Week 4 HW: Protein Design I
Part A: Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming that approx 20-25% of the weight is protein, that leaves us with 100-125g protein in a 500 gram meat piece. The average amino acid molar mass is approx. 100 g/mol. With Avogadro’s number 6.022×1023 the number of molecules is between: 1 x 6.022×1023 = 6.022×1023 and 1.25 x 6.022×1023 = 7.5 ×1023 (approx)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because the DNA of the food doesn’t get copied into the host. Digestion breaks proteins into amino acids or small peptides. Then the human body rebuilds new (human proteins) using DNA instructions and its own cell machinery.
Why are there only 20 natural amino acids?
As with all biology it’s probably an energy equilibrium. Biological life build itself with the smallest amount of chemical diversity it could find, e.g. charged, polar, hydrophobic, aromatic, sulfur, etc, that would make accurate encoding possible.
Can you make other non-natural amino acids? Design some new amino acids.
Chemists have shown that they can make other non-natural amino acide. Some useful ones would be
- Clickable amino acid: an alanine-like side chain with an azide or alkyne group (bio-orthogonal labeling).
- Photo-crosslinker: phenylalanine-like side chain bearing a benzophenone (forms covalent links under UV to map contacts).
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids could have potentially made by
- Atmospheric / energy chemistry: classic spark/UV experiments can generate amino acids from simple gases.
- Hydrothermal / geochemical routes: mineral surfaces can catalyze formation and concentration.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
D-amino acids form the opposite handedness, so an α-helix made of D-amino acids is left-handed.
Can you discover additional helices in proteins?
Depends on the definition on the alpha-helices. One can define an alpha-helices, so that there are helices that don’t match that group. There are examples in nature like the 310-helices.
Why are most molecular helices right-handed?
The Chirality of building blocks is relevant, as biology uses mostly L-amino acids, which makes the right-handed α-helix sterically favorable and the left-handed version strained.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-strands have “sticky edges”, the backbone has H-bond donors/acceptors that like to be satisfied. Additionally β-sheets can often extend by adding another strand at an exposed edge. Many β-strands present alternating side chains, creating flat, complementary surfaces that pack well.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
As referenced in Week 2, I’m interested in the Protein Myostatin. It’s function is to downregulate new muscle growth. That makes it an attractive protein to inhibit for biohackers as well as clinicians treating patients with muscular dystrophy diseases.
Identify the amino acid sequence of your protein.
To answer these questions, I went to Uniprot, looking up the protein (Entry ID: “O14793, GDF8_HUMAN”), I copied the sequence from the subchapter “Sequence”.
>sp|O14793|GDF8_HUMAN Growth/differentiation factor 8 OS=Homo sapiens OX=9606 GN=MSTN PE=1 SV=1 MQKLQLCVYIYLFMLIVAGPVDLNENSEQKENVEKEGLCNACTWRQNTKSSRIEAIKIQI LSKLRLETAPNISKDVIRQLLPKAPPLRELIDQYDVQRDDSSDGSLEDDDYHATTETIIT MPTESDFLMQVDGKPKCCFFKFSSKIQYNKVVKAQLWIYLRPVETPTTVFVQILRLIKPM KDGTRYTGIRSLKLDMNPGTGIWQSIDVKTVLQNWLKQPESNLGIEIKALDENGHDLAVT FPGPGEDGLNPFLEVKVTDTPKRSRRDFGLDCDEHSTESRCCRYPLTVDFEAFGWDWIIA PKRYKANYCSGECEFVFLQKYPHTHLVHQANPRGSAGPCCTPTKMSPINMLYFNGKEQII YGKIPAMVVDRCGCS
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Using the Colab notebook that was provided by the HTGAA TAs (see link in the HW announcement). I copied the protein sequence into the variable protein_sequence in the notebook. After running the code block I got the answer: The length of the protein is: 375 aminoacids. The most common amino acid is: L, which appears 33 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
With a BLAST search, it was determined that the protein had 250 homologes (see Fig. 1)
Does your protein belong to any protein family?
Myostatin belongs to the Transforming Growth Factor - beta (TGF-β) protein family.
Identify the structure page of your protein in RCSB
The protein I use to answer the following questions has the ID 5JI1 | pdb_00005ji1 in RCSB. This is not a protein from Homo Sapiens, but from mus musculus (a mouse). While techincally not the same protein, both proteins are very similar. The choice for the mouse protein is mainly, that there is no isolated myostatin protein from homo sapiens in RCSB.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
According to the overview page of the protein, it was deposited on the 21 April 2016 and released on the 22 March 2017. The resolution of the structure is 2.25 Å, which is below the threshhold mentioned in the question text, therefore one can assume, that the resolution and therefore quality of the protein is good.
Are there any other molecules in the solved structure apart from protein?
As mentioned above the protein is isolated, therefore there are no other structures.
Does your protein belong to any structure classification family?
As mentioned myostatin is part of the TGF-β superfamily (growth factor, cystine-knot fold).
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
I tried to install pymol with homebrew, this failed repeatedly due to errors with the QT5 library, that manages desktop applications in python. Initializing a conda environment and installing pymol was successful though. I read through the tutorial, though i decided to use the command line interface of pymol, as its better documented.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
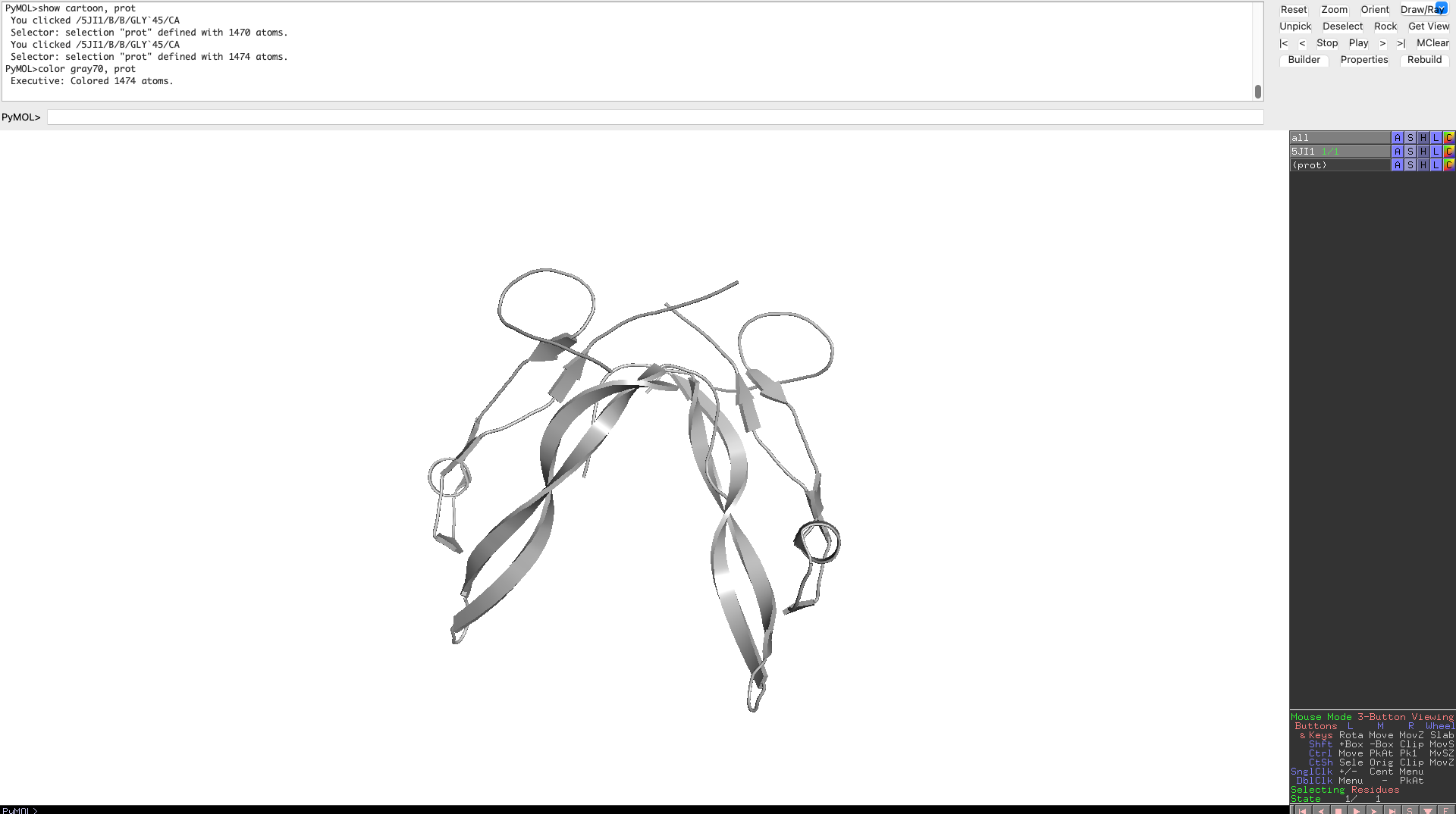

Color the protein by secondary structure. Does it have more helices or sheets?
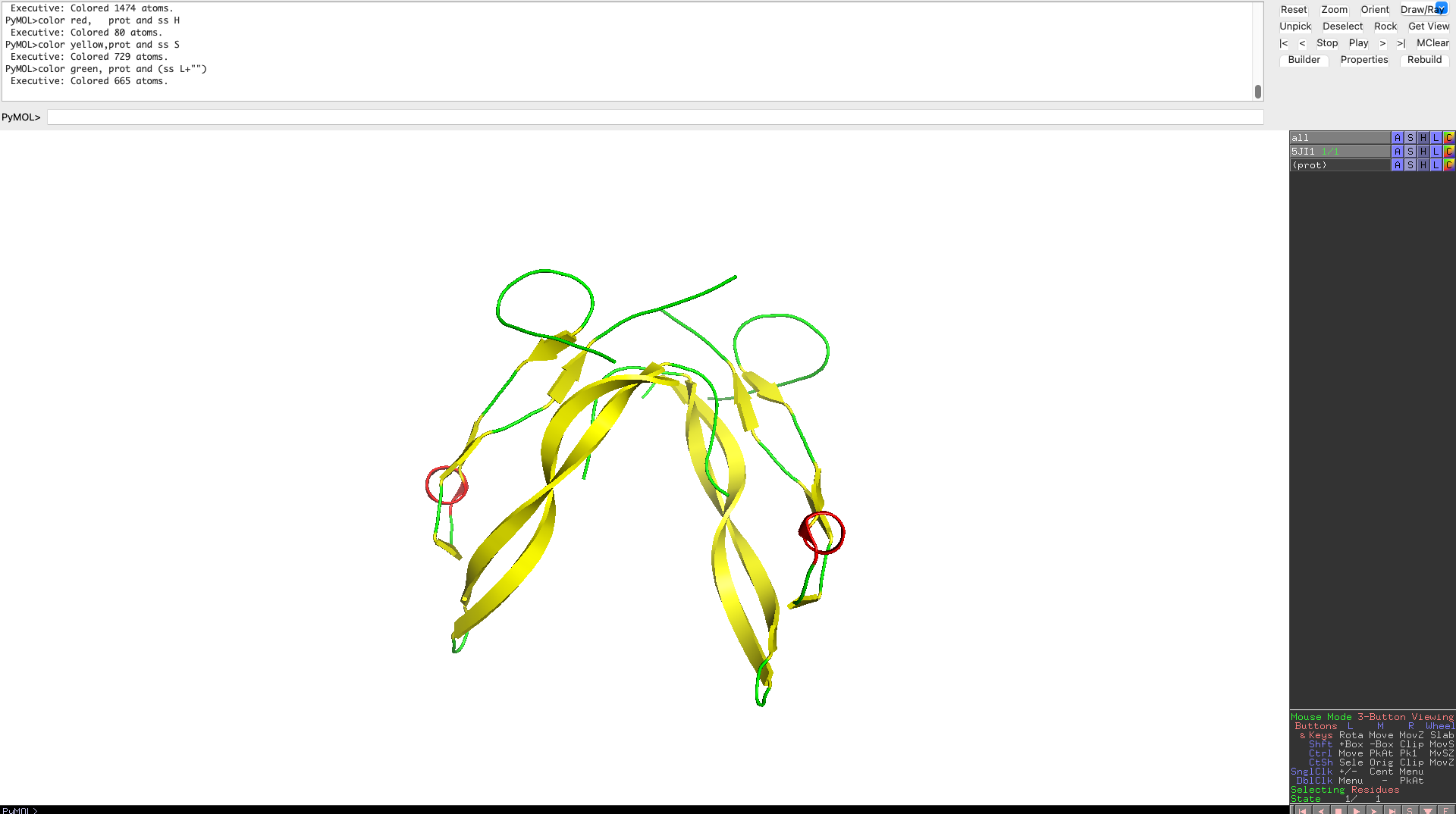 As one can clearly see the protein has more beta sheets
As one can clearly see the protein has more beta sheets
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
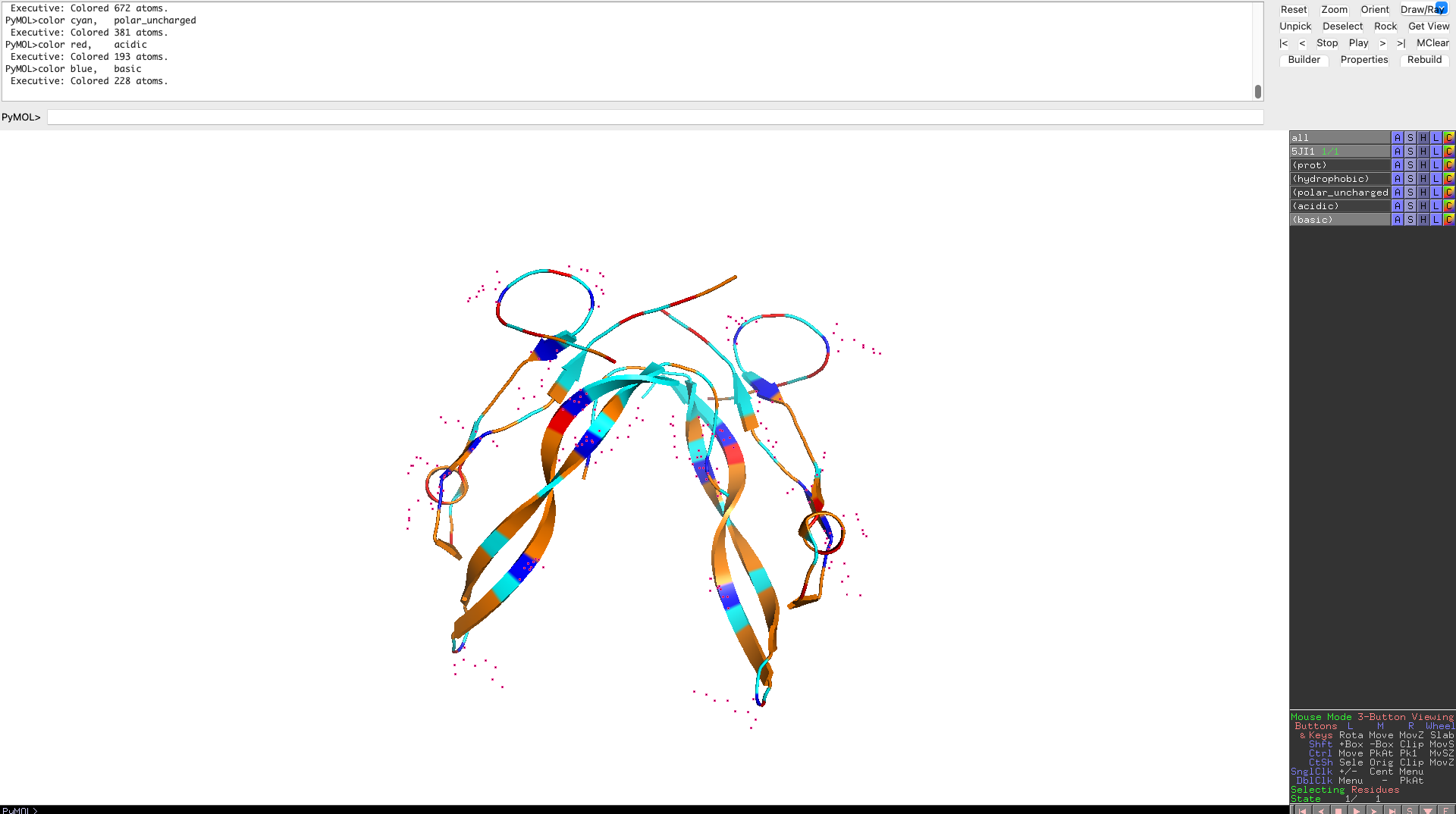 Hydrophobic residues cluster in the interior of the protein, forming a stabilizing core, while hydrophilic and charged residues are predominantly surface-exposed. This distribution reflects a well-folded, soluble signaling protein whose surface properties are optimized for molecular recognition rather than catalysis.
Hydrophobic residues cluster in the interior of the protein, forming a stabilizing core, while hydrophilic and charged residues are predominantly surface-exposed. This distribution reflects a well-folded, soluble signaling protein whose surface properties are optimized for molecular recognition rather than catalysis.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
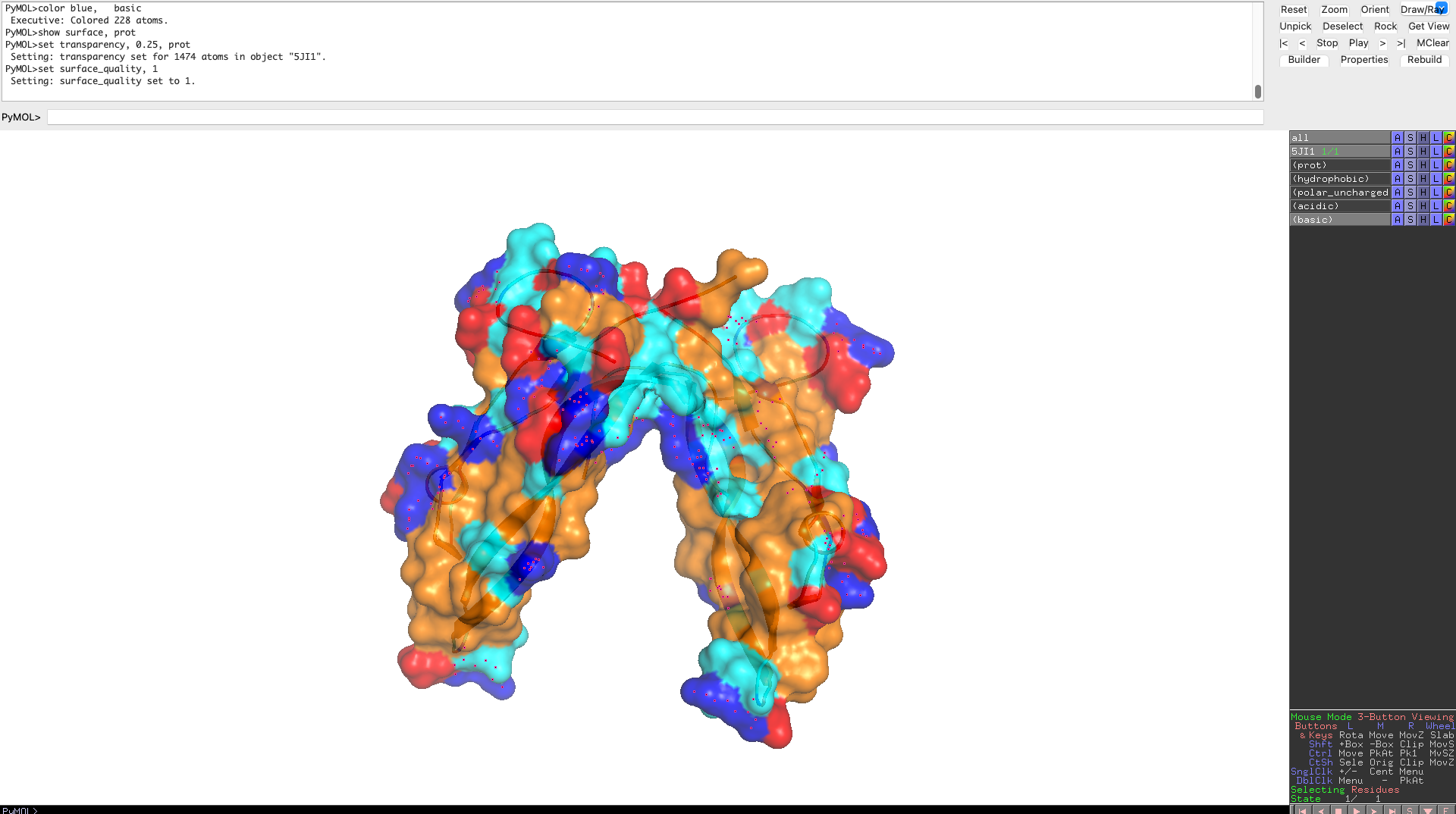
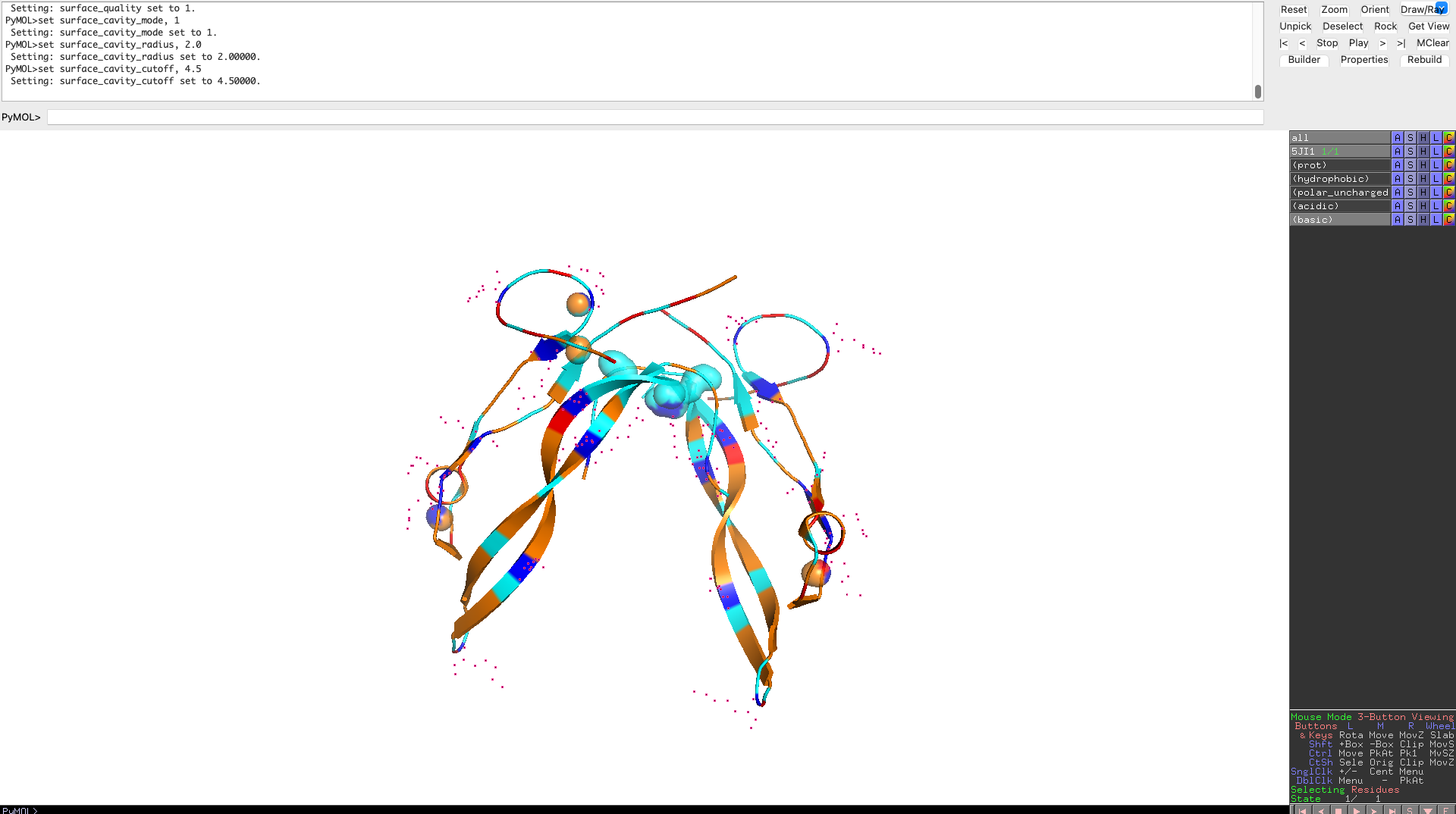 The protein surface lacks deep, well-defined binding pockets. Instead, it displays shallow surface depressions and grooves, characteristic of protein–protein interaction interfaces rather than enzyme active sites. This indicates that myostatin binds interaction partners through extended surface patches rather than classical binding pockets.
The protein surface lacks deep, well-defined binding pockets. Instead, it displays shallow surface depressions and grooves, characteristic of protein–protein interaction interfaces rather than enzyme active sites. This indicates that myostatin binds interaction partners through extended surface patches rather than classical binding pockets.
Part C: Using ML-Based Protein Design Tools
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
I set up a copy of the notebook and set it up with a Google T4 GPU.
Choose your favorite protein from the PDB.
Consistent with the rest of my homework, I chose the myostatin protein, to keep it consistent with Part B, i chose to continue to use the mus musculus sequence. We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Deep Mutational Scans
I decided to use the smallest model “esm2_t6_8M_UR50D” to conserve gpu ressources and avoid hitting rate limits, as the protein in question is rather large. Still I hit the rate limit.
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
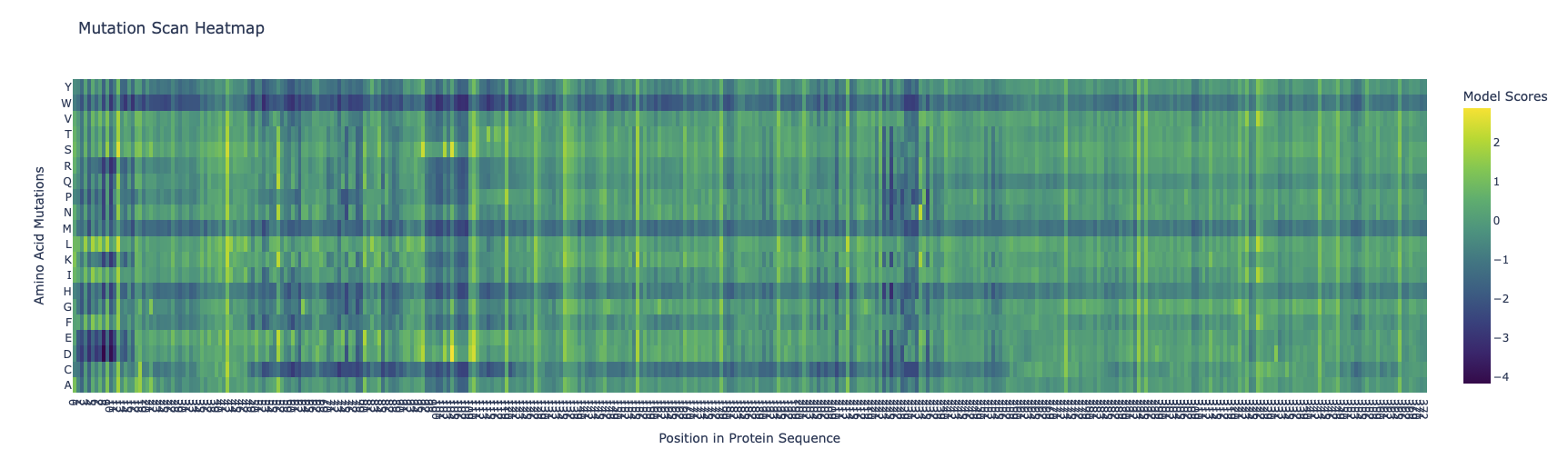
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The protein has three super columns (7-13, 97-108, 225-235), with very low scores, indicating that they are very constrained, this could indicate a lynchipin in the protein, as the areas are structurally or functionally important.
A conserved cysteine in the mature TGF-β–like domain, mutated to Ser or Ala (Cys→Ser / Cys→Ala).
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I struggled to find the data necessary ot make the comparison.
Latent Space Analysis
I downloaded the model and processed the batches:
Processing batches: 0%| | 0/15177 [00:00<?, ?it/s]
sdpaattention does not supportoutput_attentions=True. Please set your attention toeagerif you want any of these features. Processing batches: 100%|██████████| 15177/15177 [18:21<00:00, 13.77it/s] Finished forwarding sequences through the LLM and collecting mean embeddings.
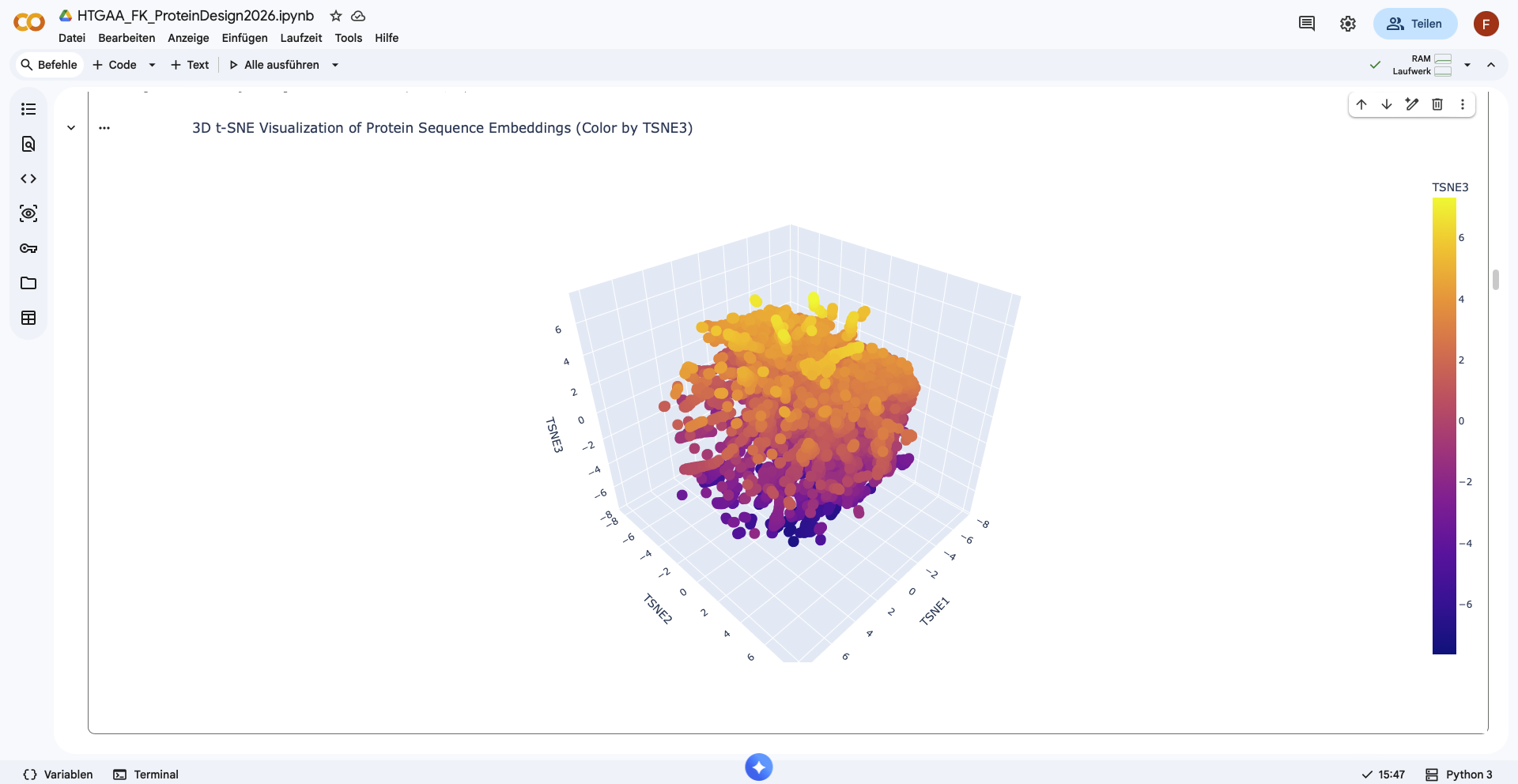
Use the provided sequence dataset to embed proteins in reduced dimensionality.
 Shape of embeddings array before 3D t-SNE: (15177, 320)
Shape of embeddings array before 3D t-SNE: (15177, 3)
Shape of embeddings array before 3D t-SNE: (15177, 320)
Shape of embeddings array before 3D t-SNE: (15177, 3)
Analyze the different formed neighborhoods: do they approximate similar proteins?
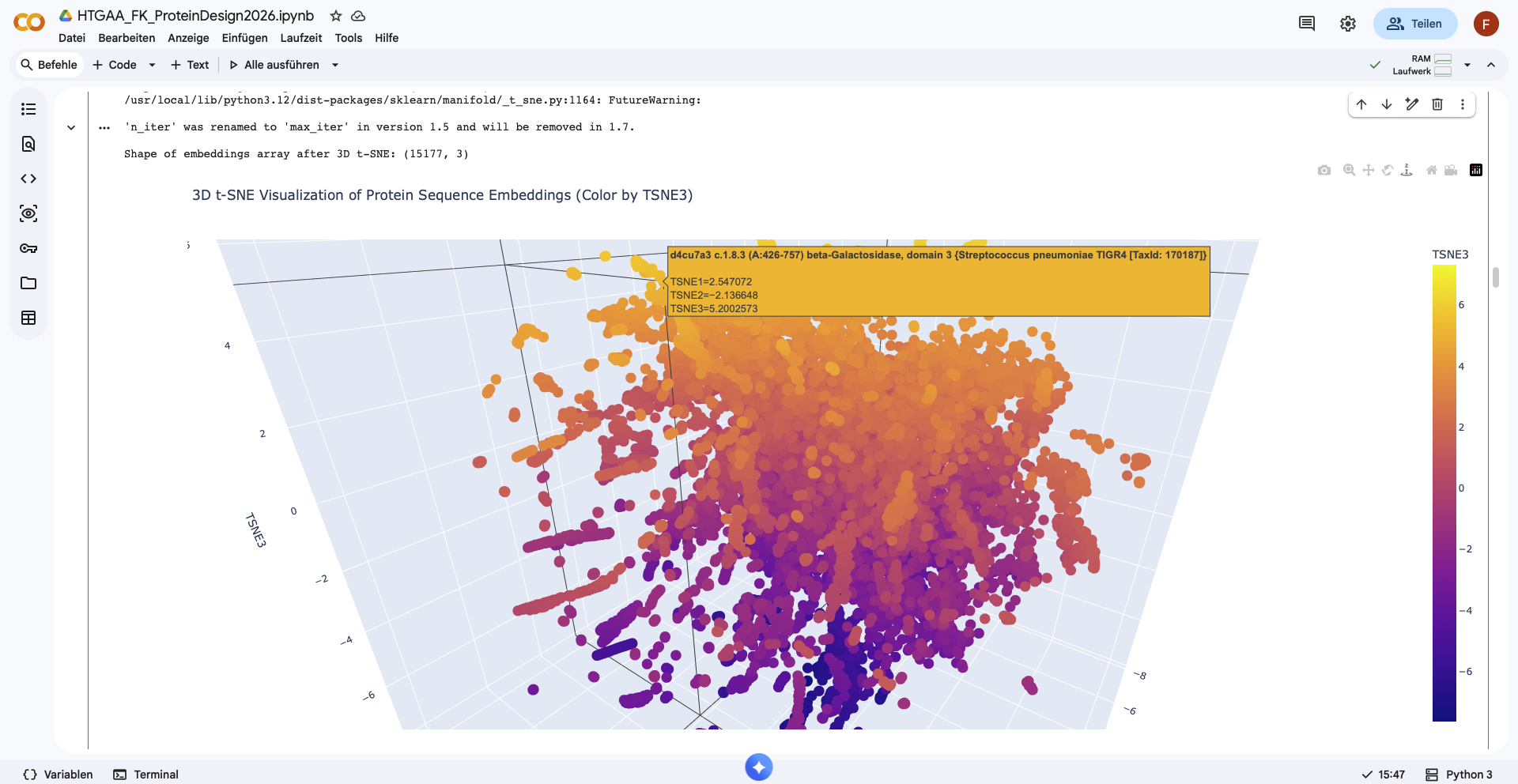 There form of the latent space looks like a sphere, with one half being densly filled, while the other one is more sparsely filled. Nevertheless no clear neighborhoods are distinguishable.
There form of the latent space looks like a sphere, with one half being densly filled, while the other one is more sparsely filled. Nevertheless no clear neighborhoods are distinguishable.
When looking at one nearest neighbor neighborhood, I still fail to see any pattern in the data, as the organisms are different, as well as the type of the protein, and the function.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
I was unable to place the myostatin in the resulting map.
C2. Protein Folding
Folding a protein I used Version 1 of ESMFold.
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I named the job “myostatin_v1”, with 1 copy and 3 num_recycles. To visualize i used the colorsheme “rainbow”
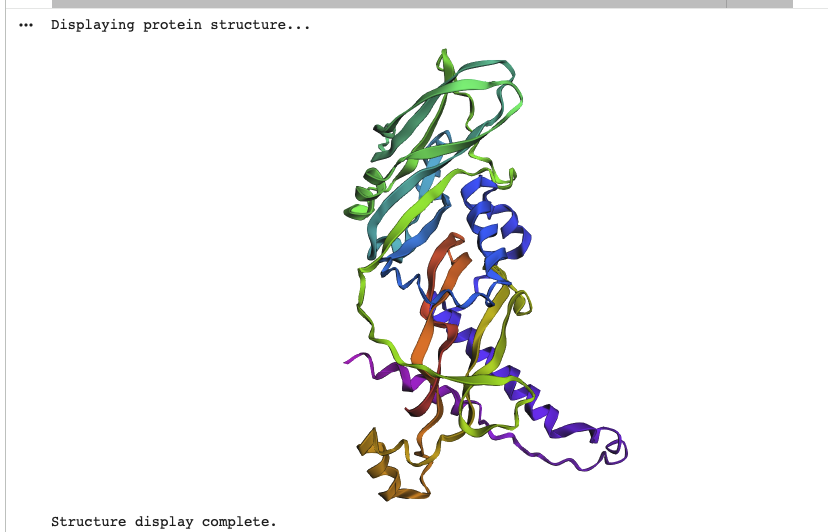 While the overall structure can be described as V or Y shaped in both predicted and actual structure, both structures don’t match on closer look. The original is mostly beta-sheets and has a destinct V shape, while the predicted model is more Y shaped, has several alpha-helices and has a more complex shape to it.
While the overall structure can be described as V or Y shaped in both predicted and actual structure, both structures don’t match on closer look. The original is mostly beta-sheets and has a destinct V shape, while the predicted model is more Y shaped, has several alpha-helices and has a more complex shape to it.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I repeatedly tried to change the structure, though i hit ratelimits with the GPU. Furthermore, as the predicted structure doesn’t match the database myostatin, either trying mutations or changing large segments will likely further remove the predicted structure from it experimental data. Proposed mutations:
- Splitting the protein in 4 equal parts and mutating the sequence at that point
- switching 20 aa for a randomly generated 20 aa section.
- Inverting the aa sequence.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN I used the model “v_48_020”, the pdb 5JI1, the homomer designed_chain A, num_seqs=1, sampling_temp=0.1
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
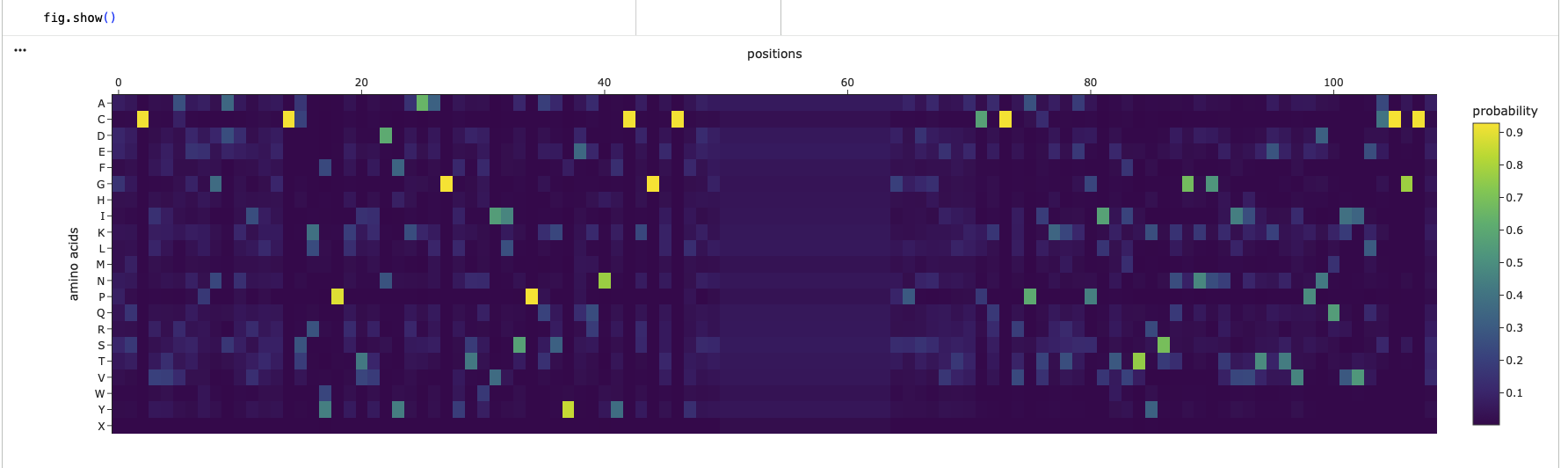 Looking at the two heatmaps, they aren’t very similar, as the predicted structure, has selectively high probabilites in random spots and else rather low probabilites. Its completely missing the 3 super clusters of low confidence, like the original one
Generating sequences…
New Sequence:GQNIVAGPGAAITECSLWPKTVDFKAAGYDWVISPKSYERNYCSGTCTSSXXXXXXXXXXXXXXGPDNVTEKRCVPTETAPITMTYSLGDGKIITETVPNQIVKACGCV
Looking at the two heatmaps, they aren’t very similar, as the predicted structure, has selectively high probabilites in random spots and else rather low probabilites. Its completely missing the 3 super clusters of low confidence, like the original one
Generating sequences…
New Sequence:GQNIVAGPGAAITECSLWPKTVDFKAAGYDWVISPKSYERNYCSGTCTSSXXXXXXXXXXXXXXGPDNVTEKRCVPTETAPITMTYSLGDGKIITETVPNQIVKACGCV
The predicted sequence isn’t similar to the originally procured sequence.
Input this sequence into ESMFold and compare the predicted structure to your original.
Trying to input the newly generated sequence, led to an error. While I cannot determine the reason for said error, my best guess is the large sequence of “X”.
Part D: Group Brainstorm on Bacteriophage Engineering
Due to later start of our Node, we had limited time to find groups and set up a meeting, therefore the drafts of our group are mainly individual, and not discussed
Goal
We target two complementary objectives: (A) Increased stability of the L protein, specifically engineering DnaJ-independent variants that fold correctly without host chaperone assistance; and (B) Higher toxicity / faster lysis, by optimizing the transmembrane oligomerization interface to accelerate pore formation. Goal A is prerequisite to Goal B: a stable, chaperone-independent L is resistant to the most documented E. coli escape mechanism (DnaJ P330Q mutation), and faster lysis narrows the window for resistance acquisition.
Scientific Rational
Three findings define our design space.
- DnaJ binds the highly basic N-terminal domain (res. 1–36) of L and relieves a steric inhibition blocking target engagement; removing this domain eliminates DnaJ dependency and accelerates lysis (Chamakura, J Bacteriol 2017).
- Near-saturating mutagenesis shows the LS motif (Leu48-Ser49) and flanking residues form a heterotypic interface with an unknown target; exquisitely conservative mutations matter (L44V = dead, L44I = functional) and all are recessive, pointing to a specific binding event rather than membrane disruption (Chamakura, Microbiology 2017).
- MS2-L oligomerizes into 10+ mers in nanodisc membranes via its TM domain; cryo-EM shows large envelope lesions starting at the outer membrane (Mezhyrova et al., 2023).
Strategy: neutralize basic charges in Domain 1 so DnaJ is no longer required, while leaving Domains 2–4 (the lytic machinery) untouched.
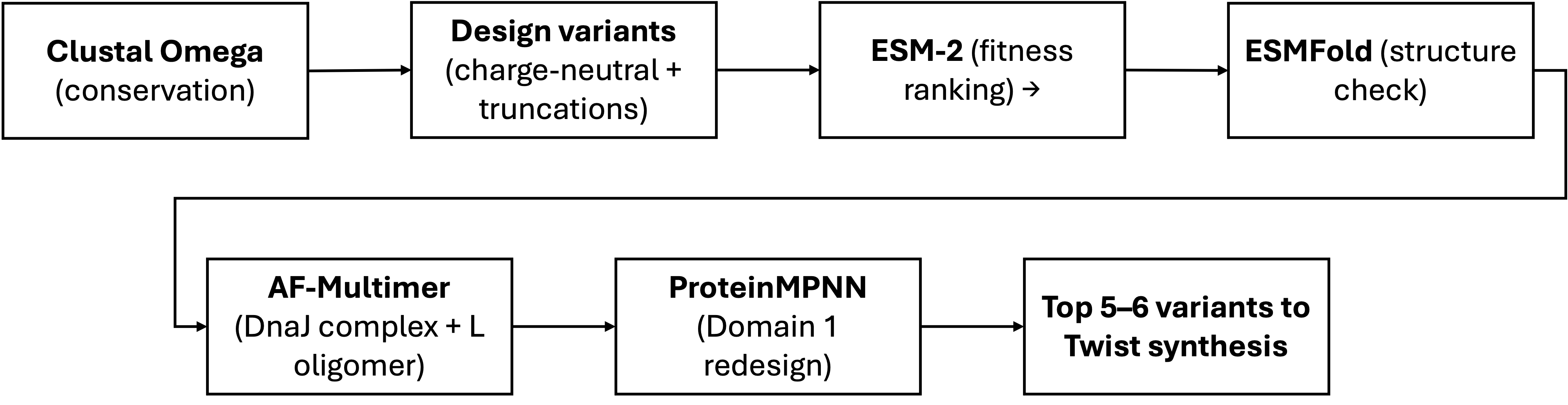
Computational Tools
| Tool | Application | Why it helps |
|---|---|---|
| Clustal Omega | Align L homologs to identify which aminoacids are freely mutable | Reproduces and extends the LS-motif alignment from Chamakura (2017). Essential first step: tells us where NOT to mutate. |
| ESMFold | Predict 3D structure and each designed variant; verify the TM helix remains intact after mutations | Fast single-sequence predictor. For a 75 aa peptide with few homologs, much more practical than full AlphaFold for screening many candidates. |
| AlphaFold-Multimer | Model the L–DnaJ complex; confirm charge-neutralized variants show reduced interface confidence. Also model L–L homodimers to check TM packing. | Key validation for Goal A: if predicted L–DnaJ interface weakens for our variants, that supports DnaJ independence. |
| ProteinMPNN | Inverse folding: redesign Domain 1 (res. 1–36) to be uncharged while fitting the ESMFold-predicted backbone. Domains 2–4 fixed as hard constraints. | new sequence for existing fold with position-specific constraints. Generates diverse candidates we can then filter with ESM-2. |
| ESM | Zero-shot fitness scoring: rank all candidate variants by pseudo-log-likelihood as a sequence-level sanity check | Independent of structure prediction. Benchmarked first against known mutants — if it captures L biology, we use it to filter; if not, we rely on conservation alone. |
Schematic
Pitfalls
We cannot model the most critical interaction (L with its unidentified host target) computationally. ML models may not capture L biology, as L is a 75 aa phage toxin with very few homologs, far outside the training distribution of ESM-2 and AlphaFold
Use of Generative AI
Generative AI was used as a conceptual drafting aid during the development of this project. Specifically, it supported the structuring and refinement of complex ideas related to protein design and use of computational tools. It was instrumental in drafting the computational strategy of engineering the MS2 Lysis Protein L, as well as clarifying the scientific concepts in the related reading. The AI was used to iteratively improve clarity of language and to explore alternative conceptual framings. All final judgments were made by the author. The link for the prompts and responses is attached in the repository.