SOD1 Binder Peptide Design
Intro from Pranam Chatterjee
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
CHALLENGE
1. Design short peptides that bind mutant SOD1
2. Then decide which ones are worth advancing toward therapy
Part 1. Generate Binders with PepMLM
SOD1 Original sequence (150 aa):
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutated sequence:
Replace A by V on position 4 (note that the initial Methionine in position 1 is not counted)
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Approach 01: Generation of 4 peptides (length 12 aa, top K 3)
| Index | Peptides | Pseudo Perplexity |
|---|---|---|
| 0 | WLSPVVAAEHKE | 15.485618 |
| 1 | WRYGAAAAEHKE | 9.098425 |
| 2 | WRYYAAAVAHKX | 8.351979 |
| 3 | HHSYAAAVALKK | 12.665942 |
Approach 02: Selection of 4 peptides out of 200 generated (length 12 aa, top K 3)
| Index | Peptides | Pseudo Perplexity |
|---|---|---|
| 47 | WRYPAAAVALKX | 5.951070210421078 |
| 135 | WRSPAAVAAHKX | 4.819682049260351 |
| 147 | WHSGVVALAHKX | 6.103797038822761 |
| 199 | WRYGAVAARLKX | 5.926568160063284 |
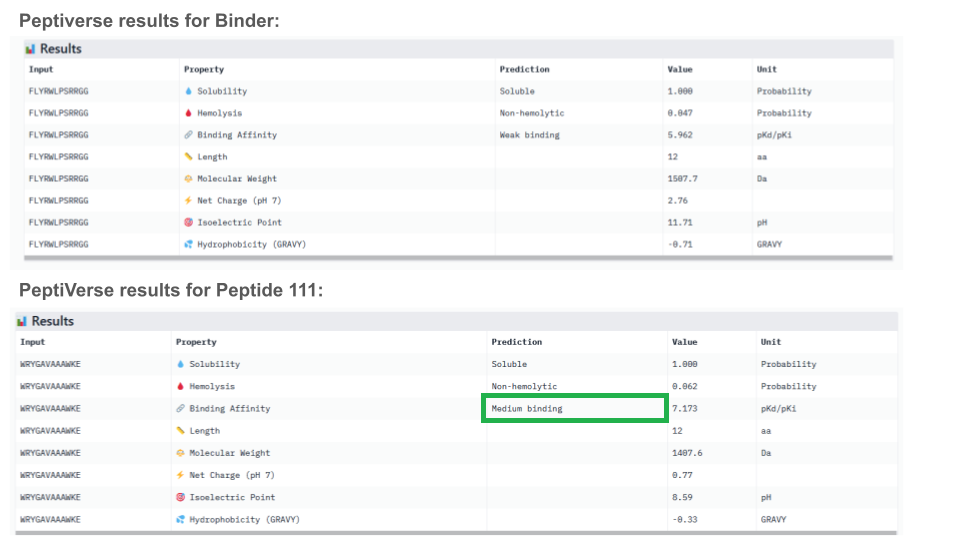
For comparison, known peptide binder FLYRWLPSRRGG:
| Index | Binder | Pseudo Perplexity |
|---|---|---|
| N/A | FLYRWLPSRRGG | 20.63523127283615 |
Pseudo Perplexity measures how confident the model is in the sequence. A lower PPL (≈ 3–6) means that the sequence fits welllearned binding patterns and thus the model is confident that the peptide will bind the target. A higher PPL (> 10–15) means that the sequence looks different than the learned patterns and thus, the model is less confident about the probability for the peptide to bind the target.
In theory, the high value of the pseudo perplexity score (>15) of the known binder FLYRWLPSRRGG can sound surprising. But in reality, the authors of the reference paper showed that known binders can have relatively high perplexity values. While the confidence scores can be useful for ranking candidates, they have their own limitations: the score evaluates familiarity, not whether the peptide truly binds the protein of interest. See documentation for more details on outliers.
UPDATE: AlphaFold basic functions don’t allow the integration of undefined amino acids (X) so a new series of peptides was generated with Colab.
| Reference | Peptides | Pseudo Perplexity |
|---|---|---|
| Binder | FLYRWLPSRRGG | 20.64 |
| 0 | WLSPVVAAEHKE | 15.49 |
| 1 | WRYGAAAAEHKE | 9.10 |
| 3 | HHSYAAAVALKK | 12.67 |
| 15 | WLSGAVGAAHKK | 8.10 |
| 43 | WRYGAAAAEHGK | 6.95 |
| 94 | WRYPAAAARLGK | 7.06 |
| 111 | WRYGAVAAAWKE | 7.94 |
2. Evaluate Binders with AlphaFold3
AlphaFold Visualization:
Except for peptides 3 and 43 which form an alpha helice secondary structure, all peptides present an unfolded structure that can appear more or less floppy or following the shape of the protein. None of the peptides are buried in the mutant SOD1: if binding to the protein, this appear to be a surface-bound for all of them. None of the peptide appear to interact with the N-terminal nor the dimer interface. All peptides seem to interact, at least partially with the beta-barrel. Beside the known binder, peptides 43, 94 and 111 appear to also interact with the electrostatic loop (loop containing a small portion of alpha helice).
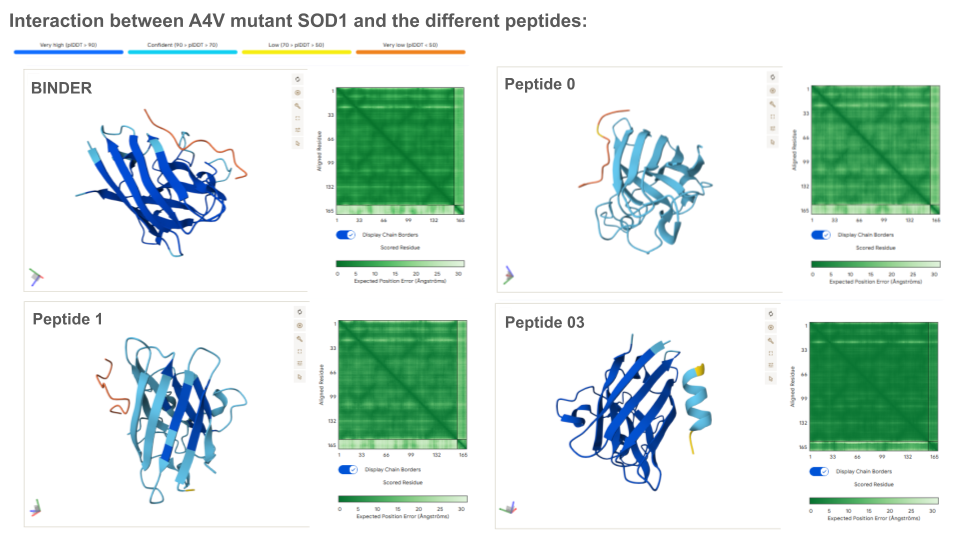

Prediction of the different A4V Mutant SOD1-peptide Bindings:
| Reference | ipTM |
|---|---|
| Binder | 0.3 |
| 0 | 0.27 |
| 1 | 0.28 |
| 3 | 0.73 |
| 15 | 0.46 |
| 43 | 0.35 |
| 94 | 0.42 |
| 111 | 0.31 |
ipTM indicates the probability of binding. Typically, if ipTM is >0.8, the probability of binding is high, and low if it’s <0.6. Thus, it is surprising that the binder only exhibits a ipTM of 0,3 with SOD1, which is either lower or similar to all generated peptides. This can maybe be explained by the fact that the peptide binds the surface vs being typically buried inside the protein, or/and because the peptide establish an usual bound with the electrostatic loop. With an ipTM = 0.73, peptide 3 exhibits the highest binding score: it might be interesting to predict its therapeutic property to evaluate if it is worth advancing toward therapy.
3. Predicting Therapeutic Properties with PeptiVerse
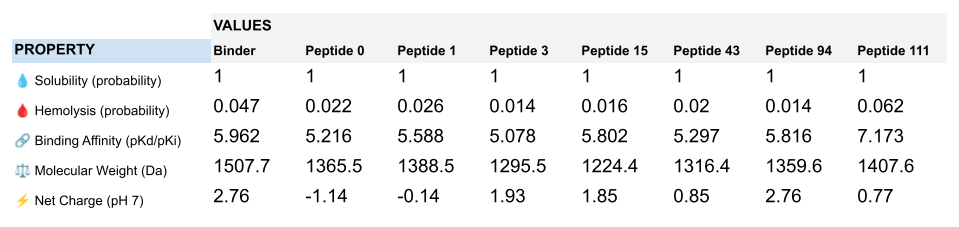
All peptides evaluated exhibit soluble and non-hemolytic properties.
Except for peptide 111, they all appear to bind only weakly to the mutant SOD1 target.
Interestingly, Peptiverse prediction about binding affinity:
- confirm previous surprising results indicating a weak binding of the known binder (FLYRWLPSRRGG) to mutant SOD1
- don’t corroborate AlphaFold results about the binding of peptides with higher ipTM, not even peptide 3 (ipTM=0.73)
- newly identify peptide 111 as a potential binding candidate (medium binding to mutant SOD1)
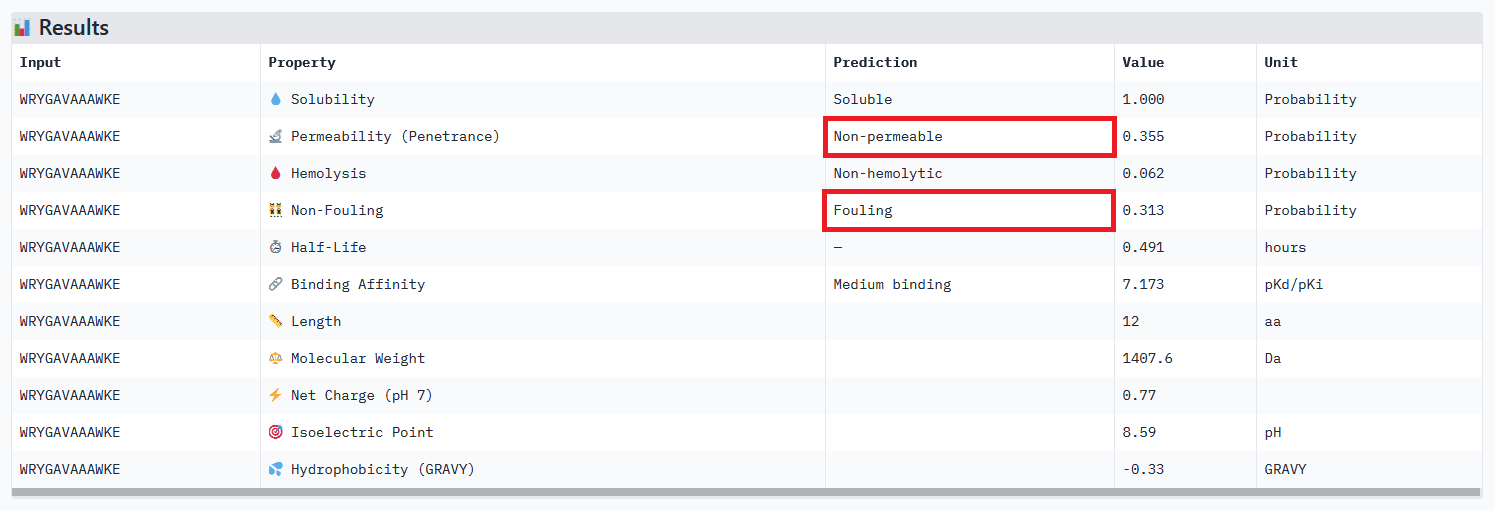
When considering how strongly the peptide binds to the target, peptide 111 seems to be the best candidate to proceed with. However, one can first question the discrepancy between the results obtained with the different prediction tools. And second, when checking more properties needed for the peptide to become a real drug, PeptiVerse indicates that this peptide presents a low membrane permeability and a fouling behavior. Thus, peptide 111 might not be the ideal candidate to be advanced for therapy. This result aligns with the purpose of PeptiVerse explained in the reference paper, which is to filter out bad candidates early and possibly, explains compromises that may have been made in the process: e.g. weaker binding property of the known SOD1 binder.
Peptide 111 extended analysis:
My strategy to find a potential candidate would be to (1) repeat the steps of peptides generation and select hundreds of candidates, (2) narrow down the selection by ensuring coherent results between the different prediction tools, checking all therapeutic properties and evaluating how the binding site may affect therapeutic efficacy (see next section).
4. Generating Optimized Peptides with moPPIt
Colab moPPIt was used to generate peptides binding to a 8 aa length motif focused on the residue 4 corresponding to the mutation site (motif: 1-8, specificity: off). Affinity guidance, solubility and hemolysis guidance parameters were enabled.
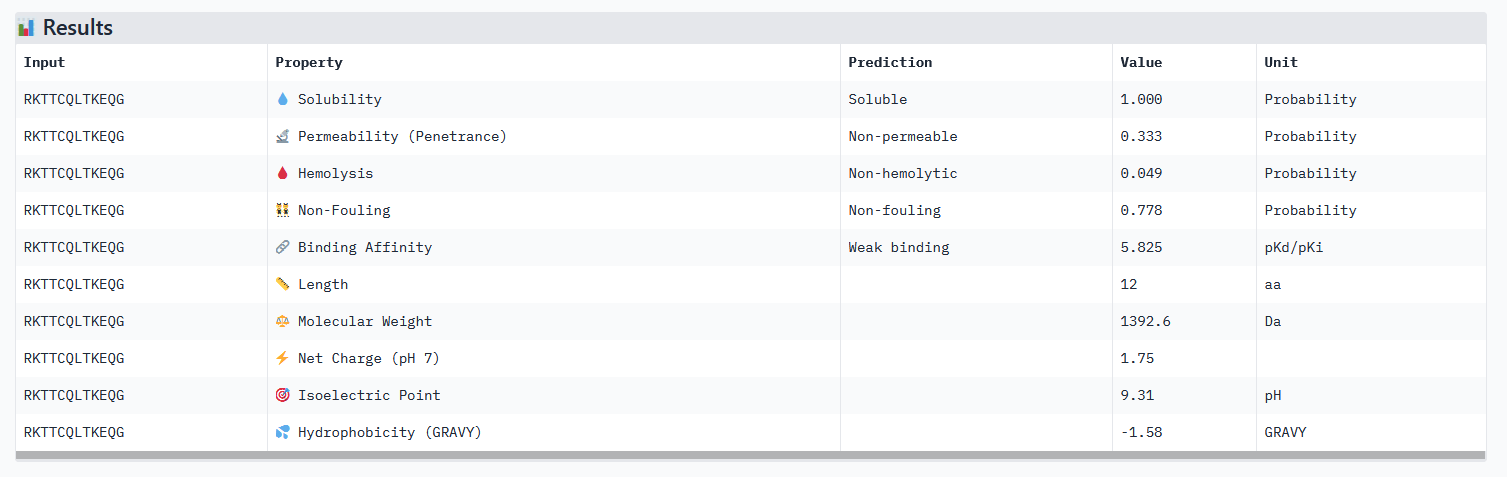
Optimized Peptide 01: RKTTCQLTKEQG
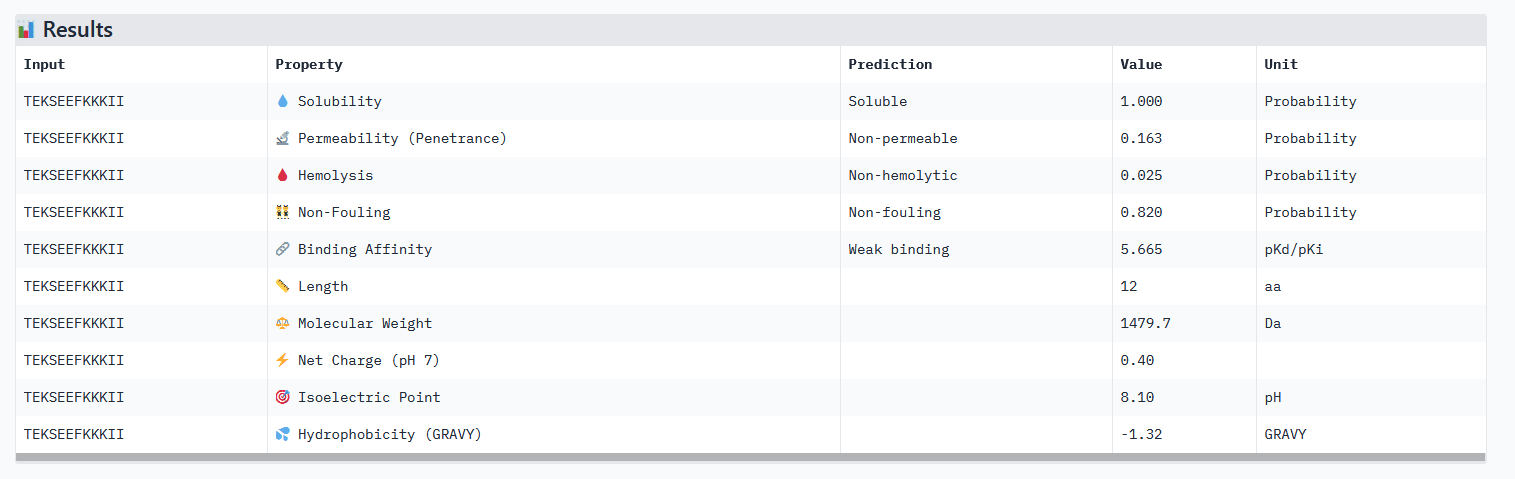
Optimized Peptide 02: TEKSEEFKKKII
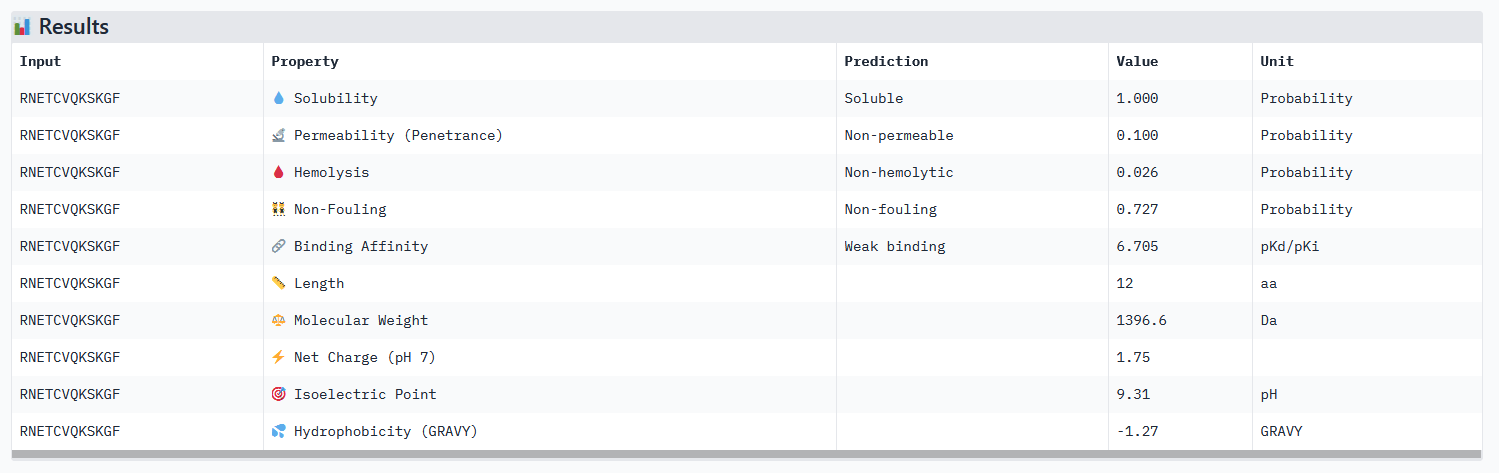
Optimized Peptide 03: RNETCVQKSKGF
moPPIt values:
| Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| RKTTCQLTKEQG | 0.97 | 0.92 | 5.82 | 0.88 |
| TEKSEEFKKKII | 0.98 | 0.75 | 5.67 | 0.42 |
| RNETCVQKSKGF | 0.97 | 0.83 | 6.70 | 0.88 |
Analysis of the optimized peptides in PeptiVerse
RKTTCQLTKEQG
TEKSEEFKKKII
RNETCVQKSKGF
Interpretation:
- All peptides only bind weakly to the target protein and are not permeable.
- However all peptides are soluble and present non-hemolytic, as well as non-fouling therapeutic properties.
- The hemolysis results of moPPIt and PeptiVerse are opposite: the “real” meaning of the moPPIt “hemolysis” values needs to be checked.
Analysis of the optimized peptides in AlphaFold
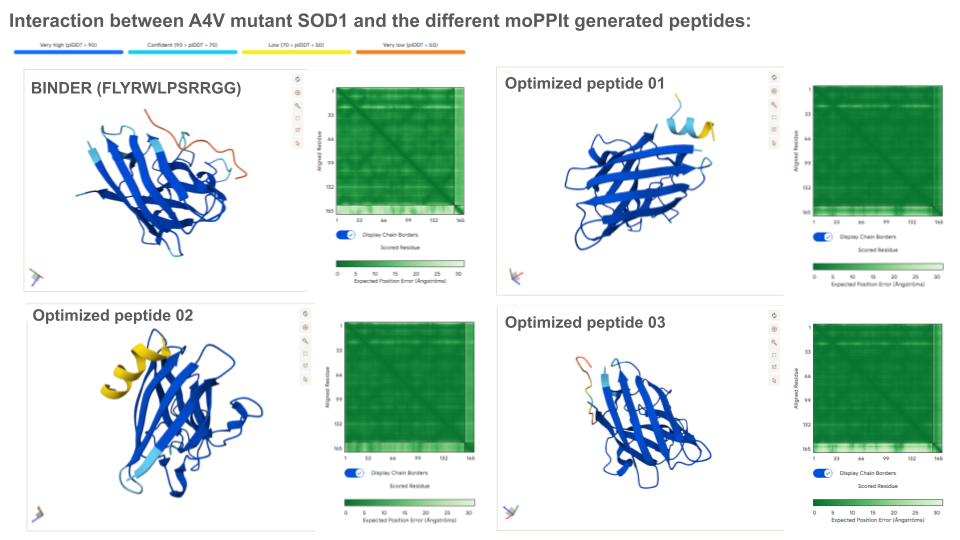
Interpretation:
- moPPIt-generated peptide 02 doesn’t interact with the N-terminal, which is coherent with the scores.
- The binding probability of the optimized peptide 01 and 02 are higher than the known binder.
CONCLUSION
While its binding affinity is evaluated as weak in PeptiVerse, 5.96 seems sufficient for it to bind to the target protein. Besides, the know binder exhibits all therapeutic characteristics needed: soluble, permeable, non-hemolytic and non-fouling.
EVALUATION OF THE KNOWN BINDER (FLYRWLPSRRGG) IN PEPTIVERSE:
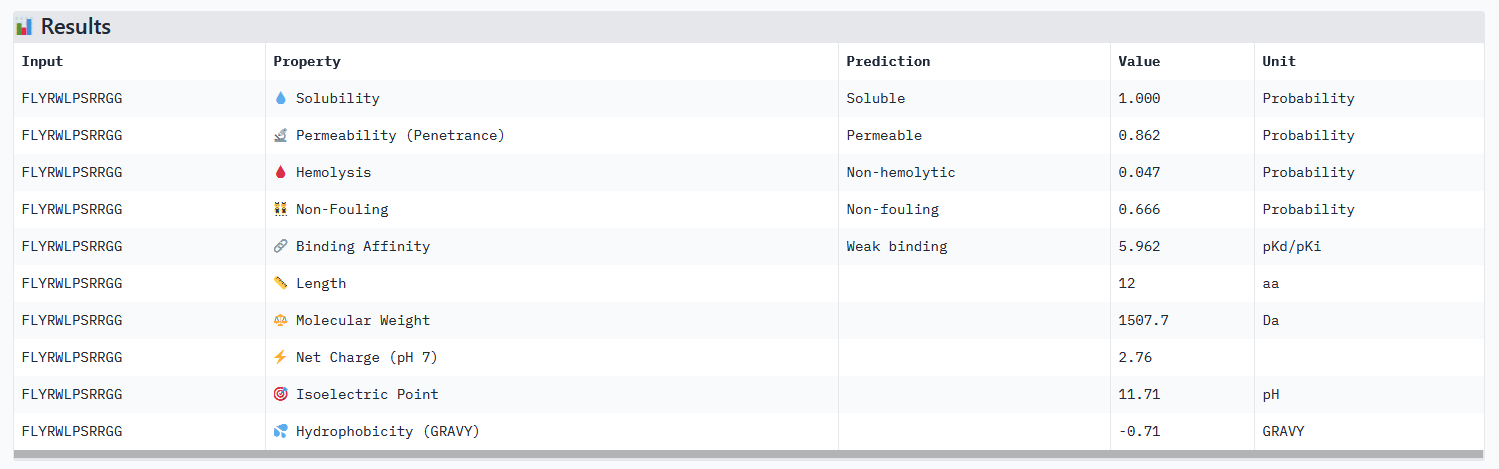
None of the generated peptides in this assignement combined all parameters at once. Concerning optimized peptide 01: a weaker permeability is not a deal breaker for potential therapeutic use, but given that the binding affinity isn’t strong I would exclude it from the list of potential therapeutic candidates for now. My strategy for the next steps would be to generate more peptides using moPPIt while testing different motifs: testing different length of the motif on a specific site (e.g. around the mutation site) and different site location. As a matter of fact, the electrostatic loop has been reported to be involved in the misfolding of SOD1 and the formation of toxic oligomers. Because it has a highly exposed surface and structural flexibility, this site is a motif top candidate (see documentation).
DOCUMENTATION
Colab PepMLM
Source Protein Sequence: UniProt P00441
Simple explanations of the reference paper:
https://chatgpt.com/s/t_69b036610424819197b52653c5b55f6b
https://chatgpt.com/s/t_69b03ad48f748191aa89855f9d3830e4
https://chatgpt.com/s/t_69b03d68d6908191a0c3e4b251c01972
Strategies for creating peptides + explanation top K value:
https://chatgpt.com/s/t_69b0555069408191ba5e1253c8c345f2
https://chatgpt.com/s/t_69b05b9e75bc8191876d8d01f138fbf8
Perplexity score: https://chatgpt.com/s/t_69b05b9e75bc8191876d8d01f138fbf8
Creation of the peptides: Peptides were created using PepMLM Colab The slider functions were blocked so the parameters (length, top K and number of binders) were changed manually in the code. Peptides were selected according to perplexity values using the interactive table function in the Generate Peptide block.
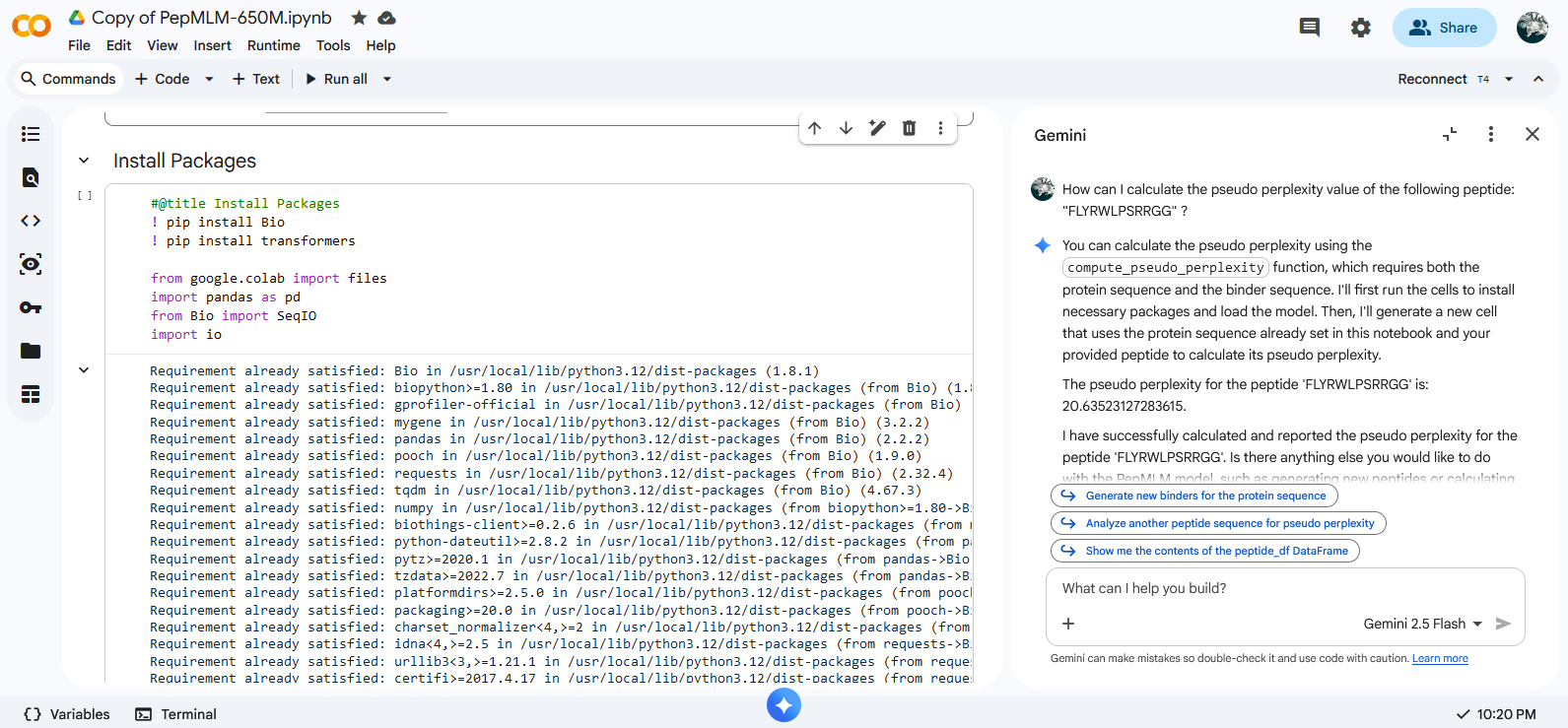
The pseudo perplexity value of the known SOD1-binding peptide FLYRWLPSRRGG was calculated by Gemini within Colab using compute_pseudo_perplexity function.
More about outliers binders: https://chatgpt.com/s/t_69b09f5842848191ad68e48392e7c919
AlphaFold
Mutant A4V SOD1 sequence and the different peptide sequences were submitted as separate chains to model the protein-peptide complex in AlphaFold. SOD1 structure
Explanation parameters of the analysis results in AlphaFold:
https://chatgpt.com/s/t_69b1d7639604819191417d9ae3069f9f
https://chatgpt.com/s/t_69b486d55cf48191b082ed6264b81c3a
Data
Mutant SOD1 - Peptide 00 complex https://alphafoldserver.com/fold/43ecc9ff963fde21
Mutant SOD1 - Peptide 01 complex https://alphafoldserver.com/fold/86c286d21a2176a
Mutant SOD1 - Peptide 03 complex https://alphafoldserver.com/fold/58da5ac311db6c0a
Mutant SOD1 - Peptide 15 complex https://alphafoldserver.com/fold/19055fa61aaf71a5
Mutant SOD1 - Peptide 43 complex https://alphafoldserver.com/fold/556af6d37be44d8f
Mutant SOD1 - Peptide 94 complex https://alphafoldserver.com/fold/3dba002c05b228b1
Mutant SOD1 - Peptide 111 complex https://alphafoldserver.com/fold/4018c707b09f0f5a
PeptiVerse
Peptides were evaluated using the PeptiVerse platform.
Simple explanations of the reference paper: https://chatgpt.com/s/t_69b489a6aa048191bbbe0e45a0227899
Colab moPPIt
Optimized peptides were generated using Colab moPPIt.
Simple explanations of the reference paper: https://chatgpt.com/s/t_69b53a1913b08191bbeffd3d085f973d
Run time: 30 min for 3 peptides (no access to A100 or L4 GPU).
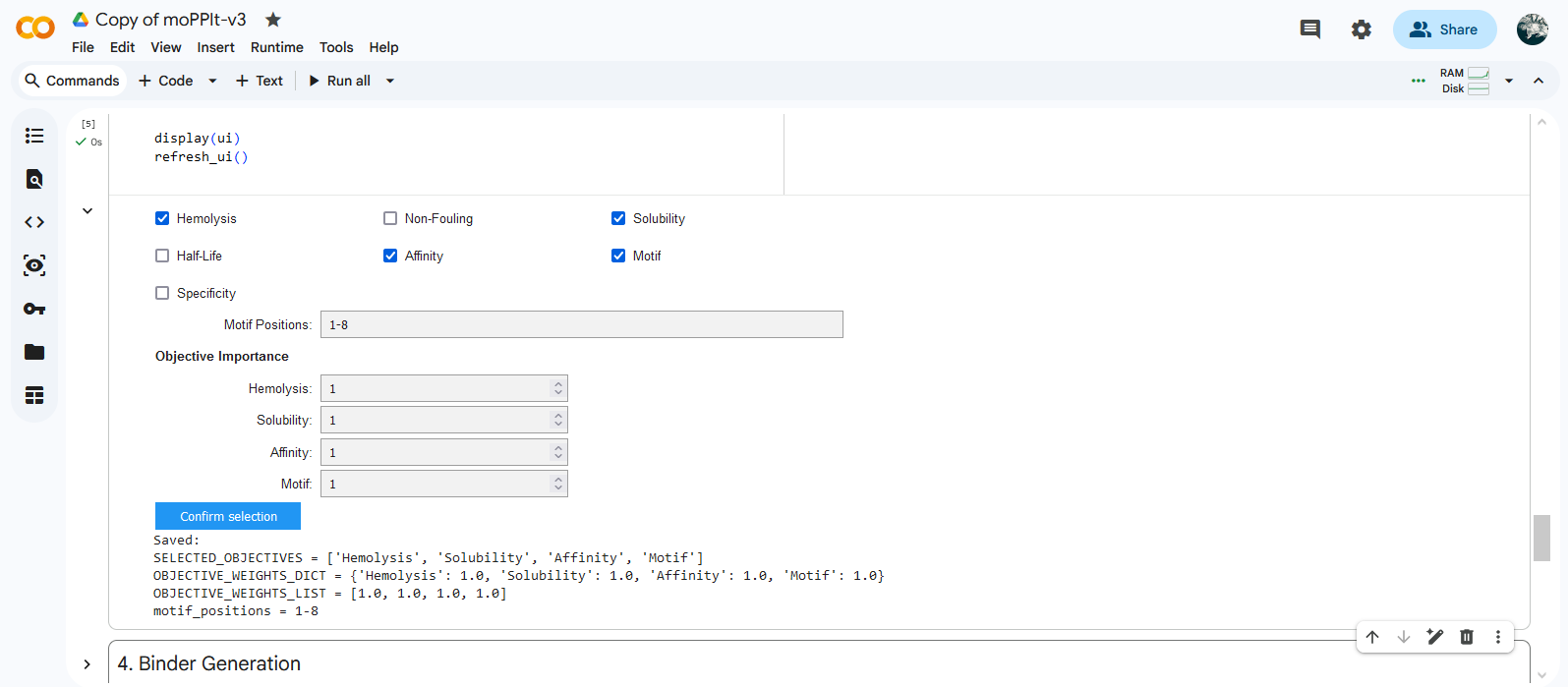
Gemini coded an extra cell to download results.
Feedback ChatGPT on motif: https://chatgpt.com/s/t_69b7054bb99881918c31e0f77b6662d4