Subsections of WEEK 02
Gel Electrophoresis
1. Principle
Gel electrophoresis is a laboratory technique used to separate molecules such as DNA, RNA, or proteins. Samples are loaded into wells and begin migrating through the gel when an electric current is applied. The speed of migration varies depending on the charge, mass, and length of the molecule. The smaller and more highly charged the molecule, the faster it moves.
Restriction enzymes (REs) are “molecular scissors” that cut DNA at specific recognition sequences. When a DNA sequence is digested by REs, it is cut into fragments of different lengths depending on the enzyme used, leading to a different “ladder pattern.” This concept has been used by Paul Vanouse to create artwork.
Reference Recitation Week 02

Image source: Gel Electrophoresis
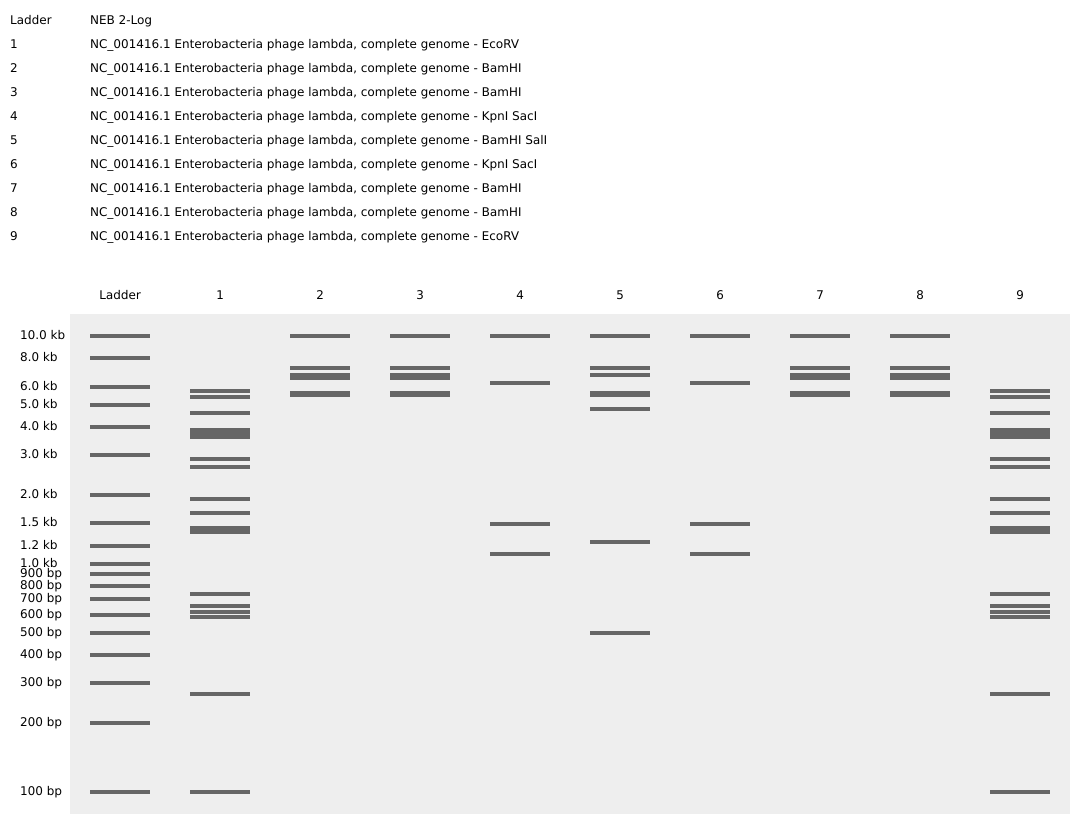
2. Benchling
Step 01: Retrieving DNA of Escherichia phage Lambda (complete genome) from Database

Step 02: Import Lambda fasta file in Benchling

Step 03: Create a visual design using the “ladder patterns” from Ronan’s DNA Gel Art Interface.
Process: I first tried to create the shape of a uterus but the pattern options available were limited so I switched to a Kawaii animal face. When running the digests, I accidentally switched the ERs for wells 3-4 and 7-8, which turned the visual into a M shape instead.

Step 04: Running the digests for each well to create the visual pattern in Benchling

3. In-silico Gel Art
DNA Design Luciferase
FIREFLY LUCIFERASE
Bioluminescent art is based on the organic production of visible light by living organisms. This light is produced through the oxidation of luciferin, which is catalyzed by an enzyme called luciferase. For this week’s assignment, we will focus on the protein coding for this enzyme, which was first identified in the firefly species (Photinus Pyrlis) [1].
[1] De Wet J.R. et al. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol (1987). https://pmc.ncbi.nlm.nih.gov/articles/PMC365129/
1. Protein Sequence
Sources
NCBI database search: https://www.ncbi.nlm.nih.gov/protein/BAF48396.1
Uniprot database search: https://www.uniprot.org/uniprotkb/P08659/entry#sequences
550 amino acids
1 medaknikkg papfypledg tageqlhkam kryalvpgti aftdahievn ityaeyfems 61 vrlaeamkry glntnhrivv csenslqffm pvlgalfigv avapandiyn erellnsmni 121 sqptvvfvsk kglqkilnvq kklpiiqkii imdsktdyqg fqsmytfvts hlppgfneyd 181 fvpesfdrdk tialimnssg stglpkgval phrtacvrfs hardpifgnq iipdtailsv 241 vpfhhgfgmf ttlgylicgf rvvlmyrfee elflrslqdy kiqsallvpt lfsffakstl 301 idkydlsnlh eiasggapls kevgeavakr fhlpgirqgy gltettsail itpegddkpg 361 avgkvvpffe akvvdldtgk tlgvnqrgel cvrgpmimsg yvnnpeatna lidkdgwlhs 421 gdlaywdede hffivgrlks likykgyqva paelesillq hpnifdagva glpdddagel 481 paavvvlehg ktmtekeivd yvasqvttak klrggvvfvd evpkgltgkr darkireili 541 kakkggkskl
2. Reverse Translate
Source
NCBI database search for P.pyralis (firefly) luciferase gene: https://www.ncbi.nlm.nih.gov/nuccore/M15077
Firefly Luciferase DNA sequence
1 ctgcagaaat aactaggtac taagcccgtt tgtgaaaagt ggccaaaccc ataaatttgg 61 caattacaat aaagaagcta aaattgtggt caaactcaca aacattttta ttatatacat 121 tttagtagct gatgcttata aaagcaatat ttaaatcgta aacaacaaat aaaataaaat 181 ttaaacgatg tgattaagag ccaaaggtcc tctagaaaaa ggtatttaag caacggaatt 241 cctttgtgtt acattcttga atgtcgctcg cagtgacatt agcattccgg tactgttggt 301 aaaatggaag acgccaaaaa cataaagaaa ggcccggcgc cattctatcc tctagaggat 361 ggaaccgctg gagagcaact gcataaggct atgaagagat acgccctggt tcctggaaca 421 attgcttttg tgagtatttc tgtctgattt ctttcgagtt aacgaaatgt tcttatgttt 481 ctttagacag atgcacatat cgaggtgaac atcacgtacg cggaatactt cgaaatgtcc 541 gttcggttgg cagaagctat gaaacgatat gggctgaata caaatcacag aatcgtcgta 601 tgcagtgaaa actctcttca attctttatg ccggtgttgg gcgcgttatt tatcggagtt 661 gcagttgcgc ccgcgaacga catttataat gaacgtaagc accctcgcca tcagaccaaa 721 gggaatgacg tatttaattt ttaaggtgaa ttgctcaaca gtatgaacat ttcgcagcct 781 accgtagtgt ttgtttccaa aaaggggttg caaaaaattt tgaacgtgca aaaaaaatta 841 ccaataatcc agaaaattat tatcatggat tctaaaacgg attaccaggg atttcagtcg 901 atgtacacgt tcgtcacatc tcatctacct cccggtttta atgaatacga ttttgtacca 961 gagtcctttg atcgtgacaa aacaattgca ctgataatga attcctctgg atctactggg 1021 ttacctaagg gtgtggccct tccgcataga actgcctgcg tcagattctc gcatgccagg 1081 tatgtcgtat aacaagagat taagtaatgt tgctacacac attgtagaga tcctattttt 1141 ggcaatcaaa tcattccgga tactgcgatt ttaagtgttg ttccattcca tcacggtttt 1201 ggaatgttta ctacactcgg atatttgata tgtggatttc gagtcgtctt aatgtataga 1261 tttgaagaag agctgttttt acgatccctt caggattaca aaattcaaag tgcgttgcta 1321 gtaccaaccc tattttcatt cttcgccaaa agcactctga ttgacaaata cgatttatct 1381 aatttacacg aaattgcttc tgggggcgca cctctttcga aagaagtcgg ggaagcggtt 1441 gcaaaacggt gagttaagcg cattgctagt atttcaaggc tctaaaacgg cgcgtagctt 1501 ccatcttcca gggatacgac aaggatatgg gctcactgag actacatcag ctattctgat 1561 tacacccgag ggggatgata aaccgggcgc ggtcggtaaa gttgttccat tttttgaagc 1621 gaaggttgtg gatctggata ccgggaaaac gctgggcgtt aatcagagag gcgaattatg 1681 tgtcagagga cctatgatta tgtccggtta tgtaaacaat ccggaagcga ccaacgcctt 1741 gattgacaag gatggatggc tacattctgg agacatagct tactgggacg aagacgaaca 1801 cttcttcata gttgaccgct tgaagtcttt aattaaatac aaaggatatc aggtaatgaa 1861 gatttttaca tgcacacacg ctacaatacc tgtaggtggc ccccgctgaa ttggaatcga 1921 tattgttaca acaccccaac atcttcgacg cgggcgtggc aggtcttccc gacgatgacg 1981 ccggtgaact tcccgccgcc gttgttgttt tggagcacgg aaagacgatg acggaaaaag 2041 agatcgtgga ttacgtcgcc agtaaatgaa ttcgttttac gttactcgta ctacaattct 2101 tttcataggt caagtaacaa ccgcgaaaaa gttgcgcgga ggagttgtgt ttgtggacga 2161 agtaccgaaa ggtcttaccg gaaaactcga cgcaagaaaa atcagagaga tcctcataaa 2221 ggccaagaag ggcggaaagt ccaaattgta aaatgtaact gtattcagcg atgacgaaat 2281 tcttagctat tgtaatatta tatgcaaatt gatgaatggt aattttgtaa ttgtgggtca 2341 ctgtactatt ttaacgaata ataaaatcag gtataggtaa ctaaaaa
3. Codon Optimization
According to the genetic code, there are fewer amino acids than codon possibilities (see chart below, image credit cdn.prod.website-files.com). Thus, in theory, multiple codons can encode for the same amino acid. But in practice, spatial configuration and kinetics factors affect the translation process. For instance, the use of some codons ressembling the STOP codons can interrupt prematurely the translation process. Thus, codon optimization is an important step when designing a nucleotide sequence.

Firefly Luciferase Optimized DNA sequence
Twist Bioscience add-ons:
Flank 5’: AGTACGCGTCTACGG
Flank 3’: TCCGATGACGTTAGC
ATGGAAGATGCAAAAAATATTAAAAAAGGCCCGGCGCCGTTTTATCCGCTGGAAGATGGCACAGCCGGTGAGCAGCTGCACAAAGCGATGAAGCGCTATGCGCTGGTTCCGGGCACCATTGCCTTCACCGATGCGCACATCGAAGTCAACATCACCTATGCTGAGTACTTTGAAATGTCTGTGCGTCTGGCGGAAGCGATGAAACGCTATGGTCTGAACACCAACCACCGTATTGTGGTCTGCTCTGAAAACAGCCTGCAGTTCTTCATGCCGGTACTGGGTGCGCTGTTTATCGGTGTTGCGGTAGCGCCGGCGAACGACATCTATAATGAGCGTGAACTGCTGAACTCCATGAACATCAGCCAGCCAACCGTTGTTTTTGTCAGCAAAAAAGGCCTGCAGAAAATCCTCAACGTTCAGAAAAAACTGCCGATCATTCAGAAAATCATCATCATGGACAGCAAAACCGATTATCAGGGTTTCCAGAGCATGTACACCTTTGTCACCAGCCACCTGCCGCCGGGTTTCAACGAATATGATTTTGTTCCGGAGAGCTTTGACCGTGATAAAACCATTGCGCTGATCATGAACAGCTCTGGCTCCACTGGTCTGCCGAAAGGTGTAGCGCTGCCGCACCGCACTGCCTGTGTGCGTTTCAGCCATGCGCGTGATCCGATTTTCGGTAACCAGATCATTCCGGACACCGCAATTCTGTCAGTGGTGCCGTTCCATCACGGTTTTGGTATGTTTACCACCCTGGGCTACCTGATCTGCGGTTTCCGCGTAGTGCTGATGTACCGCTTTGAAGAAGAGCTGTTCCTGCGCAGCCTGCAGGACTACAAAATCCAGTCTGCGCTGCTGGTACCGACCCTGTTCAGCTTCTTTGCCAAATCCACCCTGATCGATAAATATGACCTGAGTAACCTGCACGAGATTGCCTCTGGTGGTGCACCGCTGAGCAAAGAAGTTGGTGAAGCGGTGGCGAAACGTTTCCATCTGCCGGGTATCCGTCAGGGTTATGGTCTGACTGAAACCACCTCTGCGATTCTGATCACCCCGGAAGGTGATGACAAACCGGGTGCGGTGGGCAAAGTGGTACCGTTCTTCGAAGCGAAAGTGGTGGATCTCGACACCGGTAAAACGCTGGGTGTGAACCAGCGTGGTGAACTGTGTGTACGTGGCCCGATGATCATGTCTGGTTATGTCAACAACCCGGAAGCGACCAATGCGCTGATCGACAAAGATGGTTGGCTGCACAGCGGCGACATCGCCTATTGGGATGAAGATGAGCACTTCTTTATCGTTGACCGCCTGAAAAGCCTGATCAAATATAAAGGCTATCAGGTAGCACCGGCGGAACTGGAGTCGATCCTGCTGCAGCATCCGAACATCTTCGATGCCGGCGTGGCGGGTCTGCCGGATGATGATGCAGGTGAGCTGCCGGCAGCGGTGGTGGTGCTGGAGCACGGTAAAACCATGACCGAGAAAGAGATTGTTGATTATGTGGCCAGCCAGGTGACCACTGCGAAGAAACTGCGCGGTGGCGTGGTGTTTGTTGATGAAGTGCCGAAAGGTCTGACCGGTAAACTGGATGCGCGTAAAATCCGCGAGATTCTGATTAAAGCGAAAAAAGGCGGTAAAAGCAAACTG
Analysis of Twist’s Optimizations by Claude: Out of 550 codons, 526 were changed (95.6%). Removed restriction sites: EcoRV codon ~515, Xbal codon ~16 (not on the list). BsaI, MluI, AatII not present. GC Content was increased from 42.8% to 52.5%. Insect genomes tend to be AT-rich. Bacteria (E. coli) and mammalian cells prefer slightly higher GC content. Rare codons eliminated and the most frequent codons in E. coli used.
4. From DNA Sequence to Firefly Luciferase
Firefly luciferase can be produced either by using living organisms (cell-dependent systems) or in a test tube (cell-free systems). In both cases, the production follows the two steps of the central dogma:
(1) Transcription of DNA into mRNA. RNA polymerase binds to a promoter and reads and copies the DNA from start to stop codons into mRNA, in which the nucleotide thymine (T) is replaced by uracil (U).
(2) Translation of mRNA into protein. Ribosomes bind to and read the mRNA codon by codon and, for each codon, incorporate the matching amino acid via a transfer RNA (tRNA). This forms a chain of amino acids bonded together (a polypeptide), which starts folding as the chain grows and is released when the ribosome reaches the stop codon. Depending on the protein, further maturation processes and/or association into a larger complex may occur afterwards.
In cell-dependent systems, the gene of interest is first cloned into an expression vector, i.e. inserted into plasmids, which are then amplified before being transferred into host organisms (e.g. E. coli), which carry out protein synthesis. In cell-free systems, the protein is produced by adding the DNA (or mRNA) directly into a mixture containing the elements required for transcription and translation (ribosomes, enzymes, cofactors, etc.). Cell-free systems are usually used when a rapid check is needed (protein production within a few hours), whereas cell-dependent systems are preferred for higher-yield production (e.g. in the industrial sector).
References Recitation Week 02

Image source: What is the central dogma?

Image source: What is a plasmid?

Images source: Bacterial transformation & selection
5. From A Single DNA Sequence to Multiple Proteins
A. Alternative Splicing (in Eukaryotes)
A gene is made of exons (actively coding parts) and introns (silent parts). After the transcription, there are many other steps before the translation, including the processing of the pre-mRNA. The cell’s machinery cuts out the introns and can rearrange the exons in different combinations. This process called alternative splicing explains why a single gene can code for multiple proteins with have different shapes and functions (isoforms). This evolutionary mechanism allows for instance the human body to create hundreds of thousands different proteins using only around 20,000 genes.

Image source and reference: All About Alternative Splicing
B. Polycistronic RNA
6. Documentation
Claude’s guidance for the optimization of the Firefly Luciferase gene (Photinus pyralis):
1. Understanding the Flank Sequences
Flanking sequences are short DNA segments added to the 5’ and 3’ ends of your optimized insert. They serve as:
- Cloning handles for restriction enzyme digestion or Gibson Assembly
- Reading frame aligners to ensure proper in-frame fusion with a vector
- Spacers to protect restriction sites from incomplete digestion
Flank 5’ template “AGTACGCGTCTACGG” decoded: Buffer bases AGT protect the restriction site from incomplete digestion, MluI restriction enzyme site (A | CGCGT cut) and the linker CTACGG serves as a spacer before the ATG start codon.
Flank 3’ template “TCCGATGACGTTAGC” decoded: The linker TCCG serves as a spacer after the stop codon, AatII restriction enzyme site (GACGT | C cut), and buffer bases TAGC for protection.
2. Restriction Sites to Remove During Optimization
Internal occurrences of EcoRV GATATC and BsaI GGTCTC(N)1 restriction sites must be removed from within the luciferase coding sequence without changing the aa sequence (synonymous codon substitutions).
(N)1 means the RE cuts outside the recognition sequence, in this case one random nucleotide downstream.
3. DNA Regions Excluded from Optimization
Some regions should not be codon-optimized:
- Known functional RNA elements (e.g., internal ribosome entry sites, regulatory motifs)
- Regions with validated mutagenesis you want to preserve exactly
- His-tags, linkers, or fusion sequences if already codon-optimized elsewhere
DNA Construct Benchling
Design 01 : Substitution within Template
Following the steps demonstrated during recitation Week 02, I cut the sfGFP insert from the plasmid template ColE1-AmpR-sfGFP and replaced it with the optimized firefly luciferase sequence (including flanks 5’ and 3’).
Step 01: Load plasmid template ColE1-AmpR-sfGFP in Benchling

Step 02: Copy plasmid template ColE1-AmpR-sfGFP into a new project page, cut sfGFP and paste the optimized luciferase sequence.

Step 03: Run a digest of the entire construct to verify that the MluI and AatII restrcition enzymes (REs) specifically cut it within the restriction sites domains. i.e. flanks 5’ (A | CGCGT cut) and 3’(GACGT | C cut).
RESULT: Fail. The digest looked promising (2 bands of different lengths), but when checking the bp values, the numbers (~4 kb and ~150 bp) did not match the construct. Zooming in on flanks 5’ and 3’, I realized that flank 3’ did not contain the AatII restriction site, as Claude had claimed.
Presence of MluI in Flank 5’ validated

Presence of AatII in Flank 3’ not confirmed

Lesson learned Double-check every single AI suggestion and strengthen the prompts.

Step 04: Fast troubleshooting. (1) Exchange one base (C instead of T in position 1'739) to create AatII restrcition site within flank 3’. (2) Exchange one base (C instead of T in position 4'286) to remove MluI restrcition site in another part of the plasmid.


RESULT: Success. One can see on the digest one band corresponding to the backbone vector (2683bp) and another one, lower, corresponding to the Firefly Luciferase insert (1672 pb).
Final Step: Experimental validation of the DNA construct to determine whether it leads to the production of luciferase or not.

DNA read/write/edit
1. DNA Read
I want to sequence the DNA of the firefly luciferase because I want to create a bioart installation that displays bioluminescent menstrual blood.
For sequencing a single known gene like firefly luciferase, Sanger sequencing is recommended. Sanger is first-generation, adapted for small, defined targets. It is fast and cost effective.
The input would be the firefly luciferase gene from a plasmid containing it or amplified directly from a DNA library.
Essential preparation steps: PCR amplification, purification, quantification, and primer selection.
Sanger sequencing steps: (1) Sequencing PCR reaction (2) Capillary electrophoresis (3) Base calling.
The output is a chromatogram file containing: the raw fluorescence trace (coloured peaks, one per base) a called nucleotide sequence (~600–900 usable bases per read) a quality score for each base position
2. DNA Write
I would like to synthesize a genetic construct based on the firefly luciferase gene from Photinus pyralis.
Rationale
Luciferase is one of the most widely used reporter genes in molecular biology because it catalyzes a light-producing reaction. I am interested in using this gene because bioluminescence creates a striking visual effect while also symbolically transforming menstrual blood, something culturally stigmatized, into a glowing, living artwork.
The project sits at the intersection of synthetic biology, feminist bioart, and molecular sensing. Rather than treating menstrual blood as medical waste, the installation would reframe it as biologically active and aesthetically meaningful material.
DNA Construct Design
I would synthesize a plasmid containing:
- Firefly luciferase gene (luc2)
- Promoter
- Biosensor regulatory elements responsive to hormones
Firefly luciferase sequence:
1 ctgcagaaat aactaggtac taagcccgtt tgtgaaaagt ggccaaaccc ataaatttgg 61 caattacaat aaagaagcta aaattgtggt caaactcaca aacattttta ttatatacat 121 tttagtagct gatgcttata aaagcaatat ttaaatcgta aacaacaaat aaaataaaat 181 ttaaacgatg tgattaagag ccaaaggtcc tctagaaaaa ggtatttaag caacggaatt 241 cctttgtgtt acattcttga atgtcgctcg cagtgacatt agcattccgg tactgttggt 301 aaaatggaag acgccaaaaa cataaagaaa ggcccggcgc cattctatcc tctagaggat 361 ggaaccgctg gagagcaact gcataaggct atgaagagat acgccctggt tcctggaaca 421 attgcttttg tgagtatttc tgtctgattt ctttcgagtt aacgaaatgt tcttatgttt 481 ctttagacag atgcacatat cgaggtgaac atcacgtacg cggaatactt cgaaatgtcc 541 gttcggttgg cagaagctat gaaacgatat gggctgaata caaatcacag aatcgtcgta 601 tgcagtgaaa actctcttca attctttatg ccggtgttgg gcgcgttatt tatcggagtt 661 gcagttgcgc ccgcgaacga catttataat gaacgtaagc accctcgcca tcagaccaaa 721 gggaatgacg tatttaattt ttaaggtgaa ttgctcaaca gtatgaacat ttcgcagcct 781 accgtagtgt ttgtttccaa aaaggggttg caaaaaattt tgaacgtgca aaaaaaatta 841 ccaataatcc agaaaattat tatcatggat tctaaaacgg attaccaggg atttcagtcg 901 atgtacacgt tcgtcacatc tcatctacct cccggtttta atgaatacga ttttgtacca 961 gagtcctttg atcgtgacaa aacaattgca ctgataatga attcctctgg atctactggg 1021 ttacctaagg gtgtggccct tccgcataga actgcctgcg tcagattctc gcatgccagg 1081 tatgtcgtat aacaagagat taagtaatgt tgctacacac attgtagaga tcctattttt 1141 ggcaatcaaa tcattccgga tactgcgatt ttaagtgttg ttccattcca tcacggtttt 1201 ggaatgttta ctacactcgg atatttgata tgtggatttc gagtcgtctt aatgtataga 1261 tttgaagaag agctgttttt acgatccctt caggattaca aaattcaaag tgcgttgcta 1321 gtaccaaccc tattttcatt cttcgccaaa agcactctga ttgacaaata cgatttatct 1381 aatttacacg aaattgcttc tgggggcgca cctctttcga aagaagtcgg ggaagcggtt 1441 gcaaaacggt gagttaagcg cattgctagt atttcaaggc tctaaaacgg cgcgtagctt 1501 ccatcttcca gggatacgac aaggatatgg gctcactgag actacatcag ctattctgat 1561 tacacccgag ggggatgata aaccgggcgc ggtcggtaaa gttgttccat tttttgaagc 1621 gaaggttgtg gatctggata ccgggaaaac gctgggcgtt aatcagagag gcgaattatg 1681 tgtcagagga cctatgatta tgtccggtta tgtaaacaat ccggaagcga ccaacgcctt 1741 gattgacaag gatggatggc tacattctgg agacatagct tactgggacg aagacgaaca 1801 cttcttcata gttgaccgct tgaagtcttt aattaaatac aaaggatatc aggtaatgaa 1861 gatttttaca tgcacacacg ctacaatacc tgtaggtggc ccccgctgaa ttggaatcga 1921 tattgttaca acaccccaac atcttcgacg cgggcgtggc aggtcttccc gacgatgacg 1981 ccggtgaact tcccgccgcc gttgttgttt tggagcacgg aaagacgatg acggaaaaag 2041 agatcgtgga ttacgtcgcc agtaaatgaa ttcgttttac gttactcgta ctacaattct 2101 tttcataggt caagtaacaa ccgcgaaaaa gttgcgcgga ggagttgtgt ttgtggacga 2161 agtaccgaaa ggtcttaccg gaaaactcga cgcaagaaaa atcagagaga tcctcataaa 2221 ggccaagaag ggcggaaagt ccaaattgta aaatgtaact gtattcagcg atgacgaaat 2281 tcttagctat tgtaatatta tatgcaaatt gatgaatggt aattttgtaa ttgtgggtca 2341 ctgtactatt ttaacgaata ataaaatcag gtataggtaa ctaaaaa
In practice, I would likely use a commercially optimized luciferase variant.
Potential Extensions
The project could evolve beyond a simple art piece into a collaborative science platform sensing perimenopause.
Future versions might include:
- Genetic circuits that modulate brightness based on hormonal markers -Cell-free expression systems
- Multi-color bioluminescent systems using luciferases from marine organisms
- CRISPR-regulated expression patterns synchronized with hormonal cycles
What technology or technologies would you use to perform this DNA synthesis and why?
The primary technology I would use is phosphoramidite DNA synthesis, which is currently the standard industrial method used by DNA synthesis companies such as Twist Bioscience. I would use this technology because: it is highly precise and commercially accessible, it allows custom-designed DNA sequences, it supports codon optimization and it is scalable for synthetic biology applications.
Essential Steps of the DNA Synthesis Process:
- Digital Sequence Design
- Oligonucleotide Synthesis : the DNA synthesis machine chemically adds nucleotides sequentially using phosphoramidite chemistry. Because this process is most reliable for short sequences, the construct is synthesized as many smaller oligonucleotides.
- DNA Assembly. the short DNA fragments are assembled into a larger construct using methods such as Gibson or Golden Gate Assembly
- Sequence Verification: after assembly, the construct would be verified using DNA sequencing to confirm that the synthesized sequence matches the intended design.
Although modern DNA synthesis is powerful, there are several limitations:
- Sequence Length : chemical synthesis becomes less accurate with longer DNA strands because each nucleotide addition introduces a small probability of error.
- Error Rates : errors such as deletions, insertions, or substitutions can occur during synthesis and assembly.
- Speed: although synthesis is much faster than older cloning methods, complex constructs can still require several days to weeks for commercial synthesis, as well as additional time for verification and troubleshooting.
- Cost: longer or more complex DNA constructs can become expensive.
3. DNA Edit
I want to edit the firefly luciferase gene from Photinus pyralis in order to alter the color of its bioluminescence from the natural yellow-green wavelength toward red emission. I am interested in editing this DNA because red bioluminescence would create a more visceral and blood-like visual effect for my bioart installation involving menstrual blood. Conceptually, the color shift would reinforce themes of embodiment, menstruation, and intimacy.
The exact color of the emitted light depends on the structure of the luciferase protein and the chemical environment of the reaction. I would engineer mutations associated with red-shifted luminescence. Previous studies have shown that certain amino acid substitutions in luciferase can shift emission wavelengths significantly toward orange and red light.
What Technology Would I Use to Perform These DNA Edits and Why?
To edit the luciferase gene, I would use CRISPR-Cas9 combined with site-directed mutagenesis and synthetic DNA assembly techniques.
These technologies are appropriate because they allow precise modification of specific nucleotides within a gene, making it possible to engineer targeted amino acid substitutions.
Essential steps:
- Guide RNA Design: a guide RNA is designed to target a specific region of the luciferase gene near the amino acid residues to be edited.
- Cas9 DNA Cleavage: the Cas9 protein binds the guide RNA and cuts the DNA at the target location.
- DNA Repair with Desired Mutation.
- Verification: the edited DNA is sequenced to confirm the mutations were introduced correctly.
Site-Directed Mutagenesis
Because luciferase is relatively small, another efficient strategy is PCR-based site-directed mutagenesis. This method uses specially designed primers containing the desired mutations, amplifies the plasmid DNA, and creates a new edited version of the gene. This approach is commonly used in protein engineering because it is fast, inexpensive, and highly precise for small edits.
Preparation
- Identify Target Mutations to shift luciferase emission toward red wavelengths.
- Computational Sequence Design: the edited DNA sequence need to be designed digitally to maintain protein stability, preserve enzymatic activity, and optimize mammalian codon usage.
- Guide RNA or Primer Design
Although CRISPR and mutagenesis are powerful technologies, they have several limitations:
- Off-Target Effects: CRISPR-Cas9 can sometimes cut unintended DNA sequences if the guide RNA partially matches other regions.
- Editing Efficiency: Precise edits using HDR are often less efficient than simple gene disruption.
- Protein Stability Tradeoffs: Mutations that shift light color can reduce enzyme brightness, thermal stability, or overall activity.
- Scalability: editing a few mutations is relatively straightforward, but engineering complex genetic circuits becomes increasingly difficult due to cloning complexity, regulatory interactions, and sequence optimization challenges.
- Biological Context Dependence: Luciferase color can also depend on pH, temperature, co-factors, and intracellular environment. This means the same edited luciferase may produce different colors in different systems.
Due to time limitation, this part of the weekly assignments has been generated by ChatGPT without further research work.