Subsections of WEEK 04
Conceptual Questions
All questions were answered using the feedback of Gemini - and ChatGPT for Question 10 - as a research starting point. Questions were used as prompts with an oriented approach (what would Shuguang Zhang answer, how would you explain a 10 year old etc.). Sources were checked and prompts refined from the larger picture into details of interest, as well as when needed if the content was unclear to me.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
According to Gemini, meat generally contains approx 25% (+/- 5%) of protein by weight when cooked. Thus, for this calculation, we assume that a 500g piece of meat contains 125g of protein (5x25g). By convention, 1g = 6.022 x 10 ^ 23 Da , so 125g = 7,528 x 10 ^ 25 Da.
Thus, our piece of meat contains 7,528 x 10^23 molecules of amino acids (7,528 x 10^25 /100) : a lot of work awaits pepsin, trypsin and peptidase at the burger shop 🍔
2. Why do humans eat beef/fish but do not become a cow/fish?
Although the idiom “You are what you eat” suggests food directly becomes part of us, proteins ingested during meals are not directly incorporated into the human body: rather, they are first broken down into amino acids through the digestive process and then serve as building blocks for human proteins according to the DNA code. Proteins are species-specific and their production depends on both the genetic code and environmental factors.
3. Why are there only 20 natural amino acids?
The existence of only 20 primary natural amino acids is generally explained by an evolutionary optimization. Organisms evolve to optimize the balance between energy/resource consumption and the benefits derived from biological functions. These specific building blocks were selected over 4 billion years ago for their ability to form stable, functional, and soluble proteins. They provide enough diversity to support all necessary biological functions while remaining cost-efficient and easier to handle by the cell machinery than a larger set of building blocks.
Why twenty amino acid residue types suffice(d) to support all living systems
Teaching the principle of biological optimization
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, Shuguang Zhang and George Church have pushed the boundaries of molecular design by developing methods to produce non-natural amino acids. Amino acids are composed of an amino group (-NH2), a carboxyl group (-COOH) and a residue group (-R) that varies. To create new amino acids, one needs to design new residue groups. Example of new residues and how to synthesize them: SKIP for now ⏱
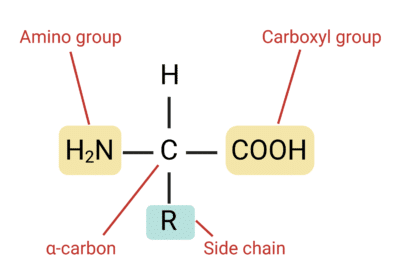
5. Where did amino acids come from before enzymes that make them, and before life started?
Scientists think that life originated from a “primordial soup” in deep-sea hydrothermal vents. Despite extreme heat and high pressure, these environments are teeming with life. These vents emit hot, mineral-rich fluids, creating environments where simple molecules could undergo chemical reactions to form more complex organic compounds, including amino acids. In 1952, Miller and Urey simulated early Earth conditions in an experiment and demonstrated that amino acids can form spontaneously through non-enzymatic pathways. It is also been proposed that amino acids may have an extraterrestrial origin. Amino acids have been found within meteorites that have crashed to Earth and in samples returned directly from asteroids, indicating that the chemical ingredients for life as we know it are widespread in the solar system 👽
A Short Tale of the Origin of Proteins and Ribosome Evolution
Insights into the formation and evolution of extraterrestrial amino acids from the asteroid Ryugu
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
In humans, the right-handed α-helix is the most common structural arrangement in the secondary structure of proteins. These helices are composed of L-amino acids. L-amino acids and D-amino acids are stereoisomer mirror images of each other, differing in the placement of the amino group (-NH2) on the alpha-carbon (see paper given in reference below). In a Fischer projection, L-amino acids have the amino group on the left (left handed), while D-amino acids have it on the right (right handed). Thus, if building an α-helix using D-amino acids, one would expect the helix to be oriented in the opposite direction and obtain a left-handed α-helix.
Structure and Function of Proteins
7. Can you discover additional helices in proteins?
After decades of intensive focus on the study of the α-helices, other helicoidal structures have been and keep being discovered, or rather reclassified after slowly regaining a certain relevance in protein science:
- 310 helices are typically observed as extensions of α-helices. They have been proposed to be intermediates in the folding/unfolding of the alpha helices and possibly involved in the initiation of the folding.
- π helices are evolutionarily derived by the insertion of a single residue into an α-helix and typically found near functional sites of proteins.
- Polyproline helices are mainly composed of proline polymers but can also form in sequences rich in other residues such as glycine, lysine, glutamate, and aspartate. They exist in two forms, either left-handed (PPII) or right-handed (PPI, rarer). Because of the rigidity and the restricted conformational space of the proline, the PPII has no internal hydrogen bonding and is relatively open as compared with the other helices. The PPII structure has been demonstrated to be essential to biological activities such as signal transduction, transcription, cell motility, and immune response.
- Collagen helices are formed by 3 PPII helices that twist together into a right-handed supercoil, also known as tropocollagen. They are the primary structural component of connective tissues (skin, bone, tendons, cartilage) providing high tensile strength.
- β-helices are formed by the supramolecular association of β-sheets in a helical pattern.
- γ-helices have been predicted but not observed yet in natural proteins.
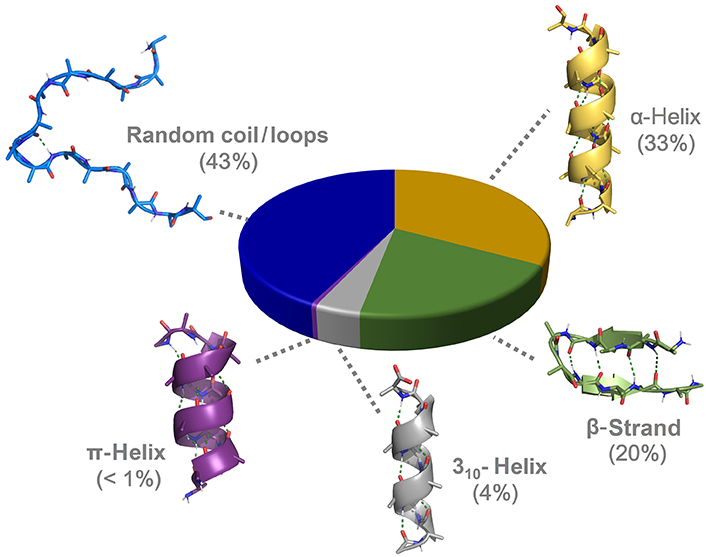
8. Why are most molecular helices right-handed?
During evolution, L-amino acids were preferred for protein synthesis and main metabolism: ribosomes possess a remarkable ability for chiral selectivity and are designed to use L-amino acids for protein synthesis. This explains why most molecular helices found in nature are right-handed (see Question 6 for reference).
To go a bit further: In the early stage of amino acids discovery, scientists actually believed that L-amino acids were solely found in nature and D-amino acids are artificial products. However, with the development of analytical methods in the past decades, D-amino acids have been found in a wide variety of living organisms both in their free form and as isomeric residues in many proteins. Their various biological functions are closely relevant to human physiology and diseases, including cancer. Although not typically formed by the translation machinery, left-handed helices can still be found in nature and are typically built through post-translational modification, non-ribosomal peptide synthetases (NRPS), and the incorporation of glycine and achiral residues.
Natural Occurrence, Biological Functions, and Analysis of D-Amino Acids
d‐amino acids: new functional insights
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The aggregation of β-sheets is a phenomenon of molecular self-assembly that is due to hydrophobicity and structural complementation. A β-sheet consists of β-strands that are connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. Because the structure of their edges is complementary (unpaired amino and carboxyl groups just waiting to find their match 💘), adjacent β-sheets naturally bond via their “sticky edges”. But what drives adjacent β-sheets to pile up like pancakes is linked to the amphiphilic nature of the β-sheets. Indeed, one of their sides is hydrophilic and the other is hydrophobic. In the same way that oil droplets gather together when added to water, β-sheets tend to aggregate in order to “hide” their hydrophobic sides from hydrophilic groups or aqueous environment (nucleoplasm, cytoplasm and ground substance).
The Supramolecular Chemistry of β-Sheets
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloids have been linked to the development of various neurodegenerative diseases such as Alzheimer’s and Parkinson’s. Pathogenic amyloids form when previously healthy proteins misfold: these proteins lose their normal structure and physiological functions, and start forming fibrous deposits within and around cells that cause the progressive disruption of brain functions. Amyloid fibrils form from different proteins, each associated with a particular disease, but they all contain a distinctive dysfunctional β-sheet pattern known as cross-β spine. When the peptides misfold, they align next to each other forming extended β-sheets that present highly stable and ordered structures. Once a small cluster of misfolded peptides is formed, a nucleation effect causes the misfolding and aggregation of other adjacent peptides, leading to an accelerated amplification of the self-propagating aggregation process. Furthermore, the aggregation of cross-β-sheets leads to the creation of steric zippers that makes the amyloids resistant to enzyme degradation (proteolysis) allowing them to accumulate in tissues and organs over time.
.
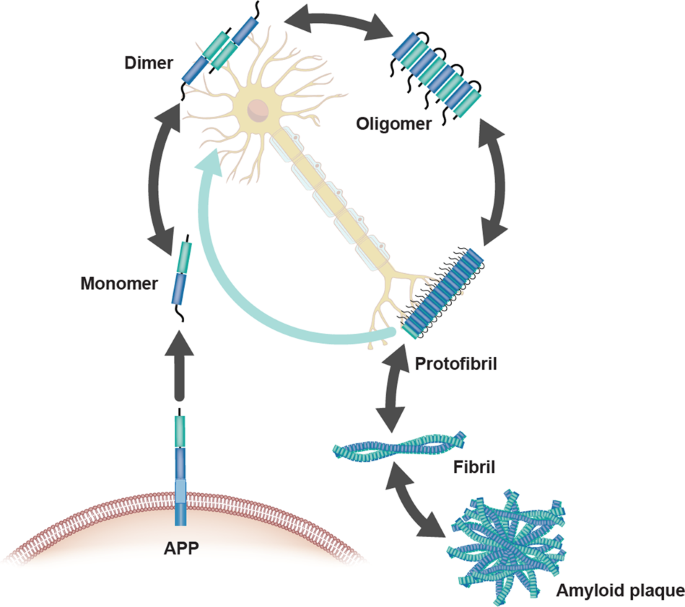
The Amyloid-β Pathway in Alzheimer’s Disease
.
One man’s loss is another man’s gain… The nanocrystal properties of cross-β sheet aggregates make them suitable for the engineering of biomaterials that can better withstand thermal stress and chemical denaturation such as:
- Hydrogels and Scaffold Engineering: Due to their ability to form nanofibrils, cross-beta peptides are used to create supramolecular hydrogels for tissue engineering and regenerative medicine.
- High-Strength Biomaterials: Synthetic polymeric amyloid fibers, containing cross-beta nanocrystals, can be engineered to exhibit exceptional mechanical strength, surpassing some natural spider silk fibers with high tensile strength and toughness.
- Nanowires and Conductive Materials: Fibrils can serve as templates for creating metallic nanowires for applications in nano-electronics, such as biosensors, actuators, and memory devices.
- Nanoporous Matrix Formation: Cross-beta aggregates are used to construct highly stable, porous, and rigid materials for specialized applications.
.

Amyloid-induced mineralization: From biological systems to biomimetic materials
.
11. Design a β-sheet motif that forms a well-ordered structure.
SKIP for now ⏱
Protein Visualisation
1. PROTEIN OF INTEREST: FIREFLY LUCIFERASE
For this week’s assignments, we will keep focusing on the first final project idea of creating bioluminescent menstrual blood. While it would be interesting to have a closer look at the structure of hemoglobin, the protein that facilitates the transportation of oxygen in erythrocytes (red blood cells) and gives its red color to human blood in visible light, we will keep studying the luciferin 4-monooxygenase. This enzyme, commonly known as firefly luciferase, catalyses the production of light through the oxidation of luciferin. The structure of this protein is simpler than hemoglobin, so this seems to be an ideal option to apply what has been covered in class this week. We might go back to hemoglobin, myoglobin and other proteins determining blood color later depending on how the project develops.
2. IDENTIFICATION OF AMINO ACIDS SEQUENCE
Length: 550 amino acids
Most frequent amino acid: Leucine
Homology: Luciferase has 250 homologs across insects (endopterygota, 95% ; polyneoptera 3%; paraneoptera %) and bacteria (allobacillus, 1%). VISUALISATION
Family: Protein families refers to groups of closely related proteins with high sequence/functional similarity and common ancestry. The firefly luciferase belongs to the acyl-adenylate/thioester-forming superfamily of enzymes, also known as the ANL superfamily or the ATP-dependent AMP-binding enzyme family.
Documentation Sequence Length and Amino Acids Frequency
AA sequence obtained from NCBI and Uniprot database (see WEEK 02 HW)
Colab code used: https://colab.research.google.com/drive/1vlAU_Y84lb04e4Nnaf1axU8nQA6_QBP1
Output: Sequence Length: 550 Amino Acid Frequencies: l: 52 g: 46 v: 44 a: 42 k: 40 i: 37 e: 33 d: 30 f: 30 p: 29 t: 29 s: 29 r: 21 n: 19 y: 19 q: 16 m: 14 h: 14 c: 4 w: 2
Documentation Homology Calculation runned with BLAST: FULL REPORT
3. FIREFLY LUCIFERASE STRUCTURE
RCSB page: https://www.rcsb.org/structure/1LCI
Structure Deposition Date: 1996-06-01
Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes
Resolution: 2.00 - 2.20 Å
The resolution is <2.70 Å and therefore considered as good.
Additional molecule(s) in the representation: none
Here is a page which presents the structure of the luciferase complexed with oxyluciferin and AMP (products of the reaction catalyzed by the luciferase): https://www.rcsb.org/structure/2D1R
Associated protein families:
- AMP-binding enzyme-like
- AMP-binding enzyme C-terminal domain-like They are both superfamilies too
Documentation Family Attribution
Calculation with SCOP
Output SCOP search for P08659 uniprot ID: 4 domains: 8025243 1BA3 A:3-434 8037622 1BA3 A:3-434 8055416 1BA3 A:435-520 8055417 1BA3 A:435-520
Output SCOP search for luciferas keyword: 4001964 Dinoflagellate luciferase repeat 4003312 Bacterial luciferase (alkanal monooxygenase)
AI GEMINI Feedback on protein family: AMP-dependent synthetase/ligase family :cross_mark:; Adenylate-forming enzyme superfamily :cross_mark:; AMP-binding enzyme C-terminal domain :check_mark_button:
4. LUCIFERASE VISUALIZATION IN PYMOL
All the visualizations and counts were carried out using the step-by-step guidance of ChatGPT (indeed, very useful). Questions were used as prompts. Once familiar with the software and tasks, further explorations were carried out on my own. Detailed documentation can be found at the end of this section.
DATASETS
PDB 1LCI: first luciferase crystal structure, open conformation
For luciferin ligand and exploration of other conformations: PDB 2D1S and PDB 4G36
3D STRUCTURE BASIC FORMATS

SECONDARY STRUCTURES

According to ChatGPT, the Firefly Luciferase is an α/β protein, but it is helix-dominated, which is typical for enzymes in the adenylate-forming enzyme family. This fact was surprising to me since I counted 17 helices and 21 sheets on the model when navigating it manually. However, the counts confirmed that the secondary structure of the Firefly Luciferase has more helix than sheets (count_atoms helices: 1367 atoms; count_atoms sheets: 817 atoms).
RESIDUES: HYDROPHATHY PROFILE
There are discrepancies in the classification of the amino acids depending on the sources that can be find online. Thus, I used the amino acids reference chart published by the pharma company Merck KGaA (Darmstadt, Germany) as reference. There are also many ways residues can be represented depending on their classification, so I opted for a binary hydrophobic/hydrophilic approach to better fit the direction of the question.
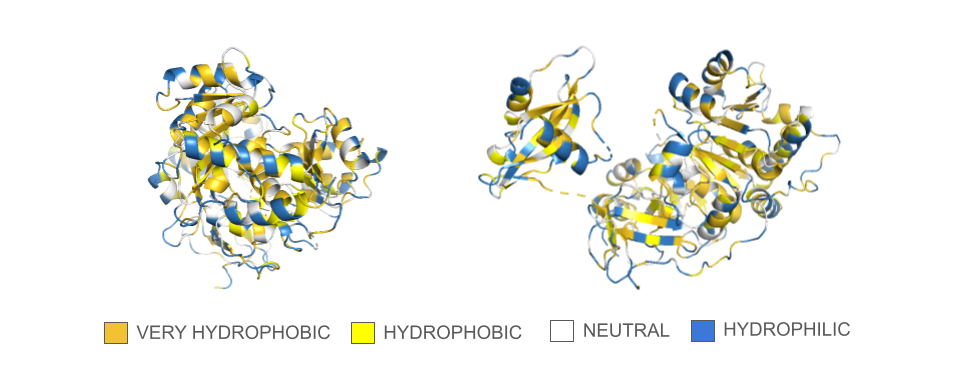
In the spin animation below, one can observe how the protein follows the hydrophobic effect, i.e. how the hydrophobic residues cluster inside the protein. The presence of hydrophilic residues on the surface of the luciferase is coherent with the fact that this enzyme is soluble.
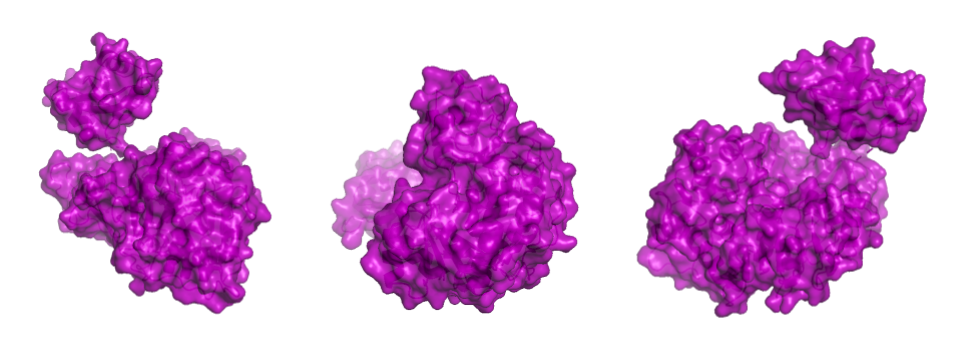
PROTEIN SURFACE
Showing the surface of the protein allows us to see more clearly that the protein is composed of two domains that have a globular shape and present small surface grooves. A large cleft between the N-terminal (residues 1-436, bottom part in the animation) and C-terminal (residues 440–550, on top) domains of the protein is clearly visible when rotating the structure: one can assume that it accommodates the luciferin and the ATP molecule (reference chemical reaction).
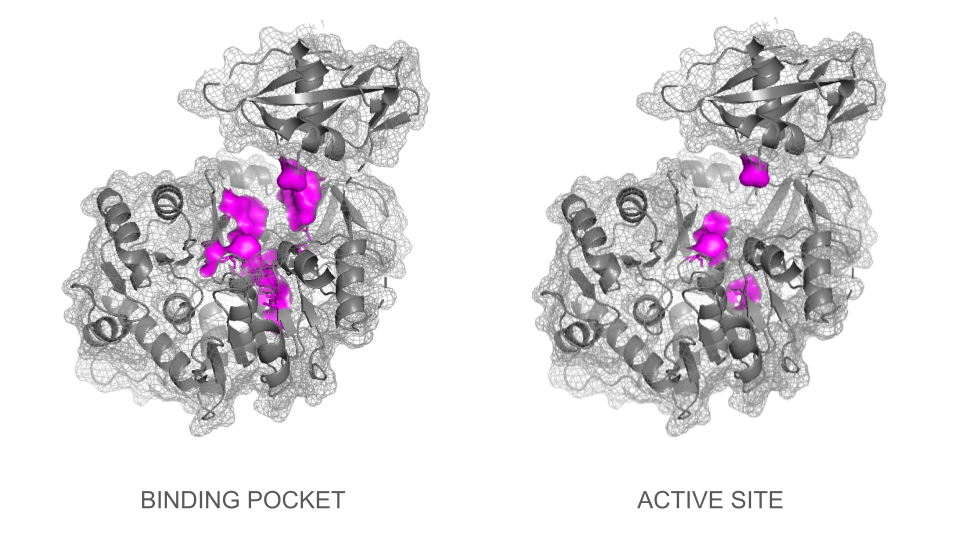
BINDING POCKET & ACTIVE SITE
In a second step, the ligand was added into the visualization. Residues reported to be present in the binding pocket and active site of the firefly luciferase were highlighted using the coloring function. One can observe how the ligand and the key residues match beautifully the binding pocket and how inside the cleft, the ligand enters a tunnel-like cavity forming inside the N-terminal domain.
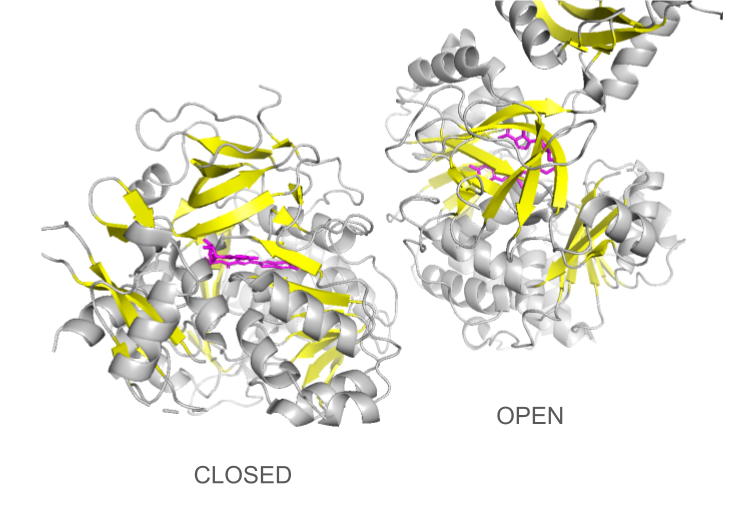
CONFORMATIONAL CHANGES
Loading the data set 2D1S containing both luciferase and ligand was confusing at first: the protein looked different than the 1LCI model. This can be explained by the fact that the enzyme undergoes large conformational changes during the different catalysis states: when the conformation is open, the enzyme allows the ligands to enter the cleft and when it is closed, the catalysis can occur. The C-terminal domain rotates perpendicularly when switching from one state to another, making the structures look quite different even though they are the same protein. Only the open conformation is presented here.
DOCUMENTATION
Documentation Visualization in PyMol: Basics
Import dataset: File > Get PDB > Enter 1LCI (PDB ID for Firefly Luciferase) or </> Python command
Useful commands recommended by ChatGPT:
- fetch 1lci (import dataset)
- hide everything
- show cartoon / show ribbon / show sticks / show spheres
- set sphere_scale, 0.25 / set stick_radius, 0.15 (adjust scale to improve 3D visualisation)
- util.cbag (coloring of the “stick and ball” according to PyMol default color coding: carbon, green; basic residues, blue; acidic residues, red; gray for others)
Documentation Visualization in PyMol: Secondary Structures
Useful commands recommended by ChatGPT:
- select helices, ss h / select sheets, ss s (select secondary structures)
- color color_01, ss h / color color_02, ss s (specific coloring of secondary structures)
- count_atoms helices / count_atoms sheets
Documentation Visualization in PyMol: Hydropathy
Useful (adapted) commands recommended by ChatGPT:
- select veryhydrophobic, resn phe+ile+trp+leu+val+met
- color gold, veryhydrophobic
- select hydrophobic, resn tyr+cys+ala
- color yellow, hydrophobic
- select neutral, resn thr+his+gly+ser+gln
- color white, neutral
- select hydrophilic, resn arg+lys+asn+glu+pro+asp
- color sky, hydrophilic
Documentation Visualization in PyMol: Surface
Useful commands recommended by ChatGPT:
- show surface, selection
- set transparency, 0.2
- clip slab, 20 (view of the surface + inside)
- set surface_cavity_mode, 1
- show mesh
Documentation Visualization in PyMol: Binding pocket and active site
Several strategies were tested to visualize the binding pocket and active site. First, visualization of the types of residues generally involved in binding pockets / active sites (charged, hydrophobe, polar, aromatic etc.) and then, types of residues involved specifically in the firefly luciferase. However, all the representations tested were not specific to the binding site.
Useful commands recommended by ChatGPT for part 01:
- select active, resn lys+arg+his+asp+glu
- color color_01, active
- select inactive, resn ala+ile+leu+met+val+gly+pro+cys+ser+phe+trp+tyr+thr+asn+gln
- color colo_02, inactive
In a second step, visualization of the individual residues involved in the active site of the firefly luciferase gave much better results although one needs to mention that the lists provided by Gemini and ChatGPT were different, and varied depending on iterations.
Commands recommended by ChatGPT to visualize binding pocket:
- select luciferin_binding_site, resi 198+214+218+222+244+245+247+286+340+343+344+347+420+421+422+529
- show sticks, luciferin_binding_site
- color color_01, luciferin_binding_site
- show surface, luciferin_binding_site
Commands recommended by ChatGPT to visualize active site:
- select luciferase_catalytic, resi 218+245+343+529
- show sticks, luciferase_catalytic
- color color_01, luciferase_catalytic
Documentation Visualization in PyMol: Adding ligand
Useful commands recommended by ChatGPT:
- fetch 1LCI
- fetch 4G36
- align 4G36, 1LCI
- select luciferin, 4G36 and organic (organic defines the ligand)
- create luciferin_copy, luciferin
- disable 4G36
- show sticks, luciferin_copy
- color color_01, luciferin_copy
Visual tools
Useful commands recommended by ChatGPT:
- mplay/mstop (play and stop movie)
- mclear (delete movie data to save memory when export is done)
- zoom object_of_interest, 8 (zoom power)
- bg_color white (change background color to white)
- set ray_trace_fog (better image quality)
- Color list
Spin animation
Useful (adapted) commands recommended by ChatGPT:
- mset 1 x360 (number of frames)
- util.mroll(1,360,1)
Zoom-through animation
Useful commands recommended by ChatGPT:
- mset 1 x360
- frame 1
- mview store
- frame 360
- move z, -360
- mview store
- mview interpolate
Video export Videos were exported in mp4 format using the screen recording function.
LEGAL ASPECTS: To use the official version of PyMol for free as a student, I had to sign the agreement copied below.
Addendum: 1- By declaring that I’m a ‘full time student’, I understand that being registered as a committed listener of the HTGAA course which is not an internship and involves 15 to 30 hours of weekly academic work fits the required criteria. 2- “Builds” should not be shared publicly. I understand that “builds” are a specific functionality offered by PyMol and that the visualizations that I have shared on this page are not “builds”.
PyMOL Educational Use Declaration for Flo Razoux
- I, Flo Razoux, am either a full-time student or am engaged in teaching full-time students. After being granted access, I will only apply Education-Use-Only PyMOL Builds (“Builds”) for education purposes and specifically including the following: COURSES or DEGREE: MIT Media Lab, Synthetic Biology, Other, 2026
- I will only share the Builds and their download access credentials with my fellow students and/or teachers, and only via private means.
- I will not post the Builds or their download access credentials in a publicly-accessible location, such as a web page, email list, or blog.
- If I apply PyMOL in any for-profit commercial activity or in any non-profit academic research, then I will compile my own builds from the open-source code or purchase an appropriate PyMOL Subscription in order to access the official PyMOL Builds not limited to educational use only.
- Except as otherwise set forth in Sections 1 through 4, I shall not: (i) modify, translate, adapt, create derivative works from or decompile the Builds, or any portion thereof, or create or attempt to create, by disassembling, reverse engineering or otherwise, the source code from the object code supplied hereunder, (ii) rent, lease, sell, transfer, publish, display, distribute, disclose or make the Builds available to third parties or use the builds, or any portion thereof, in a service bureau, time-sharing or outsourcing service or (iii) remove or alter any proprietary rights notices on the Builds. I acknowledge that the restrictions set forth in clauses (i) through (iii) of the immediately preceding sentence shall apply to distributions by Schrodinger, LLC of any third party software or other materials with the Builds.
ML-Based Protein Design
1. Protein Language Modeling
1a. Mutational Scans

The heat map was generated using the ESM-2 t6 8M UR50D model.
One can observe darker rows for tryptophan (W) and cysteine (C). These lower model scores (< -5) across all residues indicate that these two amino acids have a low probability of being used as substitutions in a mutational model without altering the spatial configuration of the protein. This may be due to the larger size of W and the unique chemical properties of C, which make them difficult to substitute into positions not specifically adapted for them.
Another observable pattern is the presence of darker columns around positions 197–208 and 338–344, indicating that these regions are highly conserved throughout evolution and that any mutations introduced there may lead to critical alterations in the structure and function of the luciferase. In fact, the positions 338–344 correspond to residues directly involved in the binding of the enzyme to the cofactor ATP and the substrate luciferin.
Reference: A View on the Active Site of Firefly Luciferase
1b. Latent Space Analysis

In this representation, each node corresponds to a protein, and the t-SNE axes represent a multidimensional matrix reduced to only three dimensions (t-SNE1, t-SNE2, and t-SNE3). Proteins positioned close together share similar sequence features and often exhibit related structural, functional, or evolutionary properties. They form clusters of proteins that belong to the same class or family, or that share similar structural folds.
Process: I first tried to identify a cluster of oxidoreductases (same class as luciferase) and the 4 luciferase proteins from the dataset by manually navigating the map using the t-SNE3 color coding, but this proved to be too time-consuming (see Documentation below).
Next step: Incorporate additional code in Colab to highlight the Firefly Luciferase and related proteins on the map.
2. Protein Folding
2a. Native protein
Luciferase structure determined experimentally

Image source: RCSB PDB 1LCI
Luciferase structure predicted ESMFold:

Result: The structure predicted by ESMFold looks similar to the one determined experimentally (RCSB PDB).
2b. Mutated proteins
Original Sequence
MEDAKNIKKGPAPFYPLEDGTAGEQLHKAMKRYALVPGTIAFTDGHIEVNITYAEYFEMSVRLAEAMKRYGLNTNHRIVVCSENSLQFFMPVLGALFIGVAVAPANDIYNERELLNSMNISQPTVVFVSKKGLQKILNVQKKLPIIQKIIIMDSKTDYQGFQSMYTFVTSHLPPGFNEYDFVPESFDRDKTIALIMNSSGSTGLPKGVALPHRTACVRFSHARDPIFGNQIIPDTAILSVVPFHHGFGMFTTLGYLICGFRVVLMYRFEEELFLRSLQDYKIQSALLVPTLFSFFAKSTLIDKYDLSNLHEIASGGAPLSKEVGEAVAKRFHLPGIRQGYGLTETTSAILITPEGDDKPGAVGKVVPFFEAKVVDLDTGKTLGVNQRGELCVRGPMIMSGYVNNPEATNALIDKDGWLHSGDLAYWDEDEHFFIVGRLKSLIKYKGYQVAPAELESILLQHPNIFDAGVAGLPDDDAGELPAAVVVLEHGKTMTEKEIVDYVASQVTTAKKLRGGVVFVDEVPKGLTGKRDARKIREILIKAKKGGKSKL
Total sequence length: 550 ptm: 0.910 plddt: 90.645
Confidence native Firefly Luciferase:

Mutation 01 : A45G
MEDAKNIKKGPAPFYPLEDGTAGEQLHKAMKRYALVPGTIAFTDGHIEVNITYAEYFEMSVRLAEAMKRYGLNTNHRIVVCSENSLQFFMPVLGALFIGVAVAPANDIYNERELLNSMNISQPTVVFVSKKGLQKILNVQKKLPIIQKIIIMDSKTDYQGFQSMYTFVTSHLPPGFNEYDFVPESFDRDKTIALIMNSSGSTGLPKGVALPHRTACVRFSHARDPIFGNQIIPDTAILSVVPFHHGFGMFTTLGYLICGFRVVLMYRFEEELFLRSLQDYKIQSALLVPTLFSFFAKSTLIDKYDLSNLHEIASGGAPLSKEVGEAVAKRFHLPGIRQGYGLTETTSAILITPEGDDKPGAVGKVVPFFEAKVVDLDTGKTLGVNQRGELCVRGPMIMSGYVNNPEATNALIDKDGWLHSGDLAYWDEDEHFFIVGRLKSLIKYKGYQVAPAELESILLQHPNIFDAGVAGLPDDDAGELPAAVVVLEHGKTMTEKEIVDYVASQVTTAKKLRGGVVFVDEVPKGLTGKRDARKIREILIKAKKGGKSKL
Total sequence length: 550 ptm: 0.910 plddt: 90.645

Confidence Mutation A45G:

Mutation 02 : H76D
MEDAKNIKKGPAPFYPLEDGTAGEQLHKAMKRYALVPGTIAFTDAHIEVNITYAEYFEMSVRLAEAMKRYGLNTNDRIVVCSENSLQFFMPVLGALFIGVAVAPANDIYNERELLNSMNISQPTVVFVSKKGLQKILNVQKKLPIIQKIIIMDSKTDYQGFQSMYTFVTSHLPPGFNEYDFVPESFDRDKTIALIMNSSGSTGLPKGVALPHRTACVRFSHARDPIFGNQIIPDTAILSVVPFHHGFGMFTTLGYLICGFRVVLMYRFEEELFLRSLQDYKIQSALLVPTLFSFFAKSTLIDKYDLSNLHEIASGGAPLSKEVGEAVAKRFHLPGIRQGYGLTETTSAILITPEGDDKPGAVGKVVPFFEAKVVDLDTGKTLGVNQRGELCVRGPMIMSGYVNNPEATNALIDKDGWLHSGDLAYWDEDEHFFIVGRLKSLIKYKGYQVAPAELESILLQHPNIFDAGVAGLPDDDAGELPAAVVVLEHGKTMTEKEIVDYVASQVTTAKKLRGGVVFVDEVPKGLTGKRDARKIREILIKAKKGGKSKL
Total sequence length: 550 ptm: 0.911 plddt: 90.796
Confidence Mutation H76D:

Mutation 03: Substitution 196-206 “MNSSGSTGLPK”>“WMHWPIGFCHK”
MEDAKNIKKGPAPFYPLEDGTAGEQLHKAMKRYALVPGTIAFTDGHIEVNITYAEYFEMSVRLAEAMKRYGLNTNHRIVVCSENSLQFFMPVLGALFIGVAVAPANDIYNERELLNSMNISQPTVVFVSKKGLQKILNVQKKLPIIQKIIIMDSKTDYQGFQSMYTFVTSHLPPGFNEYDFVPESFDRDKTIALIWMHWPIGFCHKGVALPHRTACVRFSHARDPIFGNQIIPDTAILSVVPFHHGFGMFTTLGYLICGFRVVLMYRFEEELFLRSLQDYKIQSALLVPTLFSFFAKSTLIDKYDLSNLHEIASGGAPLSKEVGEAVAKRFHLPGIRQGYGLTETTSAILITPEGDDKPGAVGKVVPFFEAKVVDLDTGKTLGVNQRGELCVRGPMIMSGYVNNPEATNALIDKDGWLHSGDLAYWDEDEHFFIVGRLKSLIKYKGYQVAPAELESILLQHPNIFDAGVAGLPDDDAGELPAAVVVLEHGKTMTEKEIVDYVASQVTTAKKLRGGVVFVDEVPKGLTGKRDARKIREILIKAKKGGKSKL
Total sequence length: 550 ptm: 0.945 plddt: 93.928

Confidence Mutation 03:

4. Documentation
4a. Reference Firefly Luciferase sequence

4b.Mutation Scans

4c. Latent Space Exploration



Bacteriophage Engineering
GROUP MEMBERS
Diogo Custodio https://pages.htgaa.org/2026a-diogo-custodio/
Flo Razoux https://pages.htgaa.org/2026a-flo-razoux/
Katharine Kolin https://pages.htgaa.org/2026a/katharine-kolin/
Marisa Satsia https://pages.htgaa.org/2026a-marisa-satsia/
Main goals (tbc)
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
Proposal (to be updated on March 8th)
- Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
- Why do you think those tools might help solve your chosen sub-problem?
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
- Include a schematic of your pipeline.
Individual and group plan for engineering a bacteriophage (to be updated on March 8th)