Week 5 HW: Protein Design II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
The Protein can be found with this link The Protein sequence is:
>sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Colab Notebook was copied to my personal drive. I applied for access, even though that doesn’t seem to be necessary anymore.
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.Record the perplexity scores that indicate PepMLM’s confidence in the binders.
I checked the single-sequence checkmark and pasted the protein sequence above. I gave it the job name “SOD1_fk_v1”. Afterwards I continued with the model and generated several sequences. This had to be done in different runs as most batches contained sequences with an “X”, which later would not be accepted by AlphaFold. In the end I decided to mix and match from different batches. These are the sequences (aa1 - aa4) with the reference sequence being named aa0. The following table recodes the sequence and the perplexity score.
| ID | Sequence | Pseudo Perplexity |
|---|---|---|
| aa0 | FLYRWLPSRRGG | - |
| aa1 | AHYGVLAAAVKWRRK | 15.4397 |
| aa2 | SRYDVYVGRVKARAK | 18.3568 |
| aa3 | WRYDPVTGRYAAKKA | 9.3430 |
| aa4 | SWVPVYTAVVKLKRK | 20.8359 |
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
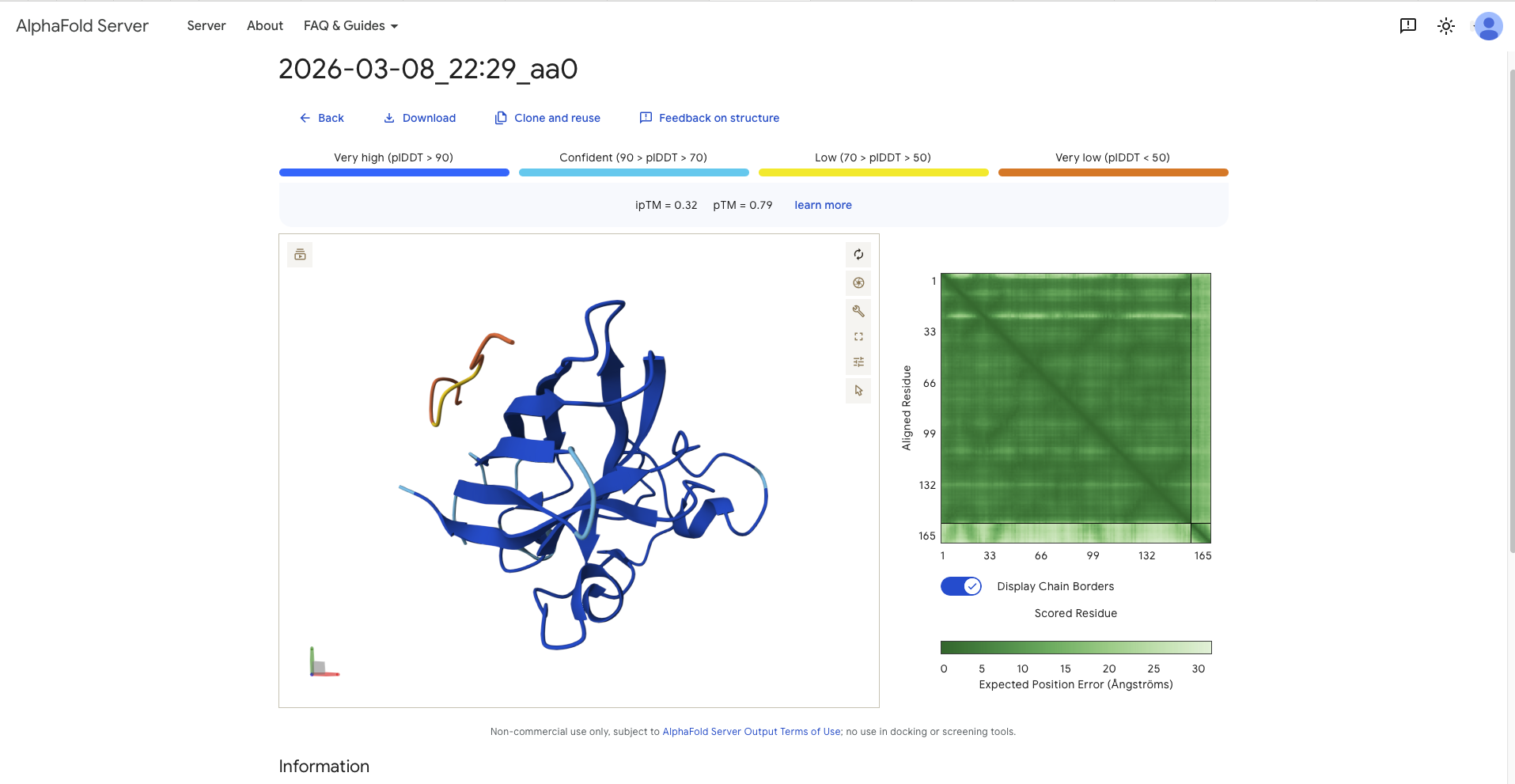
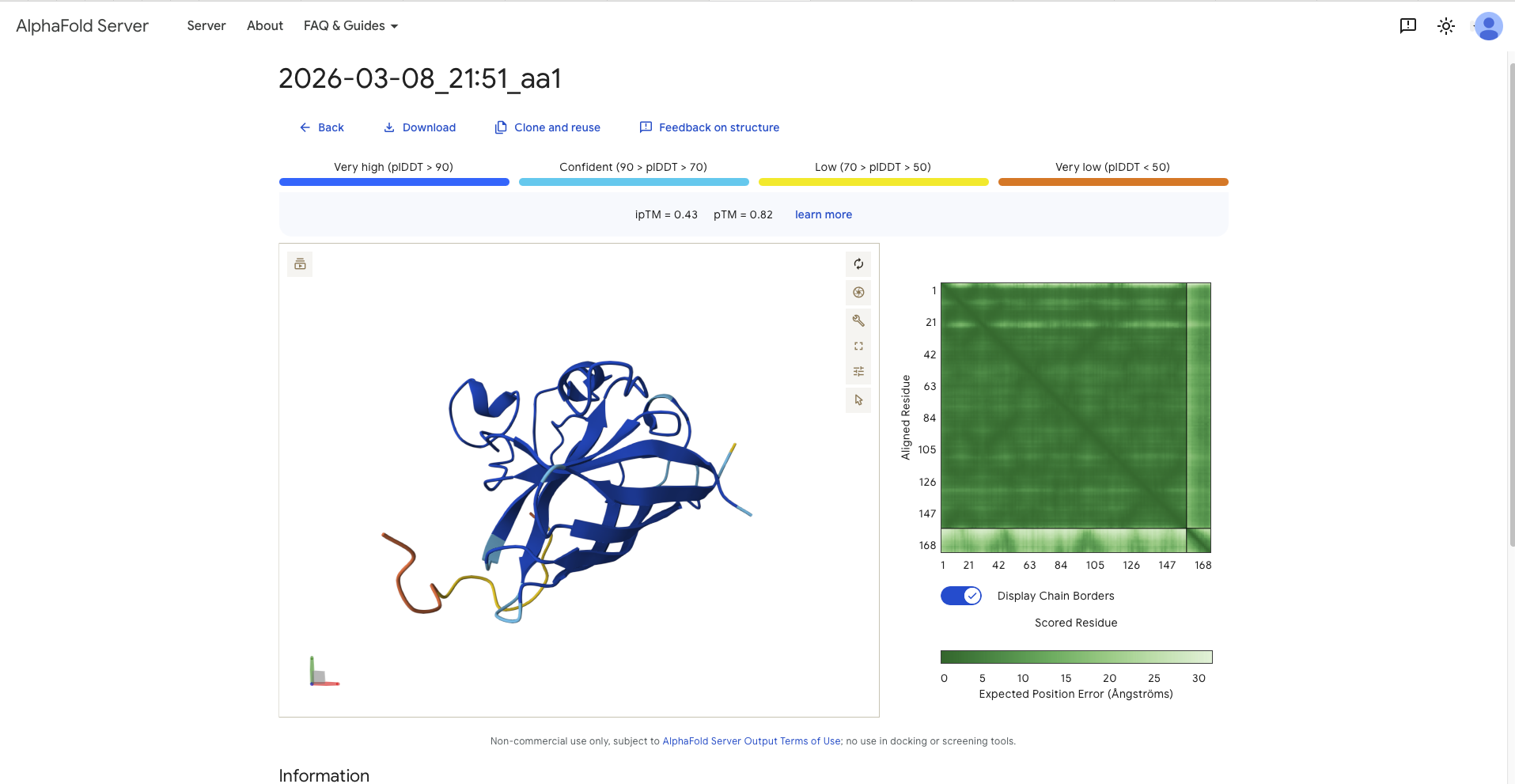
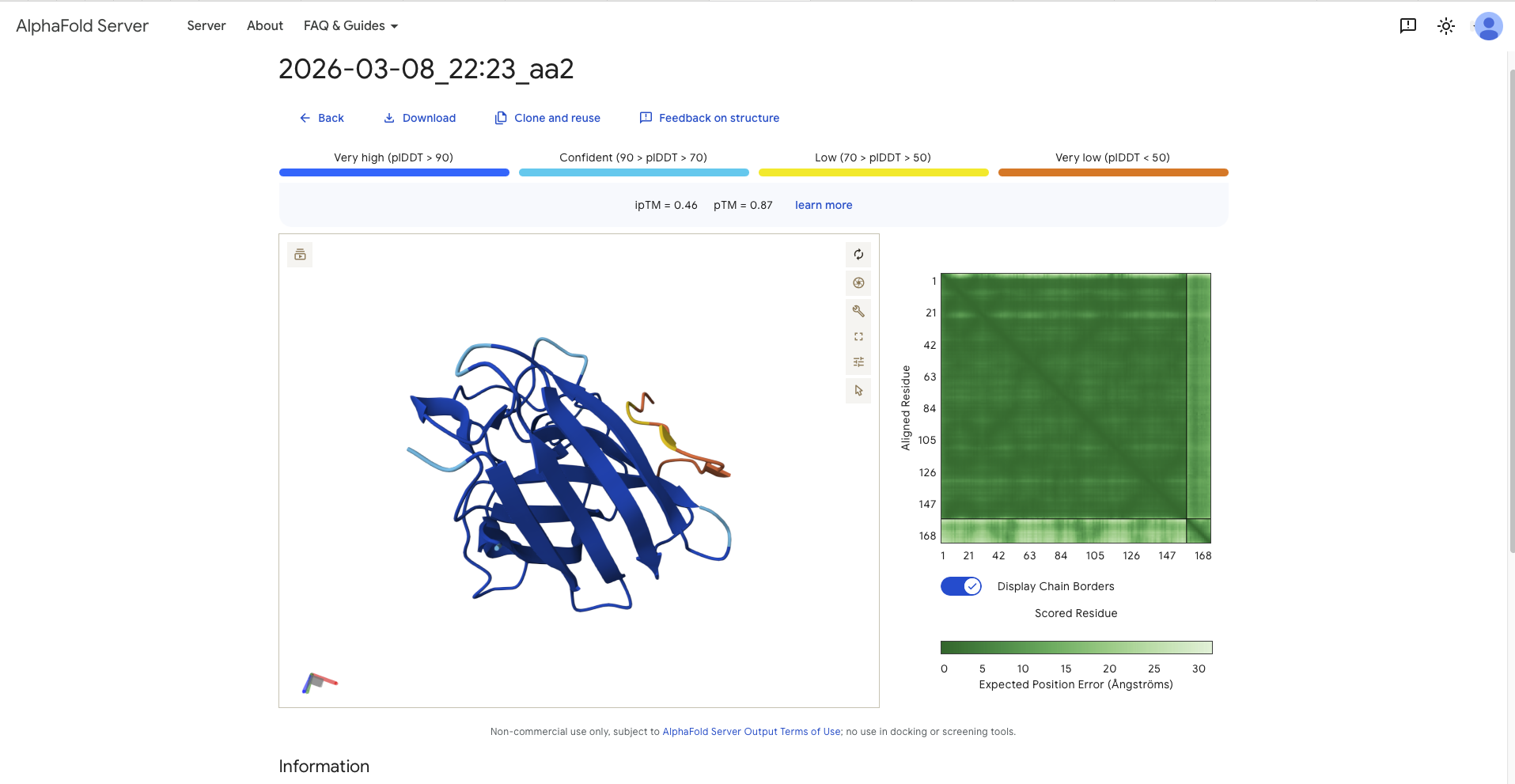

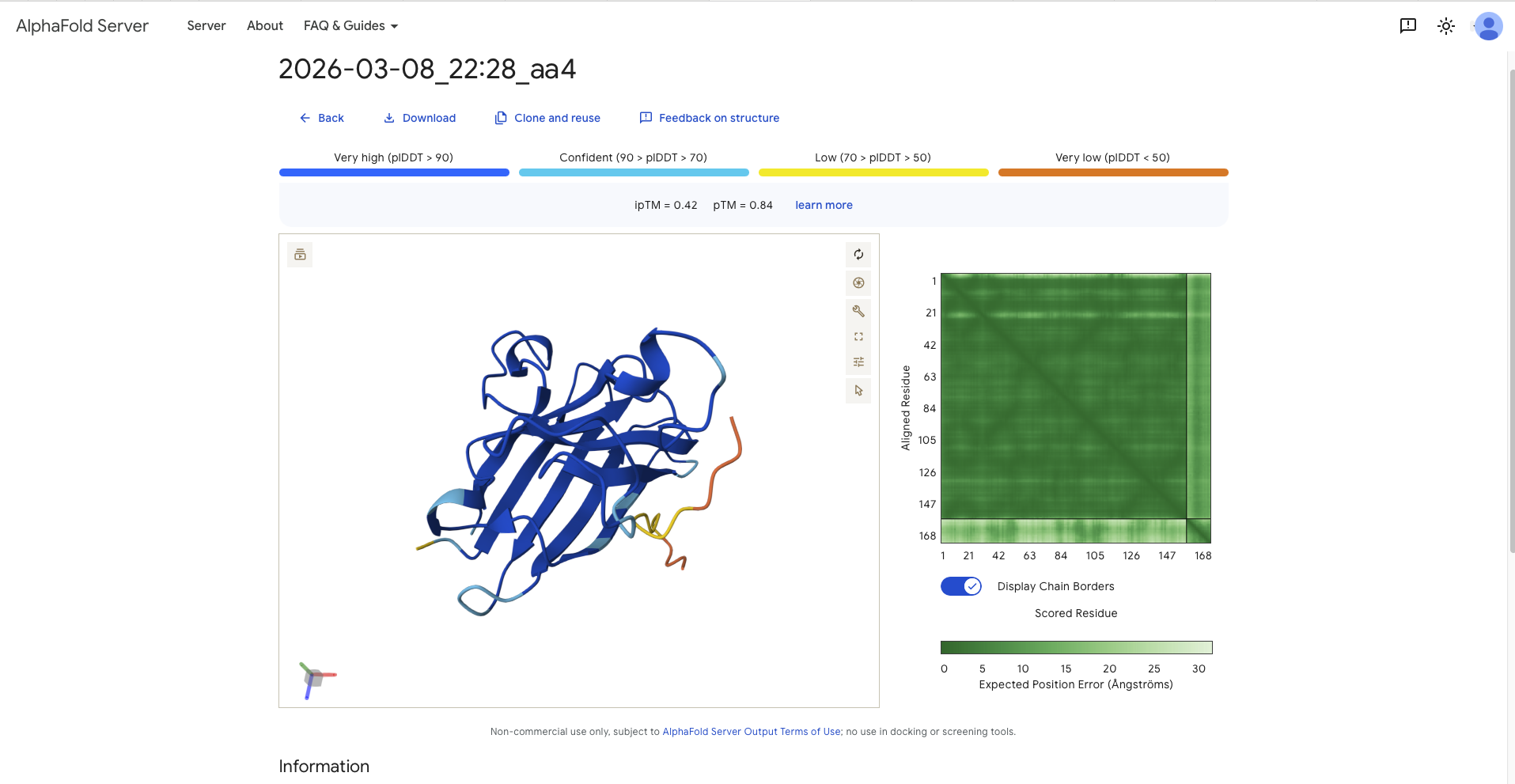
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
| ID | Sequence | Pseudo Perplexity | ipTM | pTM | Bindung Site | Binding Format |
|---|---|---|---|---|---|---|
| aa0 | FLYRWLPSRRGG | - | 0.32 | 0.79 | - In the same region as N-terminus, parallel to the beta barrel - in the same region as the dimer interface (the dimer interface is where N-Terminus and C-Terminus meet, or easier, the beginning and the end of the protein. It has the form of a strand | Surface BoundSurface Bound |
| aa1 | AHYGVLAAAVKWRRK | 15.4397 | 0.43 | 0.82 | - On the opposite side of the protein from the N-terminus - wrapping around the beta barrel from the side and the top - on the opposite side of the protein from the N-terminus | Surface Bound |
| aa2 | SRYDVYVGRVKARAK | 18.3568 | 0.46 | 0.87 | - around a 90 degree away from N-Terminus and therefore the dimer interface - paralell to the beta barrel, though it has the form of a C | Surface Bound |
| aa3 | WRYDPVTGRYAAKKA | 9.3430 | 0.23 | 0.84 | - around a 90 degree away from N-Terminus and therefore the dimer interface - perpendicular to the beta barrel - it has the form of a C, two beta sheets on either side with the belly of the C pointing towards the protein | mostly surface bound, though more burried than the others |
| aa4 | SWVPVYTAVVKLKRK | 20.8359 | 0.42 | 0.84 | - around a 120 degree away from N-Terminus and therefore the dimer interface - paralell to the beta barrel - shape is random, but wrapped into the protein in a 3D shape | mostly surface bound, though of all the generated the most burried than the others |
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
All ipTM values exceed the refrence linker, except aa3, the, though all pTM values exceed the reference sequence. After discussion with TA all values above 0.8 constitute a good confidence score that the overall structure is correct.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence. Paste the A4V mutant SOD1 sequence in the target field. Check the boxes Predicted binding affinity Solubility Hemolysis probability Net charge (pH 7) Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see.
All peptides show weak binding affinity. This is somewhat expected from the AlphaFold data, as the generated Peptides all show only surface level binding.
Do peptides with higher ipTM also show stronger predicted affinity?
According to a quick trendline analysis, the relationship is negative. The affinity scores scatter around 6.27 and with a standard deviation of around 200.
Are any strong binders predicted to be hemolytic or poorly soluble?
My predictions didn’t produce any strong binders, therefore I cannot answer the question. All predicted peptides are non-hemolytic (values range between 22 and 77) and soluable
Which peptide best balances predicted binding and therapeutic properties?
As non of my peptides have strong binding, while they have good therapeutic properties, the sequence with the highest binding affinity is the best balance. Which is aa3, that does have the lowest ipTM score.
Choose one peptide you would advance and justify your decision briefly.
I’d choose aa3, as the sample is doesn’t produce a predicted sequence that has a strong binding affinity, while the therapeutic properties are solid, the peptide with the highest binding affinity is chosen.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
Unfortunately I wasn’t able to complete this part as google didn’t allow for the use of A100 or L4 GPU. I got the tried with T4 GPUs but the first cell ran in a infinite loop till my GPU credits ran out.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
See above
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele) [optional for Committed Listeners]
Part 0: Sign-up to Boltz Lab
I signed up for access to the Boltz Lab, though as of writing the Homework i have not received any credentials
Part 1: Structural Predictions in the Sandbox
Part 2: Setting Up a BRD4 Design Project
Part 3: Running Your Virtual Screen
Part 4: Analysis and Discussion
Part C: Final Project: L-Protein Mutants
Option 1: Mutagenesis
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
I copied the Colab notebook and worked through it to the best of my ability. Any experimental work will be done in the BioPunk Node at a later time, as per discussion with Eliott Roth (Our Node Leader).
Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid. It can be positive or negative for the protein. We want to identify possible mutations that are “positive” If you run this notebook - you will see a .csv file in the sidebar. You can download it and look at it in the google sheets if that’s easier.
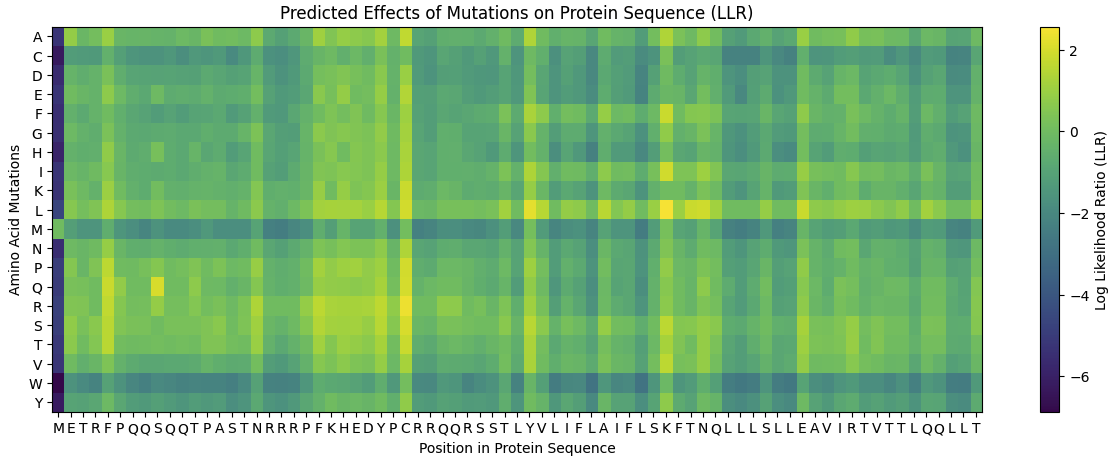
Running the Colab notebook, gave me several outputs, unfortunately they were quite badly documented therefore all information should be used with caution. The first run analyses the entire protein sequence and produces an exhaustive list of potential mutation sites: Model 1
| Position | Wild_Type_AA | Mutation_AA | LLR_Sccore |
|---|---|---|---|
| 50 | K | L | 2,561468 |
| 29 | C | R | 2,395427 |
| 39 | Y | L | 2,24178 |
| 29 | C | S | 2,04315 |
| 9 | S | Q | 2,014325 |
| 29 | C | Q | 1,997049 |
| 29 | C | P | 1,971029 |
| 29 | C | L | 1,960646 |
| 50 | K | I | 1,928801 |
| 53 | N | L | 1,864932 |
| 61 | E | L | 1,818098 |
| 52 | T | L | 1,813968 |
| 50 | K | F | 1,802069 |
| 29 | C | T | 1,797247 |
| 29 | C | K | 1,795878 |
| 5 | F | Q | 1,795244 |
| 5 | F | R | 1,659717 |
| 29 | C | A | 1,648656 |
| 27 | Y | R | 1,628061 |
| 22 | F | R | 1,602028 |
| 5 | F | P | 1,596891 |
| 50 | K | V | 1,594576 |
| 50 | K | S | 1,574557 |
| 5 | F | T | 1,559024 |
| 5 | F | S | 1,556417 |
| 45 | A | L | 1,539248 |
| 39 | Y | S | 1,517457 |
| 27 | Y | S | 1,497053 |
| 40 | V | L | 1,47763 |
| 27 | Y | L | 1,474637 |
| 22 | F | S | 1,423358 |
| 29 | C | E | 1,383281 |
| 39 | Y | A | 1,364999 |
| 29 | C | N | 1,362601 |
| 50 | K | A | 1,357795 |
| 29 | C | I | 1,344121 |
| 5 | F | L | 1,332615 |
| 17 | N | R | 1,323651 |
| 39 | Y | I | 1,320103 |
| 39 | Y | T | 1,302804 |
| 26 | D | R | 1,268762 |
| 29 | C | H | 1,246107 |
| 39 | Y | F | 1,245851 |
| 39 | Y | V | 1,24439 |
| 23 | K | R | 1,236555 |
| 25 | E | R | 1,22935 |
| 24 | H | R | 1,227779 |
| 50 | K | T | 1,222131 |
| 27 | Y | Q | 1,218851 |
| 27 | Y | T | 1,215567 |
Model 2
| ID | Amino Acid | Position | LLR_Score |
|---|---|---|---|
| 0 | L | 50 | 2,561468 |
| 1 | L | 39 | 2,24178 |
| 2 | I | 50 | 1,928801 |
| 3 | L | 53 | 1,864932 |
| 4 | L | 52 | 1,813968 |
| 5 | F | 50 | 1,802069 |
| 6 | V | 50 | 1,594576 |
| 7 | S | 50 | 1,574557 |
| 8 | L | 45 | 1,539248 |
| 9 | S | 39 | 1,517457 |
| 10 | L | 40 | 1,47763 |
| 11 | A | 39 | 1,364999 |
| 12 | A | 50 | 1,357795 |
| 13 | I | 39 | 1,320103 |
| 14 | T | 39 | 1,302804 |
| 15 | F | 39 | 1,245851 |
| 16 | V | 39 | 1,24439 |
| 17 | T | 50 | 1,222131 |
| 18 | L | 54 | 1,12086 |
| 19 | R | 39 | 1,064191 |
Model 3
| Position | Wild_Type_AA | Mutation_AA | LLR_Score |
|---|---|---|---|
| 50 | K | L | 2,561468 |
| 29 | C | R | 2,395427 |
| 39 | Y | L | 2,24178 |
| 29 | C | S | 2,04315 |
| 9 | S | Q | 2,014325 |
| 29 | C | Q | 1,997049 |
| 29 | C | P | 1,971029 |
| 29 | C | L | 1,960646 |
| 50 | K | I | 1,928801 |
| 53 | N | L | 1,864932 |
Model 4
| Position | Wild_Type_AA | Mutation_AA | LLR_Score |
|---|---|---|---|
| 50 | K | L | 2,561468 |
| 29 | C | R | 2,395427 |
| 29 | C | R | 2,395427 |
| 39 | Y | L | 2,24178 |
| 29 | C | S | 2,04315 |
| 29 | C | S | 2,04315 |
| 9 | S | Q | 2,014325 |
| 9 | S | Q | 2,014325 |
| 29 | C | Q | 1,997049 |
| 29 | C | Q | 1,997049 |
Here is the Heatmap generated from the Colab Notebook.
Use the experimental data here. This dataset contains information about mutants of the L-Protein and their effect on lysis in the lab - L-Protein Mutants
I copied the csv file from the website to the googlesheet and the Colab.
First check, does the experimental data correlate with the scores from the notebook in (b)? This should give you a clue on how well these language embeddings capture information about this protein sequence.
Let’s first look at the experimental data. Here the positions, mutations are displayed, together with a binary, whether the lysis has happened. Therefore one needs to look at the lysis = 1. These are 35 entries. Now one needs to compare, whether the positions are in the model generated mutation sites.
I’m unsure what the instructors mean with correlating the scores from the experimental data with the predicted data. The above mentioned workflow is a comparison of targets. In a next step on can check, whether the identified targets have a positive LLR score.
Using information about the effect of protein mutations at these sites - both the scores and the experimental data in the drive, come up with 5 mutations for each student along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (refer to notes above on what amino acid positions these are) and 2 of them must be in the soluble region . Remember that you can also use the pBLAST to see which residues are conserved and not mutate them if you want to. One easy way to generate sequence mutations could be to look for residue positions and mutations that have a positive mutational effect either in the experimental or have a positive score from step 1. And pick a combination of those mutations.
| ID | Model | Position | Wildtype | Mutation |
|---|---|---|---|---|
| 1 | 1 | 61 | E | L |
| 2 | 1 | 50 | K | F |
| 3 | 2 | 53 | N | L |
| 4 | 2 | 52 | L | |
| 5 | 3 | 53 | N | L |
You can utilize Af2_Multimer to generate a Multimeric Assembly; you can do this by making your query sequence as. We want to do this because - A running hypothesis for how this protein functions is that it assembles to make a perforation in the bacterial membrane.
No Colab was provided for that. If applicable the colab from option 2 could be utilized. This can be submitted at a later point.
Use of Generative AI
Generative AI was used as a conceptual drafting aid during the development of this project. Specifically, it supported the structuring and refinement of complex ideas related to protein design and use of computational tools. It was instrumental in explaining the methods used in the colab notebook to generate mutations of the lysis protein, as well as clarifying the scientific concepts in the related reading. The AI was used to iteratively improve clarity of language and to explore alternative conceptual framings. All final judgments were made by the author. The link for the prompts and responses is attached in the repository.