Subsections of WEEK 06
Gibson Assembly Protocol
THE CHROMOPHORE COLOR CLONING QUEST
1. Components of the Phusion High-Fidelity PCR Master Mix
The Phusion HF PCR Master Mix is a ready-to-use “all-in-one” cocktail designed for DNA amplification. It contains:
- Phusion DNA polymerase: copy the DNA with robust performance and high accuracy (error rate much lower than common polymerase such as Taq).
- Deoxynucleotides (dNTPs): free A, T, C and G building blocks needed to copy new DNA strands.
- Reaction Buffer: an optimized chemical buffer that keeps the PCR environment stable and optimal for the DNA polymerase to copy DNA with the highest accuracy level.
- MgCl2: salts needed for the polymerase to function.
What needs to be added before running the PCR: template, primers and water.
Sources: New England Biolabs, Fisher Scientific
2. Primer annealing temperature during PCR
Primer annealing is a crucial PCR step during which primers bind to their complementary sequences on a single-stranded DNA template. This step occurs after the high-temperature denaturation phase, when the temperature is lowered to a specific point that allows stable hydrogen bonding between the primer and the template, providing a starting point for DNA synthesis by polymerase enzyme.
The annealing temperature (Ta) must be high enough to ensure specificity (binding only to the target) and low enough to allow binding to occur efficiently. Ta is determined from the melting temperature (Tm) of the primers, which is the temperature where half of the primer–DNA duplex separates and is usually set 3-5°C below the lower primer Tm.
Key factors that influence Ta include:
- Base composition / GC Content: Guanine-Cytosine pairs have three hydrogen bonds, while Adenine-Thymine pairs have only two. Primers with higher GC content have a higher Tm and require a higher Ta.
- Primer Length: Longer primers generally have higher Tm values because they have more base pairs to stabilize the binding, allowing for a higher Ta.
- Salt and Ions Concentration: The concentration of cations in the PCR buffer, particularly Mg2+, influences the stability of the primer-template duplex. Higher salt concentrations increase the Tm, allowing for a higher Ta.
- Primer Concentration: Higher primer concentration can increase the likelihood of binding and can influence the effective annealing conditions.
- Presence of Additives: Some reagents (e.g. DMSO) can lower the melting temperature of DNA and therefore require an adjustment of the Ta.
Sources: Qiagen, ThermoFisher
3. PCR vs Restriction Enzyme Digest methods
Basic Protocol: Polymerase Chain Reaction
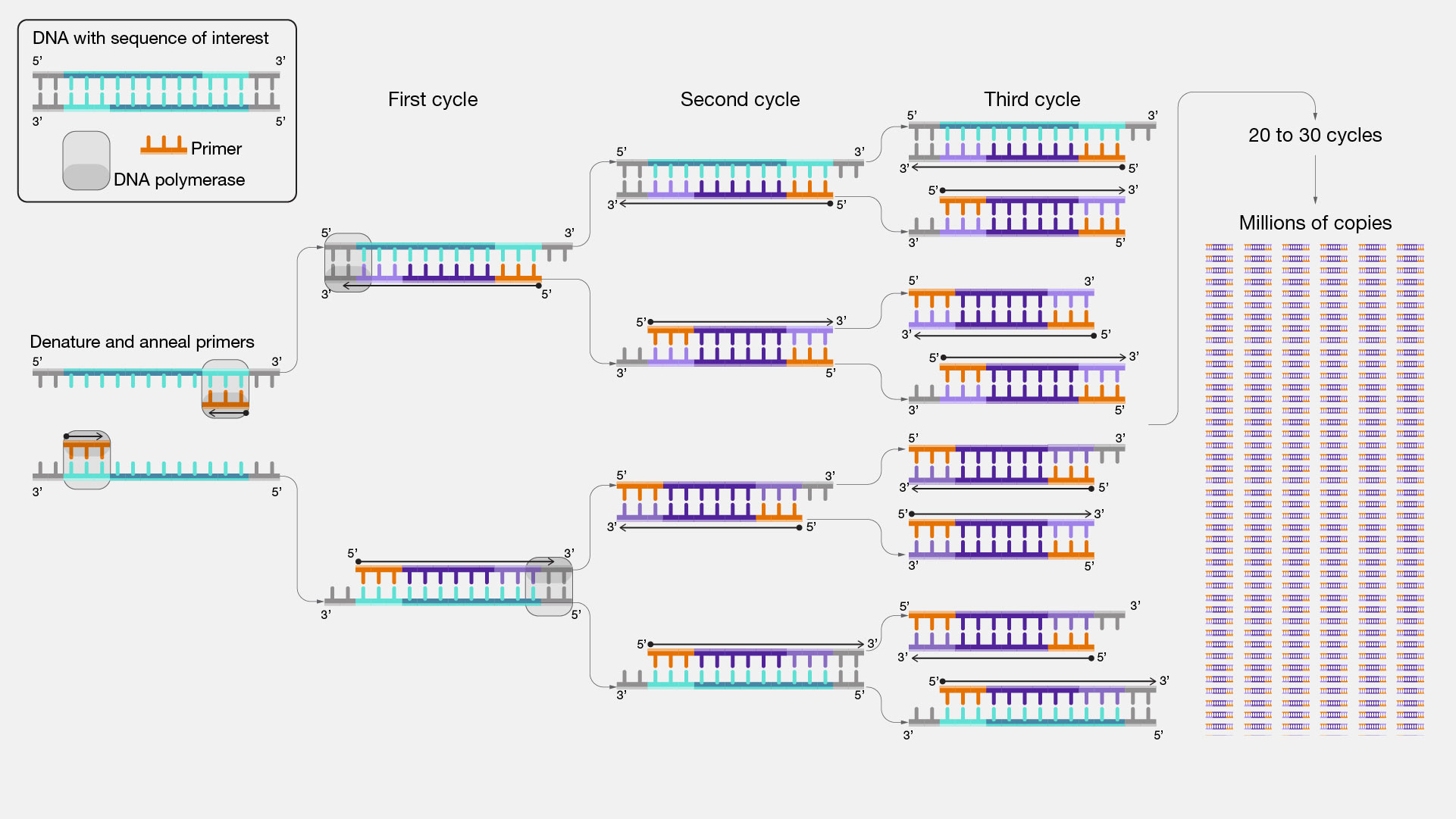
PCR creates millions of copies of a specific DNA fragment using primers(short DNA fragment) and a DNA polymerase enzyme. Key feature: The DNA fragments are defined by primer design.
Basic Protocol: Restriction Enzyme Digest

Restriction Enzyme Digest cuts DNA genome or plasmids at specific recognition sites. If a restriction enzyme cuts a plasmid, the circular DNA becomes linear. Key feature: The DNA fragments are defined by existing recognition sites in the DNA. ‘Sticky End’ vs ‘Blunt End’ Restriction Enzymes
Both methods produce linear DNA fragments, require a DNA template, use enzymes, often require gel electrophoresis afterwards to evaluate the size of the produced DNA fragments and are both commonly used in DNA cloning protocols.
However, the two methods are fundamentally different:
- PCR is used when one wants to amplify a specific gene or DNA segment. E.g. to clone a DNA sequence by using primers.
- Restriction Enzyme Digest is used when one wants to cut a DNA sequence in a specific manner. RED can be used to “clean” DNA fragments with defined ends (known restriction sites) or when one wants to linearize a plasmid before inserting a new DNA sequence.
4. Critical steps for Gibson cloning
To make sure digested and PCR-amplified DNA fragments are suitable for Gibson Assembly, one mainly needs to verify that (1) the design of the overhangs is correct and compatible, and (2) make sure to use the best quality PCR products.
(1) Overhangs Design
Gibson Assembly relies on homologous overlaps between adjacent fragments. Each primers should present a 18–22 bp core binding region, and a ~20–40 bp overlap with the next fragment. One needs to make sure that these overlaps are identical in sequence and correctly oriented (5′→3′ continuity across fragments). Furthermore, the Tm should be similar across overlaps, and primers designed to avoid cross homology, secondary structures or strong hairpins (check lab protocol for guidelines).
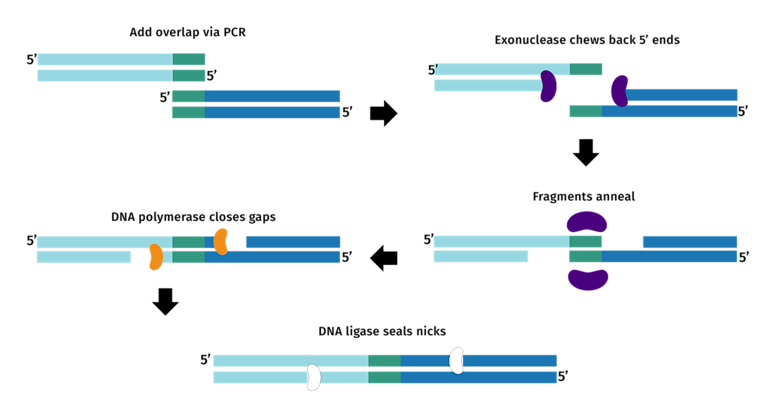
(2) Quality of the PCR Products
Template plasmid removal: the original DNA template needs to be destroyed using the enzyme Dpnl (recognition and cleavage of specific methylated sites present in the template).
DNA purification using the Zymo Research DNA Clean & Concentrator kit to remove unwanted components such as primers, enzymes, dNTPs. Contaminations such as salt and ethanol should also be avoided because Gibson enzymes (exonuclease, ligase) are very sensitive to contaminants.
(3) Verification Steps
Gel electrophoresis to confirm that the size of the fragments is correct: one clean band for the backbone and another clean one for the insert.
DNA quantification and fragment stoichiometry: one needs to make sure to respect 2:1 insert:backbone molar ratio to avoid empty plasmids or incorrect assemblies. DNA concentrations can be checked using Nanodrop/Qbit
5. Plasmid transformation
Transformation refers to the process of introducing foreign genomic material into bacterial cells (remark: one talk about transfection when working with mammalian cells). Transformation can be induced either by heat shock (like in the present protocol) or by electroporation (electrical shock).
Chemical transformation workflow
- The cells are first made chemically competent with CaCl₂ (neutralizes negative charges on DNA and bacterial cell membrane).
- Incubation on ice for 30min to allow the plasmids to bind loosely to the membrane.
- Heat shock (42°C) is applied for 45 seconds using a heat bath or thermal cycler to temporarily open the pores in the membrane: the plasmids enter the cells through the pores by diffusion.
- The cells are placed on ice for 5 min to close the pores.
- Incubation at 37°C with SOC growth media during 60 min to allow the cells to recover.
- Cells are transferred onto agar plates containing antibiotics and incubated at 37°C for 72 hours. Cells start multiplying and the ones which contain the plasmid start expressing antibiotic resistance: the cells that have successfully received the plasmid will survive and grow colonies that express the new color gene.