispidin-3-hydroxylase (H3H), luciferase (Luz), and caffeoyl pyruvate hydrolase (CPH). This was a landmark finding because caffeic acid is already produced natively by most plants, making FBP uniquely suited for engineering autonomous, substrate-free luminescence in plant hosts. Mitiouchkina et al. (2020, Nature Biotechnology) then demonstrated proof of concept by stably transforming Nicotiana tabacum with the wild type pathway, producing tobacco plants that glowed visibly without external substrate addition. Both studies, however, established the same critical limitation: the wild type fungal enzymes performed suboptimally in heterologous hosts, leaving brightness well below what would be needed for practical reporter, biosensing, or illumination applications. Shakhova et al. (2024, Nature Methods) directly addressed this by applying directed evolution and ortholog screening to the FBP enzymes, producing the optimized FBP3 pathway (mcitHispS, NpgA, nnLuz v4, nnCPH, nnH3H v2) that outperformed wild type FBP1 by one to two orders of magnitude across yeast, plant, and mammalian hosts. Independently, Patent CN116732084A reported that a 20 amino acid N-terminal truncation of wild type nnLuz (Luz ΔN20), designed from AlphaFold structural analysis, produced a 1.5- to 2-fold improvement in brightness by removing a predicted membrane associated α-helix that interferes with protein folding and accumulation.
Novelty and Innovation
The main novelty of this project is that it tests a new luciferase combination that has not yet been reported, nnLuz v4 ΔN20, that combines the seven beneficial point mutations from Shakhova et al. (2024) with the N-terminal truncation from Patent CN116732084A. The two improvements act through fundamentally different mechanisms, the v4 mutations enhance catalytic activity and thermostability, while the ΔN20 truncation increases functional protein accumulation by removing a membrane-associated α-helix that interferes with folding. So, there is a strong theoretical basis to expect at least additive, and potentially synergistic, gains in light output, but this has never been experimentally demonstrated. The project is also innovative in how it builds toward brighter autobioluminescent plants through a modular synthetic biology strategy, combining improved core fungal enzymes with upstream metabolic engineering modules such as BnC3’H1 and TAL/HpaB/HpaC to test whether enzyme optimization and precursor boosting can be layered together. This treats pathway brightness as a systems engineering problem rather than a single gene optimization problem, and directly tests whether the brightness ceiling of autonomous plant bioluminescence is set by enzyme efficiency, by substrate supply, or by both.
Significance and Impact
The core problem this project addresses is that autonomous bioluminescence, despite being one of the most visually striking achievements in synthetic biology, remains too dim to be practically useful. Current glowing plants work as proof of concept and novelty demonstrations, but their light output is far below what would be required for quantitative reporters, field deployable biosensors, or functional living light sources, and nearly every published study in the field, from Mitiouchkina et al. (2020) to Shakhova et al. (2024), explicitly calls out further brightness improvement as a priority. Solving this problem matters because autonomous bioluminescence offers a fundamentally different model for biological imaging than the dominant tools currently in use. Conventional luciferases like firefly luciferase and NanoLuc require continuous addition of expensive luciferin substrates, while fluorescent proteins require excitation light that causes phototoxicity and autofluorescence background. A bright autonomous system would eliminate both problems at once, enabling truly noninvasive, long-term monitoring of biological processes in ways no existing technology supports.
The broader societal contributions span multiple domains. In agriculture, bright autonomous reporter plants could give a real time visual readout of stress, nutrient status, or pathogen exposure without specialized equipment. In environmental sensing, engineered plants could function as living biosensors for soil contaminants or pollutants in places where electronic sensors are impractical. In basic research, brighter autonomous reporters could allow long-term imaging of gene expression and development without repeated substrate addition. And in the longest term, sufficiently bright autonomous bioluminescence could begin to challenge the paradigm of energy intensive artificial illumination. Taken together, the immediate scientific contribution (a brighter enzyme pair and a tested chimera), the methodological contribution (a screening first workflow), and the conceptual contribution (treating brightness as a systems level rather than enzyme level problem) could collectively reshape how the synthetic biology community approaches engineering of autonomous bioluminescence.
Ethical Implications
This project raises ethical issues related to environmental release, responsible genetic engineering, and unintended ecological effects. Although the immediate work is being done in controlled systems such as E. coli and laboratory grown Nicotiana, the long-term goal of brighter autobioluminescent plants creates questions about what could happen if such organisms were released outside containment. The most relevant ethical principles are non-maleficence, because the work should avoid ecological harm; responsibility, because engineered organisms should be designed and communicated carefully; beneficence, because the technology could create useful tools for research and agriculture; and justice, because the benefits of the technology should not be limited only to wealthy institutions or private companies.
To keep the project ethical, the work should proceed in a staged and containment focused way. Early testing should remain in bacterial systems, transient plant assays, or controlled greenhouse settings rather than open environmental release. If autobioluminescent plants are to be used in environmental deployment, concrete biocontainment strategies would need to be built into the organisms themselves. These could include reproductive sterility to prevent uncontrolled spread through seed or pollen, synthetic auxotrophy so that the organism depends on a nutrient not available in the wild and cannot survive outside managed conditions, or genetic kill switches that cause the organism to self terminate if containment conditions are no longer met. Any environmental release would also require staged deployment beginning with small scale controlled field trials, regulatory review, and post release ecological monitoring to detect unintended effects on local ecosystems, pollinators, or nocturnal wildlife. Beyond containment, the project should evaluate possible unintended effects on growth, metabolism, or fitness, since increasing pathway flux could create burdens on the host organism. It is also important to communicate the project honestly and not overstate near-term applications such as biological lighting when the more realistic near-term value is in imaging and biosensing. If increased brightness causes unacceptable tradeoffs, the ethical response would be to redesign the system or restrict its use to contained research settings rather than consumer or environmental deployment.
Section 4. Experimental Design, Techniques, Tools, And Technology

Step 1: DNA Design in Benchling (Timeline: Day 1, manual)
Design four pET-28a(+) plasmids, one encoding nnLuz v4 ΔN20 (with four of the Shakhova v4 substitutions T99P, T192S, A199P, I63T and a 20 amino acid N-terminal truncation) and nnH3H v2 (with four Shakhova v2 substitutions D37E, V181I, S323M, M385K). The other three pET-28a(+) plasmids will be the controls, wild type (WT) nnLuz & nnH3H WT (baseline), nnLuz v4 (full length) & nnH3H v2, and nnLuz ΔN20 (truncated, no v4 mutations) & nnH3H v2. Use codon optimization for each of the plasmids for E. coli expression. Insert both genes in each of the plasmids under T7 promoter control with a ribosome binding site (RBS) between them. Include BamHI and HindIII restriction sites flanking the insert for future cloning flexibility. Export the plasmid maps and sequence annotations. Expected result: Complete plasmid design files ready for Twist Bioscience submission.
Step 2: Order Whole Plasmid from Twist Bioscience (Timeline: Day 1–14)
Submit all four complete pET-28a(+) plasmid designs to Twist Bioscience for synthesis. Request all four plasmids in standard cloning vectors format with kanamycin resistance. Twist will synthesize all four plasmids (including backbone). Machine/Platform: Twist Bioscience synthesis platform. Expected result: Lyophilized plasmid DNA arrives within 10–14 business days. Timeline: 10–14 days turnaround.
Step 3: Resuspend and Transform Plasmid into BL21(DE3) (Timeline: Day 15, manual)
Resuspend lyophilized Twist plasmids in sterile nuclease-free water to 50–100 ng/µL. For each of the plasmids, transform 1 µL into 50 µL chemically competent BL21(DE3) cells using standard heat-shock protocol (42°C for 45 seconds). Recover in 250 µL SOC medium at 37°C for 1 hour. Plate 100 µL onto LB-agar plates supplemented with 50 µg/mL kanamycin. Incubate overnight at 37°C. Expected result: 50–200 colonies per plate.
Step 4: Colony PCR and Sanger Sequencing Verification (Timeline: Day 16–18)
Pick 3–5 colonies and perform colony PCR using NEB OneTaq 2X Master Mix with primers flanking the insert regions. Run PCR products on a 1% agarose gel to confirm correct insert size. Submit PCR products for Sanger sequencing to verify all four plasmids inserts are correct. Machine: ATC Thermal Cycler for colony PCR. Expected result: Correct insert size on gel; sequencing confirms all inserts are correct and no frame shifts exist. Timeline: 2–3 days including sequencing turnaround.
Step 5: Miniprep (Timeline: Day 18, manual)
Inoculate 5 mL LB + kanamycin (50 µg/mL) in separate tubes with sequence verified colonies. Grow overnight at 37°C with shaking (250 rpm). Perform plasmid miniprep using a commercial kit (e.g., Qiagen QIAprep). Quantify plasmids DNA by NanoDrop. Expected result: 50–200 ng/µL plasmid DNA.
Step 6: Prepare 96-Well Starter Cultures (Timeline: Day 19, automated)
Dispense 150 µL LB + kanamycin (50 µg/mL) into each well of a 96-well deep-well plate (96-v-eppendorf-951033502-deep) using Multiflo automated dispenser. Inoculate wells with sequence verified BL21(DE3) transformants for all 7 experimental conditions (wild-type nnLuz + nnH3H WT, nnLuz v4 only, nnLuz ΔN20 only, nnLuz v4 ΔN20, uninduced controls, hispidin only blank, no hispidin control). Include 6–8 replicate wells per condition. Machine: Multiflo automated dispenser. Plate type: 96-well deep-well plate. Expected result: Uniform liquid dispensing across all wells. Timeline: 30 minutes.
Step 7: Overnight Growth of Starter Cultures (Timeline: Day 19–20, automated incubation)
Seal the 96-well plate using Plateloc with a breathable A4s seal. Incubate overnight at 37°C with shaking (900 rpm) in the Cytomat shaking incubator. Machine: Cytomat (30°C shaking incubator, set to 37°C mode if available; otherwise use Inheco Plate Incubator + BioshakeD3000). Expected result: Turbid cultures at OD600 ~2–4 after 16 hours.
Step 8: Subculture into Fresh Media and Measure OD600 (Timeline: Day 20, automated)
Dilute overnight cultures 1:100 into fresh LB + kanamycin in a new 96-well deep-well plate (200 µL final volume per well) using Bravo-96 plate stamp or manual multichannel pipette. Transfer 5 µL of each well to a 96-well flat-bottom clear plate (1-flat-thermo-264728-omni-96) and add 195 µL LB using Multiflo. Measure OD600 using Spark Plate Reader to confirm starting density. Machine: Bravo-96 plate stamp, Multiflo, Spark Plate Reader. Expected result: OD600 ~0.02–0.05 post-dilution.
Step 9: Growth to Mid-Log Phase (Timeline: Day 20, 2–3 hours)
Incubate the subculture plate at 37°C with shaking until OD600 reaches ~0.4–0.6 (mid-log phase). Monitor OD600 every 30–60 minutes using Spark Plate Reader. Machine: Cytomat or Inheco + BioshakeD3000, Spark Plate Reader. Expected result: OD600 ~0.5 after 2–3 hours.
Step 10: IPTG Induction (Timeline: Day 20, automated)
Add IPTG to a final concentration of 0.5 mM using Multiflo or Echo525 acoustic liquid handler. If using Multiflo, prepare a 10 mM IPTG stock and dispense 10 µL per well. If using Echo525, prepare a 100 mM IPTG stock and transfer 1 µL per well. Mix gently by pipetting or brief shaking. Machine: Multiflo or Echo525. Expected result: Uniform IPTG addition across all wells.
Step 11: Protein Expression (Timeline: Day 20, 4–6 hours)
Incubate the induced cultures at 30°C (or 25°C for improved soluble protein expression) for 4–6 hours with shaking (900 rpm). Measure OD600 at the end of expression to confirm continued growth. Machine: Inheco Plate Incubator + BioshakeD3000. Expected result: OD600 ~1.5–2.5 post-induction; visible cell pellet.
Step 12: Transfer to 96-Well Black Assay Plate (Timeline: Day 20, automated)
Transfer 180 µL of induced culture from the deep-well plate into a 96-well black microplate with clear bottom (96-well black plates - Greiner # 655076) using Bravo-96 plate stamp or multichannel pipette. Retain 20 µL in the original plate for final OD600 measurement. Machine: Bravo-96 plate stamp or manual multichannel. Plate type: 96-well black assay plate. Expected result: Uniform transfer; black plate ready for luminescence measurement.
Step 13: Hispidin Substrate Addition (Timeline: Day 20, automated)
Prepare a 10 mM hispidin stock solution in DMSO. Dilute to 2 mM in sterile water immediately before use. Add 10 µL of 2 mM hispidin solution to each well (final concentration ~100 µM hispidin) using Multiflo or manual multichannel pipette. For no-hispidin controls, add 10 µL sterile water + DMSO vehicle. Mix gently by pipetting. Machine: Multiflo or manual multichannel. Expected result: Final hispidin concentration 100 µM; no precipitation.
Step 14: Kinetic Luminescence Measurement (Timeline: Day 20, 30–60 minutes)
Immediately transfer the 96-well black plate to the Spark Plate Reader or PHERAstar FSX. Configure the instrument for luminescence detection (open filter, no wavelength selection, integration time 1 second per well). Program a kinetic read: measure luminescence every 3–5 minutes for 30–60 minutes to capture signal rise, plateau, and decay. Machine: Spark Plate Reader or PHERAstar FSX. Expected result: Time course luminescence data for all wells; expected peak signal at ~10–20 minutes post-hispidin addition. Timeline: 30–60 minutes.
Step 15: Final OD600 Normalization (Timeline: Day 20, 10 minutes)
After kinetic luminescence measurement is complete, measure final OD600 for all wells in the original deep-well plate using Spark Plate Reader. Normalize luminescence data (RLU) to OD600 to calculate brightness per cell. Calculate integrated luminescence (area under the curve) using trapezoidal integration. Machine: Spark Plate Reader. Expected result: Normalized luminescence/OD600 values for all conditions; integrated AUC values for statistical comparison.
Plate Layout Diagram

Technique Checklist
The following techniques from the HTGAA curriculum are directly used or referenced in this project:
- ☑ DNA design (Benchling)
- ☑ Codon optimization
- ☑ DNA synthesis (Twist Bioscience)
- ☑ Plasmid transformation (E. coli)
- ☑ Colony PCR
- ☑ Sanger sequencing
- ☑ Protein expression (IPTG-inducible T7 system)
- ☑ Microplate-based assays
- ☑ Automated liquid handling (Multiflo, Bravo-96, Echo525)
- ☑ Luminescence detection (Spark Plate Reader / PHERAstar FSX)
- ☑ Data normalization and statistical analysis
Technique Expansion 1: DNA Design and Codon Optimization in Benchling
DNA design in Benchling is a foundational technique for synthetic biology that allows researchers to computationally design, annotate, and optimize genetic constructs before committing to expensive DNA synthesis. For this project, Benchling was used to design all four of the pET-28a(+) plasmids encoding nnLuz v4 ΔN20 & nnH3H v2, wild type (WT) nnLuz & nnH3H WT (baseline), nnLuz v4 (full length) & nnH3H v2, and nnLuz ΔN20 (truncated, no v4 mutations) & nnH3H v2 with appropriate ribosome binding sites and codon optimization for E. coli expression. Codon optimization is particularly important because the fungal genes (from Neonothopanus nambi) have different codon usage than E. coli, and rare codons can lead to slow translation, ribosome stalling, and poor protein expression. Benchling’s codon optimization tool identifies rare codons, replaces them with synonymous high-frequency codons, avoids introduction of unintended restriction sites or secondary structures, and maximizes the Codon Adaptation Index (CAI) for the target organism. This computational step improves protein expression yield and reduces experimental variability, making it a critical design-phase intervention for heterologous expression projects.
Technique Expansion 2: Automated Luminescence Kinetics and Normalization
Luminescence kinetics is a powerful technique for measuring time-resolved light emission from bioluminescent enzymes, providing far more information than single-endpoint measurements. In this project, the Spark Plate Reader is programmed to measure luminescence from all 96 wells every 3–5 minutes for 30–60 minutes after hispidin substrate addition, capturing the rise, peak, and decay of the signal. This kinetic approach has several advantages: it is less sensitive to timing artifacts (since peak brightness varies slightly between wells), it allows calculation of integrated luminescence (area under the curve) as a robust metric of total photon output, and it reveals enzyme kinetics such as substrate turnover rate and signal stability. Raw luminescence data (RLU) must be normalized to cell density (OD600) because brightness depends both on enzyme activity and the number of cells present. Normalization to OD600 isolates the “brightness per cell” metric, making comparisons between conditions valid even if growth rates differ slightly. This combination of kinetic measurement and normalization is standard practice in bioluminescence research and was the method used by Shakhova et al. 2024 to quantify improvements in the v4 enzyme set.
Section 5. Results & Quantitative Expectations
Validation Choice
The validation experiment for this project is Sanger sequencing of the Twist delivered plasmid to confirm that all designed mutations and the N-terminal truncation are present and correct. This validation step is the most critical quality control checkpoint in the entire project because every downstream experiment depends on the construct being exactly as designed. If even one of the v4 mutations is missing or incorrect, or if the ΔN20 truncation boundary introduces a frameshift, the luminescence data become uninterpretable and the hypothesis cannot be tested.
Validation Protocol
Step 1: Pick 3–5 colonies from the transformation plate and perform colony PCR with primers flanking the nnLuz_v4_ΔN20 and nnH3H_v2 insert. Run PCR with the following cycling conditions: 94°C initial denaturation (3 min), then 30 cycles of [94°C denaturation (30 sec), 58°C annealing (30 sec), 68°C extension (2 min)], followed by 68°C final extension (5 min).
Step 2: Run 5 µL of each PCR product on a 1% agarose gel. Confirm that the insert size matches the expected length (nnLuz_v4_ΔN20 and nnH3H_v2 insert is ~2250 bp).
Step 3: Purify the PCR product using a commercial PCR cleanup kit. Elute in 30 µL nuclease-free water.
Step 4: Submit purified PCR product for Sanger sequencing using primers that cover the full insert region.
Step 5: Align sequencing chromatograms to the reference design file in Benchling. Verify that all mutations are present, no unexpected substitutions exist, and the reading frame is intact across the ΔN20 junction.
Step 6: If sequencing confirms the construct is correct, proceed with miniprep and expression experiments. If sequencing reveals errors, re-pick colonies or re-order the plasmid from Twist.
Techniques Used
Sanger sequencing is the gold standard for sequence verification of plasmid constructs. It uses chain terminating dideoxynucleotides (ddNTPs) to generate fluorescently labeled DNA fragments of different lengths, which are separated by capillary electrophoresis and detected as a chromatogram. This technique provides single nucleotide resolution across 600–1000 bp per read, making it ideal for confirming point mutations and deletion boundaries. For this project, Sanger sequencing is essential because the nnLuz_v4_ΔN20 chimera contains single nucleotide substitutions plus a 60-nucleotide deletion, and all of these must be verified before interpreting luminescence data. Unlike next-generation sequencing (which is better for whole-genome analysis), Sanger sequencing is fast (2–3 day turnaround), affordable ($5–15 per read), and provides unambiguous base calls at every position, making it the standard validation method for synthetic biology constructs ordered from commercial vendors like Twist Bioscience.
Hypothetical Data
The following table shows hypothetical luminescence data for the key experimental conditions after normalizing to OD600 and calculating integrated luminescence (area under the curve, AUC) over a 60-minute kinetic read. Values represent mean ± standard deviation.
| Condition | Peak RLU/OD600 | Integrated AUC (RLU·min/OD600) |
|---|---|---|
| Wild-type (nnLuz + nnH3H_WT) | 1200 ± 150 | 52,000 ± 6,800 |
| nnLuz_v4 + nnH3H_v2 | 4300 ± 420 | 188,000 ± 19,000 |
| nnLuz_ΔN20 + nnH3H_v2 | 2800 ± 310 | 128,000 ± 14,500 |
| nnLuz_v4_ΔN20 + nnH3H_v2 | 7100 ± 690 | 315,000 ± 28,000 |
| Uninduced (no IPTG) | 22 ± 8 | 980 ± 350 |
| Hispidin alone (no cells) | 3 ± 1 | 140 ± 50 |
| Induced, no hispidin | 6 ± 2 | 270 ± 90 |
Interpretation: The chimeric nnLuz_v4_ΔN20 enzyme produces ~7100 RLU/OD600 at peak brightness, which is 5.9-fold brighter than wild-type and 1.65-fold brighter than nnLuz_v4 alone. The integrated AUC shows a similar trend: the chimera is 6.1-fold brighter than wild-type and 1.68-fold brighter than nnLuz_v4 alone. This suggests that the v4 mutations and ΔN20 truncation stack with modest synergy (greater than additive). All controls show minimal background signal, confirming that luminescence depends on both luciferase expression and exogenous hispidin substrate.
Hypothetical Kinetic Luminescence Graph (ASCII):
Conclusion: The chimeric enzyme reaches peak brightness at ~15 minutes post-hispidin addition and maintains elevated signal for ~40 minutes, consistent with the kinetics reported by Shakhova et al. 2024. The data support the hypothesis that the v4 mutations and ΔN20 truncation combine to produce a brighter luciferase than either modification alone.
Troubleshooting
Several experimental challenges may arise during this project. If luminescence signal is absent or far lower than expected, the most likely causes are (1) incorrect plasmid sequence (resolved by Sanger sequencing validation), (2) poor protein expression due to insoluble aggregation (testable by SDS-PAGE or Western blot), or (3) degraded or impure hispidin substrate (testable by LC-MS or purchasing fresh hispidin from a different supplier). If background signal is high in negative controls, this may indicate autofluorescence from the microplate, media components, or hispidin oxidation; switching to a different plate type or preparing fresh substrate immediately before use can resolve this. If luminescence kinetics are inconsistent between replicates, this may reflect pipetting errors during hispidin addition; using automated liquid handling (Multiflo or Echo525) instead of manual multichannel pipettes improves reproducibility. If OD600 normalization reveals high variability, this suggests uneven cell growth; pre-warming media and ensuring uniform shaking during growth can reduce this. Finally, if the nnLuz_v4_ΔN20 chimera is not brighter than nnLuz_v4 alone, this would indicate that the two improvements do not stack, which is itself a publishable negative result that informs future enzyme engineering strategies.
Section 6. Additional Information
References
Ge, J., Lang, X., Ji, J., Qu, C., Qiao, H., Zhong, J., Luo, D., Hu, J., Chen, H., Wang, S., Li, S., Li, W., Zheng, P., Xu, J., & Du, H. (2024). Integration of biological and information technologies to enhance plant autoluminescence. The Plant Cell, 36(11), 4703–4715. https://doi.org/10.1093/plcell/koae236
Kotlobay, A. A., Sarkisyan, K. S., Mokrushina, Y. A., Marcet-Houben, M., Serebrovskaya, E. O., Markina, N. M., … & Yampolsky, I. V. (2018). Genetically encodable bioluminescent system from fungi. Proceedings of the National Academy of Sciences, 115(50), 12728-12732. https://doi.org/10.1073/pnas.1803615115
Mitiouchkina, T., Mishin, A. S., Somermeyer, L. G., Markina, N. M., Chepurnyh, T. V., Guglya, E. B., … & Sarkisyan, K. S. (2020). Plants with self-sustained luminescence. Nature Biotechnology, 38(8), 944-946. https://doi.org/10.1038/s41587-020-0500-9
Shakhova, E. S., Karataeva, T. A., Markina, N. M., Mitiouchkina, T., Palkina, K. A., Perfilov, M. M., Wood, M. G., Hoang, T. T., Hall, M. P., Fakhranurova, L. I., Alekberova, A. E., Malyshevskaia, A. K., Gorbachev, D. A., Bugaeva, E. N., Pletneva, L. K., Babenko, V. V., Boldyreva, D. I., Gorokhovatsky, A. Y., Balakireva, A. V., … Mishin, A. S. (2024). An improved pathway for autonomous bioluminescence imaging in eukaryotes. Nature Methods, 21, 406–410. https://doi.org/10.1038/s41592-023-02152-y
Zheng, P., Ge, J., Ji, J., Zhong, J., Chen, H., Luo, D., Li, W., Bi, B., Ma, Y., Tong, W., Han, L., Ma, S., Zhang, Y., Wu, J., Zhao, Y., Pan, R., Fan, P., Lu, M., & Du, H. (2023). Metabolic engineering and mechanical investigation of enhanced plant autoluminescence. Plant Biotechnology Journal, 21(8), 1671–1681. https://doi.org/10.1111/pbi.14068
Patent CN116732084A. (2023). Application of fungal luciferase truncations in improving fungal or plant bioluminescence intensity. China National Intellectual Property Administration.