Homework Week 1: Class Assignment Biological Engineering Application First Steps towards “Intelligence in a (warehouse)-dish” Guided by the vision of building a biological general computing system, the goal of the proposed tool is to provide a minimal, yet replicable brain organoid based system, that can be engineered to exhibit controllable, learning-like signal processing behaviour. The system consists of 3 conceptual parts (input - computation - output), that manifest in 2 integrated physical devices.
Part 0: Basics of Gel Electrophoresis Watch Week 2 Lecture (Zoom) Watch Week 2 Recitation (Zoom) Watch BioBootcamp Day 1 - Day 3 (Zoom) Part 1: Benchling & In-silico Gel Art Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Artwork After struggling quite some time with the task of creating artwork with the limited amount of restriction enzymes, in the end decided to stick to a relatively easy and repetitive pattern that with a little imagination has a lot of versatile interpretations: It can be two friends hanging out It can be DNA (or at least a rought estimation of the firrst two loops) To help you visualize it a bit better i created some generative AI art
Python Script for Opentrons Artwork Review this week’s recitation and this week’s lab for details on the Opentrons and programming it. Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. I generated a quick design using the above mentioned tool: BioPunk Initials See the: Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons. You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept. If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead. If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that: If you use AI to help complete this homework or lab, document how you used AI and which models made contributions. As its good practice in Software Engineering, not to reinvent the wheel, I had a look at the provided examples, and figured Example 7 would be a good basis for my requirements. Nevertheless, significant updates needed to be done to make the code useful for my purposes. These include:
Part A: Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Assuming that approx 20-25% of the weight is protein, that leaves us with 100-125g protein in a 500 gram meat piece. The average amino acid molar mass is approx. 100 g/mol. With Avogadro’s number 6.022×1023 the number of molecules is between: 1 x 6.022×1023 = 6.022×1023 and 1.25 x 6.022×1023 = 7.5 ×1023 (approx)
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. The Protein can be found with this link The Protein sequence is:
DNA Assembly Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? I didn’t find anything pertaining the matter in the protocol itself, though a quick websearch (Link 1, Link 2), revealed that the PCR Master Mix contains 4 main ingreadients.
Part 1 What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? The key advantage of intracellular analog neural networks (IANNs) over traditional genetic circuits lies in the shift from discrete logic to continuous computation. Classical genetic circuits are typically engineered as Boolean systems: inputs are interpreted as “on” or “off”. This abstraction is convenient for engineering and design, but it is fundamentally misaligned with how biology actually operates, where signals exist as continuously varying concentrations and reaction rates.
Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis provides major advantages over traditional in vivo protein expression because the reaction occurs outside living cells, giving researchers direct control over reaction conditions and components. Variables such as DNA concentration, salts, energy substrates, cofactors, temperature, and additives can be adjusted independently without needing to maintain cell viability, allowing rapid optimization and faster experimental iteration. Another key advantage is flexibility: proteins can be expressed immediately after adding DNA templates, without time-consuming cloning, transformation, or cell culturing steps. Cell-free systems also allow incorporation of non-natural amino acids, toxic proteins, or synthetic circuits that would otherwise harm or interfere with living cells. Two important cases where cell-free expression is more beneficial than cell-based production are:
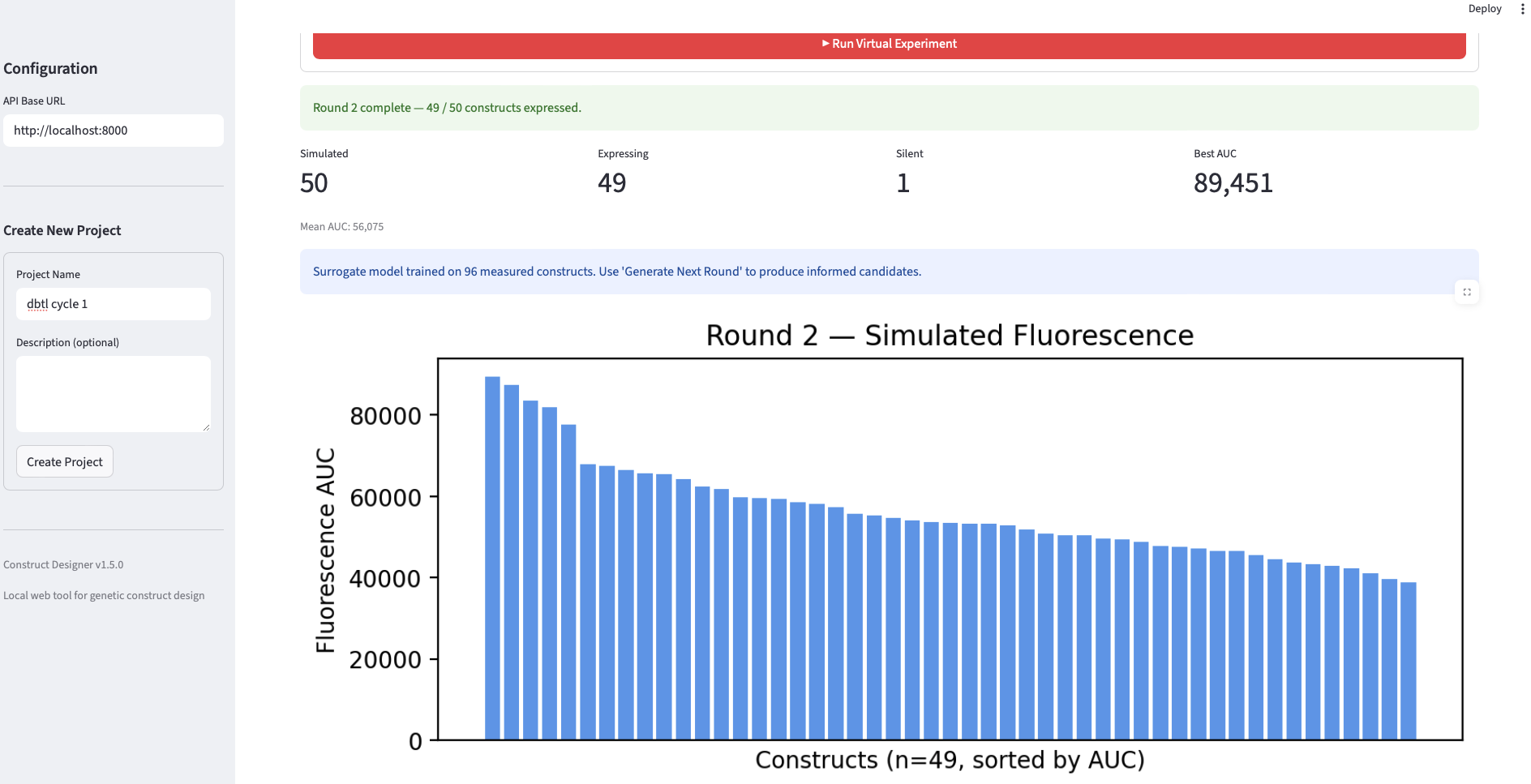
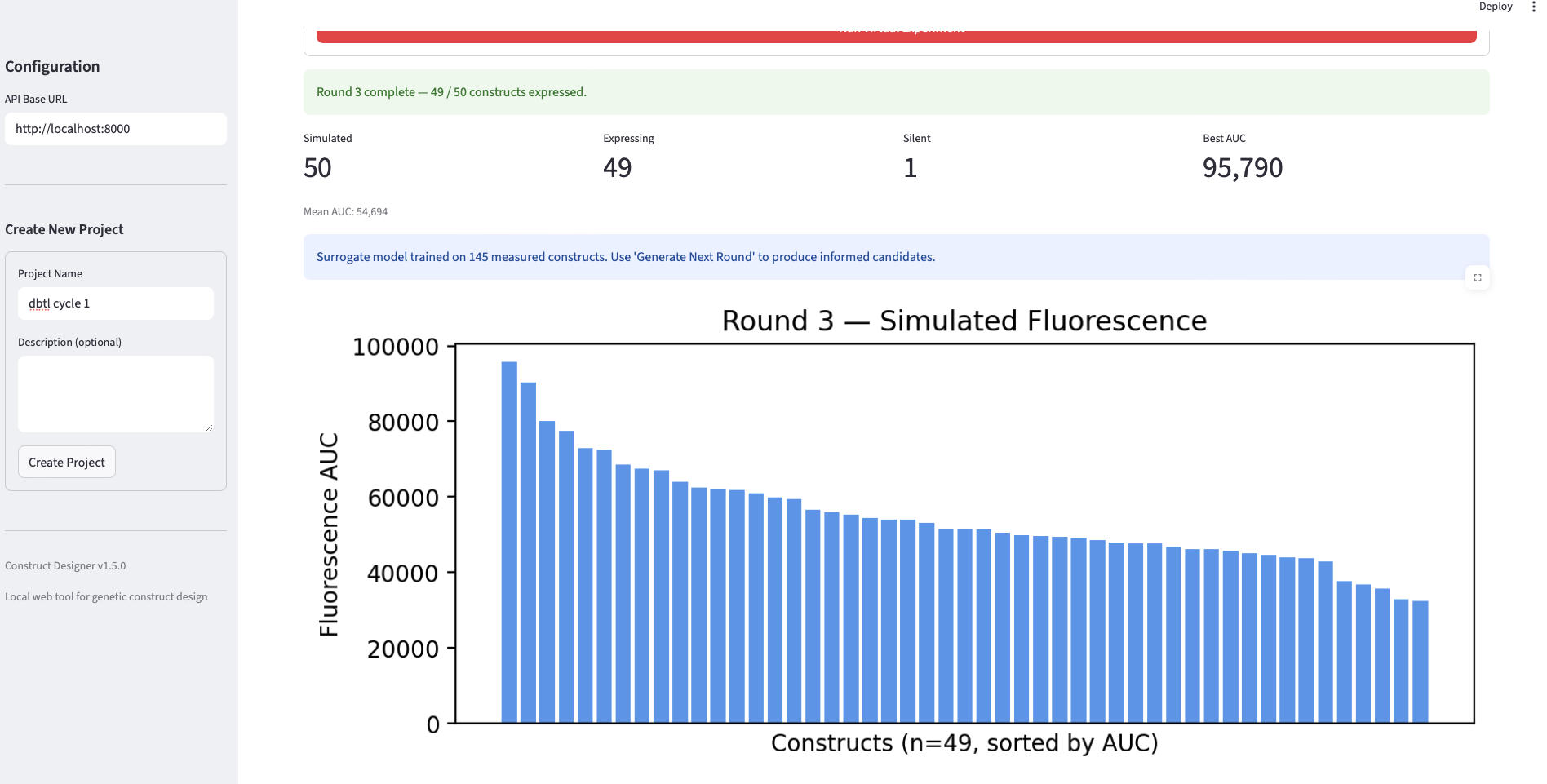
Final Project Measurement Plan The project’s central question — do AI-guided designs outperform standard, random, and unguided foundation-model designs in cell-free expression? — requires measurements at three levels: the DNA (to confirm we test what we designed), the protein output (the primary readout feeding the surrogate), and the surrogate model itself (to know whether the loop is learning).
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse. If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉 Let me try to become a TA for How to biomanufacture almost anything
Homework: Finish your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners) Done ;)
Subsections of Homework
Week 1 HW: Principles and Practices
Homework Week 1: Class Assignment
Biological Engineering Application
First Steps towards “Intelligence in a (warehouse)-dish”
Guided by the vision of building a biological general computing system, the goal of the proposed tool is to provide a minimal, yet replicable brain organoid based system, that can be engineered to exhibit controllable, learning-like signal processing behaviour. The system consists of 3 conceptual parts (input - computation - output), that manifest in 2 integrated physical devices.
Firstly, there is the 3D organoid culture chamber that handles the computation. Brain organoids, based on iPSC cultures, with a diameter below 500 micrometer, containing less than 100,000 cells can be manufactured with a high degree of standardization and scalability. More recent research allowed the long-term culture of brain organoids exceeding one year, displaying spontaneous electrophysical (re)activity, show extensive myelination, and can be enriched with several relevant cell types, e.g. oligodendrocytes, microglia, and astrocytes [1, 2, 3, 4].
Second is the input-output system that handles the input and output functions. As current research focuses on avascular brain organoids that require delivery of nutrients via diffusion, a higher order, more complex brain organoid requires brain vasculature. The microfluidic system mimics above-mentioned vasculature and contributes to the development of higher-order brain organoids. Additionally, the system can deliver and record chemical signals in a spatiotemporal manner [1, 5, 6]. Another activation mechanism lies in 3D microelectronic arrays. These allow precise stimulation and recording of spatiotemporal signals across the entire surface of the brain organoid [7].
Therefore, combining advances in 3D culture of brain organoids, with a combination of microfluidics and microelectronic arrays, poses an exciting research avenue and aims to contribute to the research topic of Organoid Intelligence (OI) [1].
Minimize the harm on the biological system through careful research design, consideration of biological responses and sensibility at the intersection of the research goal and societal norms.
Biological harm reduction emphasizes preserving the physiological integrity of the organoid system on the biological level. The intent is to avoid inducing unnecessary stress, damage or pathological stress in living tissue and to ensure that experimental interactions remain compatible with healthy biological function.
Limitation of harm arising from emergent properties seeks to prevent unintended transitions toward higher-order dynamics that could raise ethical concerns, as organoids exhibit more complex, self-organizing behavior. This includes constraining system complexity and maintaining clear boundaries on the duration and scope of experiments, as well as ensuring that organoids are not maintained beyond their justified research purpose.
Donor rights
Ensuring that individuals that contribute their biological samples for research retain fair rights, autonomy and benefits and are protected again extractive behaviour of third parties though the entire lifecycle and downstream applications.
Transparent disclosure of organoid intelligence research ensures that donors clearly understand that their biological material may be used to generate brain organoids for learning-like signal processing and computational experimentation.
Fairness in benefit sharing and protection against discrimination aims to include donors into the benefits generated by the research, nor expose them to harm through stigmatization, profiling, or inequitable use of data derived from their biological contributions. Donor participation should not create asymmetries where value is extracted without corresponding ethical consideration.
Privacy preservation protects donors from identity linkage, misuse or inappropriate inference of personal traits.
Access
Ensuring that access to the systems themselves as well as the associated knowledge and benefits are disseminated in a fair way, while extractive use attempts are actively prevented.
Equitable research access seeks to avoid concentration of organoid intelligence capabilities within a small number of research groups and institutions. The intent is to enable participation by a diverse range of (also non-scientific) communities.
Non-exclusive access tries to ensure that foundational biological systems and insights won’t be locked behind proprietary structures. The goal is to preserve openness at the level of core knowledge and enabling technologies.
Limiting extractive use ensures that access to sensitive biological data does not enable exploitation of contributing or downstream affected individuals. This goal emphasizes that organoid intelligence research should generate value that is aligned with societal benefit.
Governance Actions
Action 1: Technically Enforced Graduated Freedom
Purpose: Redefining the locus of governance from external to internal. The goal of this action is to embed harm reduction directly into the technical architecture, while still preserving scientific flexibility. Instead of imposing rigid limits, the system provides ethical “factory settings” that enable safe and broadly acceptable use by default, while allowing controlled exploration beyond these settings when justified.
Design: The organoid computing platform is developed with a set of default operating parameters, e.g. size, culture duration, stimulation intensity, and learning persistence. These can be used without additional ethical review.
At the same time, a clearly defined subset of parameters is designated as research-variable, allowing researchers to intentionally explore higher complexity, longer duration, or altered learning dynamics. Deviations beyond default settings require explicit justification and appropriate ethical oversight, but are technically supported rather than prohibited. The system logs when and how parameters are modified.
Assumptions: It is assumed that most researchers will operate within default settings unless there is a genuine scientific reason to deviate.. It also assumes that technical transparency (rather than hard locks) is an effective governance lever.
Risks of Failure & “Success”: The model fails if defaults are treated without care rather than minimum safeguards, or if parameter variation becomes routine without oversight. Successful use of the system could create a false sense of ethical safety. There is also a risk that logging is perceived as surveillance, which in turn would discourage experimentation.
Action 2: Reciprocal Donor Stewardship
Purpose: Current consent frameworks mostly are a one-time action, offering limited protection against extractive use. This action proposes a reciprocal donor stewardship model, in which the collecting institution acts as a fiduciary to protect the donor’s interests, but also to maintain a two-way informational relationship. Donors are recognized as long-term stakeholders.
Design: Universities and biobanks adopt stewardship responsibilities as a condition for ethical approval and public funding. Donors opt into a structured relationship that includes regular high-level updates on relevant scientific developments as well as personalized notifications when findings derived from their samples may have health relevance.
Assumptions: This model assumes donors want an ongoing relationship. Furthermore it assumes that institutions can responsibly manage communication. It also assumes that research findings can be meaningfully categorized into general scientific updates versus personally relevant information.
Risks of Failure & “Success”: The model may fail if institutions lack the willingness to maintain long-term engagement. There is also a risk that donors misinterpret research signals as medical diagnoses, causing anxiety or harm. Successful implementation could blur the boundary between research and clinical care.
Action 3: Simple Public-Interest Licensing
Purpose: Biological computing moves toward commercialization, this creates the option that foundational technologies become locked behind exclusive or opaque licensing arrangements. The goal of this action is to preserve the public-interest while enabling rapid and practical commercialization, ensuring that ethical constraints do not themselves become barriers to innovation.
Design: Universities and spin-outs adopt standardized, plain-language public-interest licenses. These licenses are intentionally short, unambiguous, and easy to interpret, defining only a small number of clearly prohibited applications, while leaving all other commercial uses unrestricted. Investors and companies opt in upfront, gaining predictability.
Assumptions: This approach assumes that ethical constraints can be expressed in a small number of clear, enforceable prohibitions. It also assumes that companies and investors value legal certainty and speed of commercialization enough to accept modest limits on exclusivity and application scope.
Risks of Failure & “Success”: The model fails if prohibited-use categories are defined too broadly or too minimally. Conversely, “success” could lead to widespread adoption, which may normalize these constraints.
Scoring Matrix
Does the option:
Tecnically Enforced
Reciprocal Stewardship
Simple Licensing
Harm Reduction
1
N/A
2
• Biological harm reduction
1
N/A
2
• Limitation of harm arising from emergent properties
1
N/A
3
Donor rights
N/A
1
2
• Transparent disclosure of organoid intelligence research
N/A
1
3
• Fairness in benefit sharing and protection against discrimination
N/A
1
1
• Privacy preservation
N/A
1
2
Access
N/A
1
1
• Equitable research access
N/A
2
2
• Non-exclusive access
N/A
2
1
• Limiting extractive use
N/A
2
2
Recommendation to ethics boards
For research on organoid intelligence, ethics boards should prioritize governance mechanisms that operate at the point of experimental design and focus on setting default use behaviour. Based on the policy goals of harm reduction, donor rights, and access, I recommend ethics boards focus primarily on Action 1 (Technically Enforced Graduated Freedom) for the experiment design and Action 2 (Reciprocal Donor Stewardship) ensuring a modern relationship management. Action 3 (Simple Public-Interest Licensing) will become more relevant in the near future, therefore it should be considered down the road.
First, ethics boards should require technically enforced ethical defaults for organoid intelligence systems. Rather than relying on lengthy binary approval decisions, projects should be judged on justification of any intended deviations. Second, ethics boards should transition from one-time consent towards reciprocal stewardship plans. These plans should treat donors as long-term stakeholders engaging in two-way communication when findings may be personally relevant. This strengthens donor autonomy and public trust without conflating research with clinical care. While ethics boards do not manage IP, they should recommend investigators to include public interest licensing in their research lifecycle.
The risk that overly cautious governance discourages legitimate research. These uncertainties argue for graduated, revisitable oversight rather than rigid prohibitions.
Use of Generative AI
Generative AI was used as a drafting aid throughout the development of this homework assignment. Specifically, it supported the structuring and refinement of complex ideas at the intersection of organoid intelligence, and governance, including the logical separation and articulation of policy goals and governance actions. The AI was used to iteratively clarify language and explore alternative framings, while all substantive ideas, judgments, and final decisions were made by the author.
Week 2 Lecture Prep
Prof. Dr. Jacobson
Nature’s machinery for copying DNA is called polymerase.
What is the error rate of polymerase?
The error rate is 1:10E6 (see page 8, right side “biological systhesis”)
How does this compare to the length of the human genome?
The human genome has a length of 3.2 Gbp or 3.2 x 10E9 (see page 10 diagram)
How does biology deal with that discrepancy?
The human body engages in a “proofreading” process (page 8). The error correction process, called “MutS Repair System” is described in detail in Lamers et al. Nature 407:711 (2000) (page 14)
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The average human protein is 1036 bp, equating to 1036 / 3 = 345 amino acids. On average 3 codons encode an amino acid, therefore the number of number of different DNA sequences encoding the same 345 amino acids is 3 x 10E345.
Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite chemistry. Page 2 illustrates the 4 step process. See the historical development from page 9.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Not directly found in the slides. Searching further ressources says the fundamental issue is cumulative coupling efficiency loss. Each cycle has a coupling efficiency of ~99–99.5%. The overall yield is the stepwise efficiency raised to the power of the oligo length: for a 200-mer at 99.5% coupling efficiency, the yield of full-length product is roughly 37%.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Same rational as answer above. Decay is exponential, therefore the efficiency drops to negligible levels.
Prof. Dr. George Church
Using Google & Prof. Church’s slide #4: What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Opus 4.6 Prompt: How does this relate to Lysine Deficiency?
The connection to Jurassic Park wasn’t clear to me, as i haven’t seen the movie. Therefore here is Opus answer (which was an amusing read): “The “Lysine Contingency” from Jurassic Park
In the film/novel, the engineers claim dinosaurs were designed to be lysine-deficient — unable to produce lysine — so they’d die without supplemental lysine provided by the park, serving as a biocontainment mechanism.
Why It Doesn’t Work
The “contingency” is essentially meaningless as a containment strategy because lysine is already an essential amino acid for all animals. No animal synthesizes its own lysine. The dinosaurs wouldn’t be any more dependent on external lysine than a cow, a bird, or a human. Any environment with protein-containing food sources (plants, prey, insects) would supply adequate lysine. The dinosaurs would simply eat and obtain lysine the same way every other animal does.
Connection to Church’s Work
Interestingly, slide #4’s emphasis on “knowing your monomers” and the references to non-standard amino acids (NSAAs) and xenomicrobiology (genetic code engineering, referenced from the 2016 Microb. Biotech paper) point to what actual biocontainment might look like. As shown in slides 39–42, Church’s lab has built Genomically Recoded Organisms (GROs) that depend on NSAAs like BipA — amino acids that truly do not exist in nature. This is a real lysine contingency done right: organisms that require a synthetic amino acid unavailable in any natural food web, providing genuine genetic and metabolic isolation.
So Crichton’s fictional biocontainment was flawed at the basic biochemistry level, but Church’s lab has essentially built the version that actually works.”
Week 2 HW: DNA Read, Write & Edit
Part 0: Basics of Gel Electrophoresis
Watch Week 2 Lecture (Zoom)
Watch Week 2 Recitation (Zoom)
Watch BioBootcamp Day 1 - Day 3 (Zoom)
Part 1: Benchling & In-silico Gel Art
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Artwork
After struggling quite some time with the task of creating artwork with the limited amount of restriction enzymes, in the end decided to stick to a relatively easy and repetitive pattern that with a little imagination has a lot of versatile interpretations:
It can be two friends hanging out
It can be DNA (or at least a rought estimation of the firrst two loops)
To help you visualize it a bit better i created some generative AI art
To create the loop like structure, I used the Restriction enzymees BamHI - KpnI - SalI - KpnI - BamHI
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
not relevant as I don’t have access to a lab
Part 3: DNA Design Challenge
3.1. Choose your protein.
For Week 2 Homework I choose the Growth/differentiation factor 8 (short GDF-8), also known as human myostatin protein.
I choose myostatin for the inital reason, that it was the first protein that came to mind. Being known for the viral video of Jo Zayner injecting the DIY-Gene Therapy to knock out the myostatin associated gene, or the many pictures of muscled animals, like cattle and dogs.
Digging further, myostatin seemed to be a good choice, not only for it’s fame. It is a well studied protein, with a clear function to negatively regulate muscle growth, as seen with the example of the “jacked” bagle or the cattle. Furthermore myostatin is not only interesting to biohackers and instagram scrollers, it has actual therapeutical interest, and is actively researched to combat muscular dystrophy. In the up and coming field of Longevity, myostatin is researched to mitigate age-related muscle loss.
3.2 Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Using the Reverse Translate tool from Bioinformatics.org, I got the following results:
>reverse translation of sp|O14793|GDF8_HUMAN Growth/differentiation factor 8 OS=Homo sapiens OX=9606 GN=MSTN PE=1 SV=1 to a 1125 base sequence of most likely codons.
atgcagaaactgcagctgtgcgtgtatatttatctgtttatgctgattgtggcgggcccg
gtggatctgaacgaaaacagcgaacagaaagaaaacgtggaaaaagaaggcctgtgcaac
gcgtgcacctggcgccagaacaccaaaagcagccgcattgaagcgattaaaattcagatt
ctgagcaaactgcgcctggaaaccgcgccgaacattagcaaagatgtgattcgccagctg
ctgccgaaagcgccgccgctgcgcgaactgattgatcagtatgatgtgcagcgcgatgat
agcagcgatggcagcctggaagatgatgattatcatgcgaccaccgaaaccattattacc
atgccgaccgaaagcgattttctgatgcaggtggatggcaaaccgaaatgctgctttttt
aaatttagcagcaaaattcagtataacaaagtggtgaaagcgcagctgtggatttatctg
cgcccggtggaaaccccgaccaccgtgtttgtgcagattctgcgcctgattaaaccgatg
aaagatggcacccgctataccggcattcgcagcctgaaactggatatgaacccgggcacc
ggcatttggcagagcattgatgtgaaaaccgtgctgcagaactggctgaaacagccggaa
agcaacctgggcattgaaattaaagcgctggatgaaaacggccatgatctggcggtgacc
tttccgggcccgggcgaagatggcctgaacccgtttctggaagtgaaagtgaccgatacc
ccgaaacgcagccgccgcgattttggcctggattgcgatgaacatagcaccgaaagccgc
tgctgccgctatccgctgaccgtggattttgaagcgtttggctgggattggattattgcg
ccgaaacgctataaagcgaactattgcagcggcgaatgcgaatttgtgtttctgcagaaa
tatccgcatacccatctggtgcatcaggcgaacccgcgcggcagcgcgggcccgtgctgc
accccgaccaaaatgagcccgattaacatgctgtattttaacggcaaagaacagattatt
tatggcaaaattccggcgatggtggtggatcgctgcggctgcagc
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Organisms have different procivities for using certain codons. Some codons are used more frequently in one orgamis. So for the same amino acide certain codons are “preferred” over others. If the inserted DNA matches the preferred codons of the organism more translation happens. In my case, as the protein I’m trying to express is of human origin, though expressed in e.coli (common for early experiments), many of the codons common in homo sapiens, are rare in e.coli.
Using the Codon Optimization tool from VectorBuilder, and choosing e.coli K-12 as the organism i get the following optimized codon:
ATGCAGAAACTGCAGCTGTGCGTTTACATTTATCTGTTCATGCTGATTGTGGCCGGCCCGGTGGATCTGAACGAAAACAGTGAACAGAAAGAAAACGTGGAAAAAGAAGGTCTGTGCAACGCCTGTACCTGGCGCCAGAATACCAAATCGAGCCGCATTGAAGCCATTAAAATTCAGATCCTGTCAAAACTGCGTCTGGAAACCGCGCCGAATATTAGCAAAGATGTGATCCGTCAGCTGCTGCCGAAAGCCCCGCCGCTGCGTGAACTGATTGATCAGTATGATGTGCAGCGCGATGATAGCAGCGATGGCAGCCTGGAAGATGATGATTATCACGCGACCACCGAAACCATTATTACCATGCCGACCGAAAGCGATTTTCTGATGCAGGTGGATGGCAAACCGAAATGCTGCTTCTTCAAATTTAGCTCGAAAATTCAATATAATAAAGTGGTGAAAGCGCAGCTGTGGATCTATCTGCGCCCGGTGGAAACCCCGACCACCGTGTTTGTGCAGATTCTGCGCCTGATTAAACCGATGAAAGATGGCACCCGCTACACCGGCATTCGCAGCCTGAAACTGGATATGAACCCGGGCACCGGCATCTGGCAGAGCATTGATGTGAAAACCGTTCTGCAGAATTGGCTGAAACAGCCGGAAAGCAACCTGGGCATTGAAATTAAAGCCCTGGATGAAAATGGCCATGATCTGGCAGTGACCTTTCCGGGCCCGGGCGAAGATGGCCTGAATCCGTTCCTGGAAGTGAAAGTGACCGATACCCCGAAACGCAGCCGCCGCGACTTTGGCCTGGATTGCGATGAACACAGCACCGAAAGCCGCTGCTGCCGCTACCCGCTGACCGTGGATTTTGAAGCGTTCGGCTGGGATTGGATTATTGCGCCGAAACGCTATAAGGCGAACTACTGCAGCGGTGAATGCGAATTTGTGTTTCTGCAGAAATATCCGCACACCCATCTGGTGCACCAGGCAAACCCGCGCGGCAGCGCGGGCCCGTGCTGTACCCCGACCAAAATGAGCCCGATTAACATGCTGTATTTTAACGGCAAAGAACAGATTATCTATGGCAAAATCCCGGCGATGGTTGTGGATCGCTGCGGTTGTAGC
while avoiding cleavage sites of restriction enzymes: BamHI HindIII
Whether the e.coli is the proper host for this application is debateable, as e.coli lacks the post-translational modification capabilities. Other hosts like CHO cells or human cells, could prove to be a better choice, if one aims for a properly folded, functioning protein.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Let me focus on cell-dependent methods. There are several expression systems like e.coli, yeast and mamalian cells. Each system has their pros and cons. E.coli is a procaryote, it’s cheap, fast and well established, though it lacks post translational modification abilities.
Therefore the myostatin might be misfolded. Addtionally the cells have to be lysed to get to the myostatin.
Yeast is eukaryotic, therefore it has some post-translational folding capabilities, additionally secretion is possible.
Mammalian cells offer human-like folding, and secretion, though they grow slower, are a-lot harder to handle, offer lower yields and are significantly more expensive than e.coli or yeast.
To get the DNA into the orgamism:
Perform PCR on the optimized myostatin DNA strand, to generate many copies of said gene. make sure the DNA strand is flanked with BanHI and HindIII sites.
Open the plasmid at the previously avoided restriction sites (BamHI HindIII), creating compatible sticky ends.
Cut the DNA strand with the same restriction enzymes
Mix plasmid and DNA strand, introduce ligase enzymes and ATP, to connect the matching sticky ends.
Transform the plasmid into e.coli using common transformation methods, e.g. heat-shock or electroporation
(optional) screen for uptake of plasmid
Having chosen e.coli here the DNA would transscribe into mRNA, which in turn would translate into myostating facilitated by ribosomes.
For lab-sized fermentation, a batch system is sufficient, on industrial scale a fed-batch system would be used.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
Create Twist Account
Create Benchling Account
4.2. Build Your DNA Insert Sequence
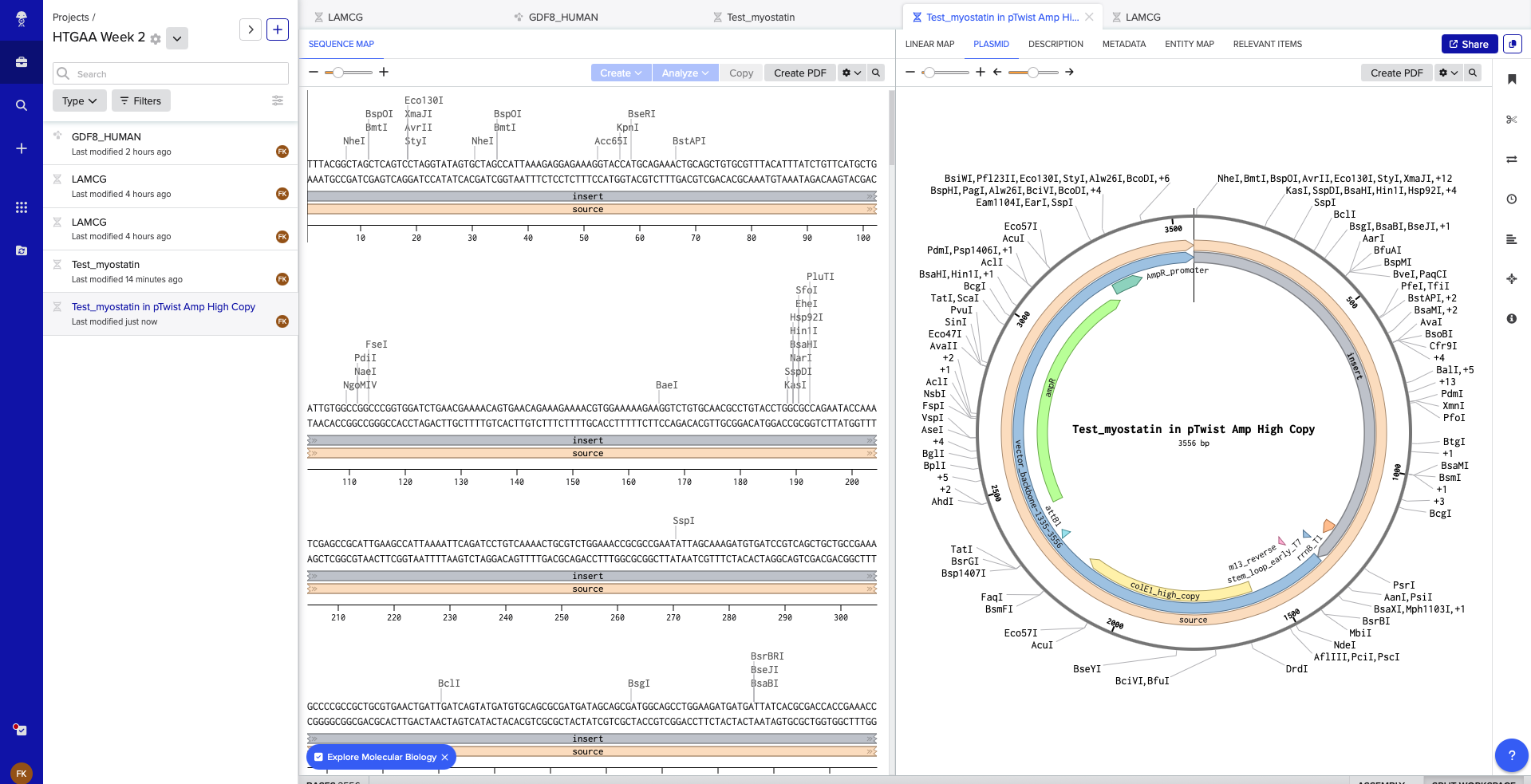
Following the example on the course site, this is the linear map of the sequence:
In Twist having chosen the Clonal Gene, with the vector: pTwist Amp High Copy, this is the output from Twist
Following is the final Clonal Gene in Benchling
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I’m interested in genetic origin for muscle growth. People have different outcomes for the similar inputs, I’m interested into the marginal influence Gene’s have on ones physiological development. While overall health is important, this also has clinical application, e.g. for patients with muscle loss diseases, age-related muscle atrophy.
Therefore a number of genes can be studied. Using search and LLMs, these are relevant proteins for muscle growth,
Myostatin: given that my Design a Gene Challenge was about this, it makes sense to study this gene. I suspect mutations in the gene lead to enhanced or reduced function.
ACTN3: Determines between “fast” or “slow” muscle fiber twitches. They determine whether one is dispositioned for heavy lifting or endurance.
IGF1: This gene expresses a growth factor that aids muscle repair and their growtn after exercise. People with different versions might respond differently to certain training styles.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I’d choose NGS sequencing, as I’m interested in targeted genes. NGS allows for a good middle ground of cost, and accuracy, and also has established protocols for variant detetction.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
Second-generation sequencing. DNA fragments are sequenced simultaniously in massively paralellized fashion.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
With NGS library prep is necessary. First DNA is extracted. Then the DNA is fragmented into smaller chunks (around 300-500 bp). Next adapter ligation is used to attache each DNA chunk to the coded oligonucleotide on the flow plate. PCR afterwards is optional
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
NGS sequencing takes DNA and fragments it, then ligates it to adapter sequences, which add to a sequencing library. The fragments bind to a flow cell, then aplified to create clusters of identical DNA strands.
The gene strand is sequenced by synthesizing a complementary strand using labeled nucleotides, and only one base is incorporated per cycle. After incorporation the flow cell is imaged, and the specific fluoresence is used to identify the added base.
What is the output of your chosen sequencing technology?
A FASTQ files, with a large number of small DNA fragment readouts. Additionally these fragements are then aligned and referenced. These can then be screened for differences.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
A synthetic DNA construct that can be injected locally, that will express miRNAs that silence myostatin mRNA, to allow for temporary enhanced muscle growth. This would pose a therapeutical intervention, either for biohackers or patients with degenerative muscle diseases.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
I’d probably choose Phosphoramidite Chemistry, as its the current gold standard and ample for smaller DNA fragments.
What are the essential steps of your chosen sequencing methods?What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
I’m a bit confused why sequencing instead of synthesis methods are mentioned. So assuming that synthesis is meant, I’ll talk about the synthesis based on Phosphoramidite Chemistry
The essential steps with PC is repeating the so called Coupling cycle, that is repeated for each base. First the protecting group gets removed (deetritylation), theen the next phosphoramidite nucleeatide is added (coupling), the unreacted 5’ groups get blocked (cappping), lastly unstable phosphite is converted to stable phosphate (oxidation). This way fragemnts up to 200bp are synthesised and later assembled.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Going with the theme a reversible knockout for myostatin expression. It could be an alternative treatment for patients with degenerative muscular diseases as well as biohackers.
(ii) What technology or technologies would you use to perform these DNA edits and why?
To ensure reversability, I’d consider base editors, introducing stop codons into the myostatin coding sequence. I’d chose base editors as they make single-nucleotide changes without cutting DNA, to reduce the chance of unwanted off-target effects, compared to classical CRISPR-Cas9 technology.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
First Guide RNA detects a specific myostatin sequence, then guides the Base editor there. Next a base conversion occures, where e.g. a cytosine base editor converts a C to a T. With this edit normal codons are converted to pre-mature stop codons. This leads to the myostatin being misformed and ideally not bioactive.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Select a target sequence to turn a normal codon into a premature stop codon, e.g. CGA to TGA. Next you have to design a Guide RNA to position the base editor at the chosen target. Next check whether there are any off target effects.
I’d need the DNA of the base editor, the DNA Template for the guide RNA, an plasmid that integrates the base editor with the guideRNA, restricition and ligation enzymes. For injection into humans, probably also the targeted muscle cells as well as culture media and transfection equipment.
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Base editing has some key limitation, the most severe one being the low efficiency compared to classical CRISPR methods.
Human cells are notoriously difficult to work with, muscle cells are some of the hardest to edit in humans. The delivery of the editor to muscle cells poses another challenge. The base editors have limited target options, based on their capability to make edits in a narrow basepair window. Off target effects are also a concern. Also it needs to be checked, that the editor only comes in contact with the intended C to T conversion as it converts Cs indiscriminantly.
Use of Generative AI
Generative AI was used as a drafting aid throughout the development of this homework assignment. Specifically, it supported the structuring and refinement of complex ideas at the of DNA Design as well as aiding the understanding of DNA Read, Write and Edit technologies. The AI was used to iteratively clarify language and explore alternative framings, while all substantive ideas, judgments, and final decisions were made by the author.
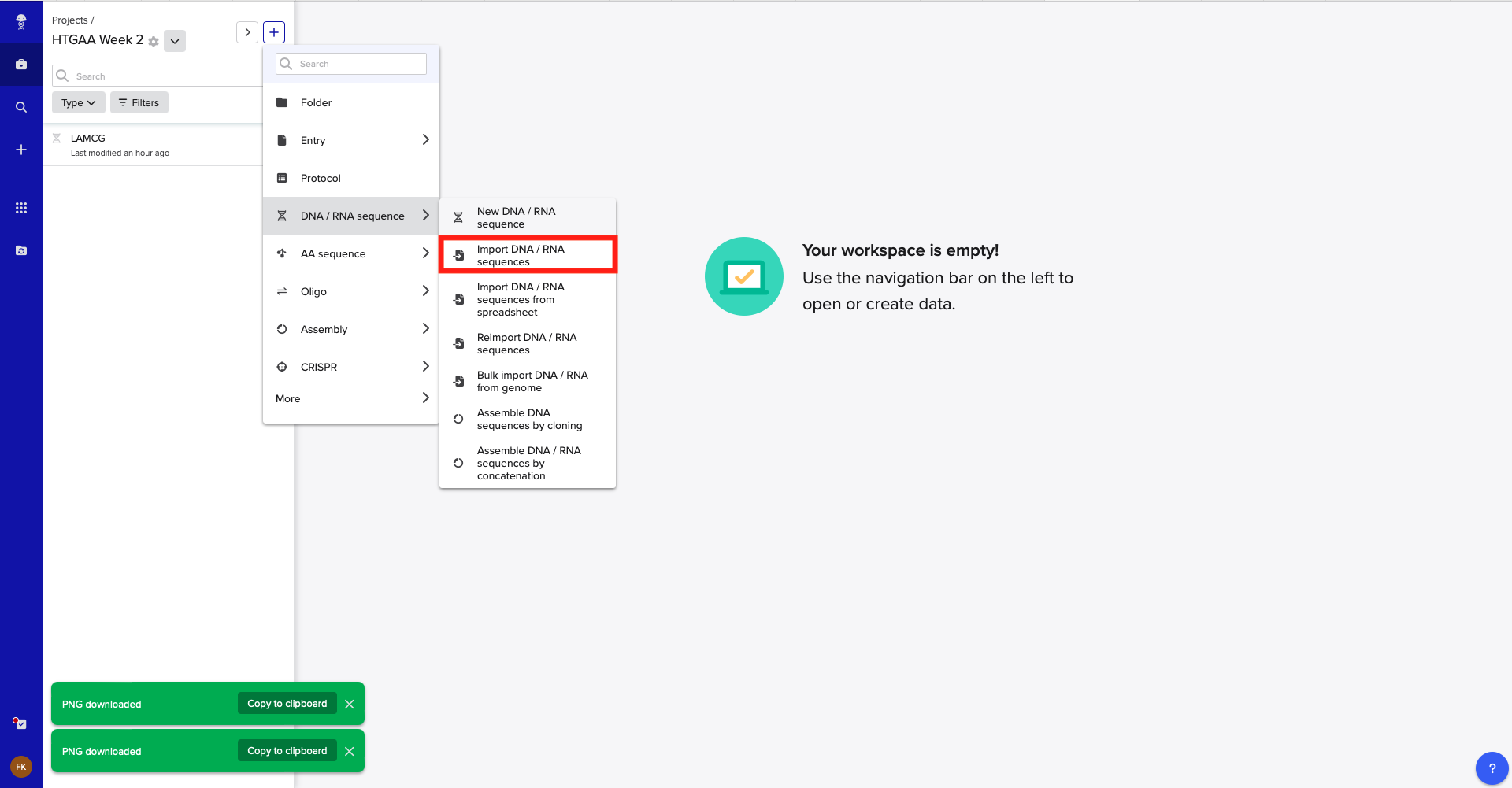
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
I generated a quick design using the above mentioned tool: BioPunk Initials
See the:
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that:
If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
As its good practice in Software Engineering, not to reinvent the wheel, I had a look at the provided examples, and figured Example 7 would be a good basis for my requirements. Nevertheless, significant updates needed to be done to make the code useful for my purposes. These include:
Importing necessary libraries
Converting CSV file reading to Pandas Dataframe
Removing inversion of y-data
Changing the color scheme
###
### YOUR CODE HERE to create your design
###
# Using nonstandard color setup - will not run on a robot with this lab's standard deck setup.
well_colors = {
'B1' : 'Green'
}
# Converting coordinates for the design to Opentrons usable dataframe
bp_coord = [(-19.8,17.6), (-17.6,17.6), (-15.4,17.6), (-13.2,17.6), (-11,17.6), (2.2,17.6), (4.4,17.6), (6.6,17.6), (8.8,17.6), (11,17.6), (13.2,17.6), (-19.8,15.4), (-8.8,15.4), (2.2,15.4), (15.4,15.4), (-19.8,13.2), (-6.6,13.2), (2.2,13.2), (17.6,13.2), (-19.8,11), (-6.6,11), (2.2,11), (17.6,11), (-19.8,8.8), (-6.6,8.8), (2.2,8.8), (17.6,8.8), (-19.8,6.6), (-6.6,6.6), (2.2,6.6), (17.6,6.6), (-19.8,4.4), (-6.6,4.4), (2.2,4.4), (17.6,4.4), (-19.8,2.2), (-6.6,2.2), (2.2,2.2), (17.6,2.2), (-19.8,0), (-8.8,0), (2.2,0), (15.4,0), (-19.8,-2.2), (-17.6,-2.2), (-15.4,-2.2), (-13.2,-2.2), (-11,-2.2), (2.2,-2.2), (4.4,-2.2), (6.6,-2.2), (8.8,-2.2), (11,-2.2), (13.2,-2.2), (-19.8,-4.4), (-8.8,-4.4), (2.2,-4.4), (-19.8,-6.6), (-6.6,-6.6), (2.2,-6.6), (-19.8,-8.8), (-6.6,-8.8), (2.2,-8.8), (-19.8,-11), (-6.6,-11), (2.2,-11), (-19.8,-13.2), (-6.6,-13.2), (2.2,-13.2), (-19.8,-15.4), (-6.6,-15.4), (2.2,-15.4), (-19.8,-17.6), (-8.8,-17.6), (2.2,-17.6), (-19.8,-19.8), (-17.6,-19.8), (-15.4,-19.8), (-13.2,-19.8), (-11,-19.8), (2.2,-19.8)]
data = pd.DataFrame(bp_coord, columns=["x", "y"])
data.columns = ["x", "y"]
#Get min and max x-/y-values from my coordinates
raw_x_min = np.amin(data['x'])
raw_x_max = np.amax(data['x'])
raw_y_min = np.amin(data["y"])
raw_y_max = np.amax(data["y"])
#Shift data, so that the centerpoint 0/0 is at the center of my design
bp_x_shifted = data['x']-((raw_x_min + raw_x_max)/2)
bp_y_shifted = data["y"]-((raw_y_min + raw_y_max)/2)
all_distances_to_center = np.sqrt(np.square(bp_x_shifted) + np.square(bp_y_shifted));
bp_x_85mm_shifted = 40/np.amax(all_distances_to_center)*bp_x_shifted;
bp_y_85mm_shifted = 40/np.amax(all_distances_to_center)*bp_y_shifted;
# Get the top-center of the plate, make sure the plate was calibrated before running this
center_location = agar_plate['A1'].top()
cell_well = color_plate['B1'] # Change to location of green transformands
# Aspirate
pipette_20ul.pick_up_tip()
for i in range(len(bp_x_85mm_shifted)):
if i%20 == 0:
# pick up more every 20 uL, but only as much as we're going to need!
pipette_20ul.aspirate(min(20, len(bp_x_85mm_shifted)-i), cell_well)
adjusted_location = center_location.move(types.Point(bp_x_85mm_shifted[i], bp_y_85mm_shifted[i]))
pipette_20ul.dispense(1, adjusted_location)
hover_location = adjusted_location.move(types.Point(z = 2))
pipette_20ul.move_to(hover_location)
# Don't forget to end with a drop_tip()
pipette_20ul.drop_tip()
Submit your Python file via this form.
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
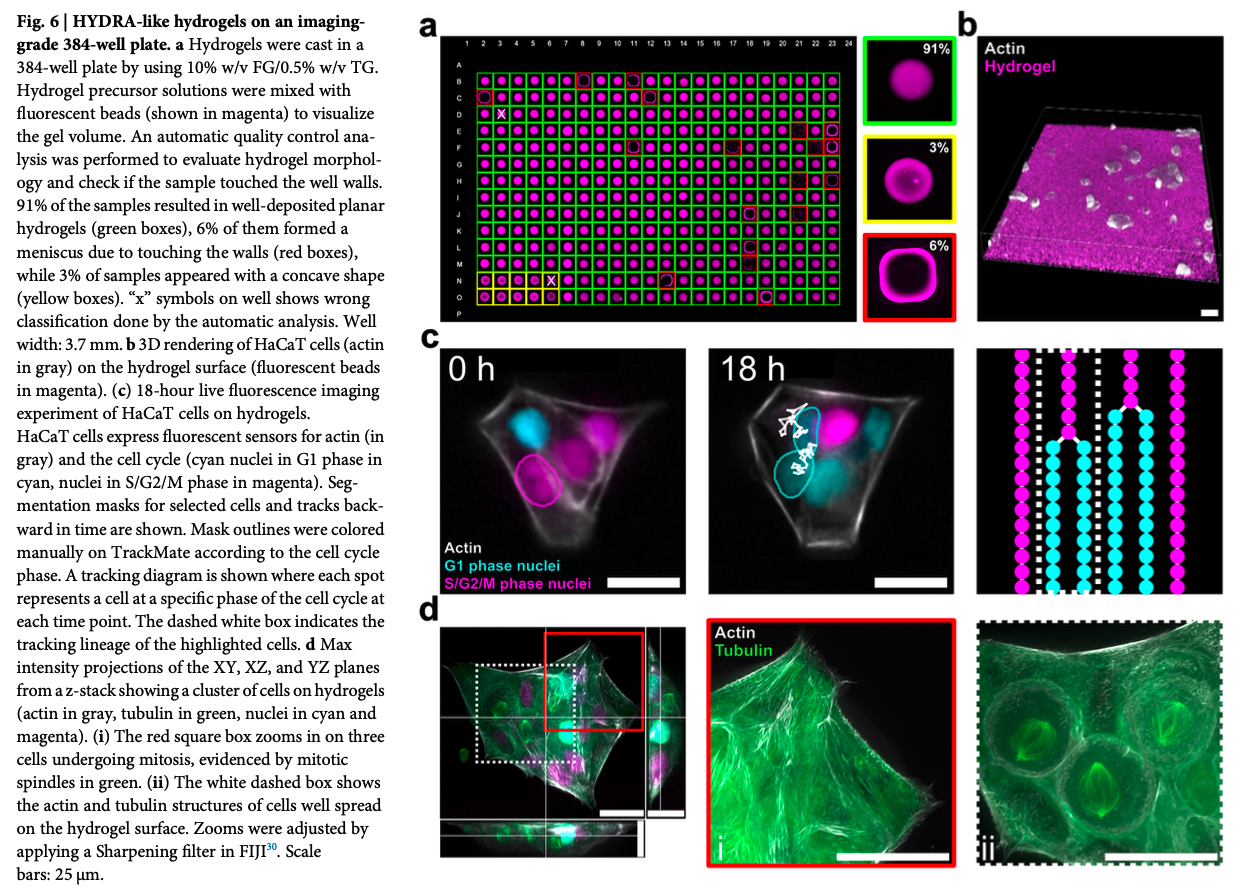
Given my background in tissue engineering I picked the paper “Fabrication of cell culture hydrogels by robotic liquid handling automation for high-throughput drug testing” published in “Communications Engineering” (2025)4:222 DOI
The paper introduces HYDRA (HYDrogels by Robotic liquid-handling Automation), a method for fabricating flat and thin hydrogel films directly in standard 96- and 384-well HTS plates using an Opentrons liquid handling machine.
The paper tackles one of the main problems in modern drug discovery. Around 50% of compounds passing preclinical assays fail human trials. This has several proposed reasons, the paper tackles one of the widely mentioned problems. In standard HTS cells are grown on plastic dishes that do not resemble the ECM structure of the human body, nor have the dynamic context (mechanical, electrical, etc.) that the ECM provides. Organ on a chip improve the missing biomimicry, but are not compatible with HTS. Additionally, they are too complex too manufacture cheaply and are incompatible with automated screening pipelines.
There are existing hydrogel coatings, but they either are so thin that they don’t provide the proper ECM environment mechanically, or so thick that they block high-resolution imaging. Both produce a curved meniscus that makes uniform cell seeding impossible.
This gap, hydrogel coatings thin enough for imaging and thick enough for mechanosensing, is tackled with HYDRA. HYDRA uses fish gelatin for its hydrogel base material, dissolved in PBS at 5-20% w/v and is cross-linked enzymatically with transglutaminase at 0.5-2% w/v. The method was demonstrated on an Opentrons, for easy accessibility, for scalability on a INTRGRA Assist Plus. The robot dispenses precalculated sub-contact volumes 96 well plates: 12 micro liter, 382 well plate: 1 micro liter), and immediately re-aspirates the volume, to archive a thin film. This is possible by using contact angle hysteresis. The volume was optimized using FE simulations. The process takes around 10 minutes for the whole plate, then the plates are gelled, and incubated at 37 degree Celsius, sterilized and swollen and rinsed in PBS. Afterwards cells can be seeded.
The hydrogel thin films are between 10-50 micrometer thickness, with tunable stiffness. Lastly the authors validated several imaging platforms (digital holographic, widefield fluorescence, and high-resolution confocal microscopy) see Fig. 1
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
Developmental biology aims to understand how tissues and cells develop from dynamic, history-dependent processes governed by interacting biochemical, electrical, mechanical cues in a temporal manner. While these properties are well established theoretically, in practice experiments lack behind by what can be executed manually. Even if automation is deployed it only addresses one of the drawbacks of manual experimentation. This forced experiments to make simplifications, such as searching only low-parameter search spaces, coarse temporal guiding and open loop design of experiments. This results in heuristic sampling and interpretation of results with lacking search spaces. Using automation directly addresses these limitations by enabling precise temporal control, systematic exploration of parameter rich search spaces, and adaptive, feedback-driven intervention. This is especially relevant in organoid and organ-on-chip systems.
Mimicking the ECM in developmental biology
Many developmental signals only have their proper effect through the timing, duration, and frequency, rather than concentration alone. Temporal regimes are rarely explored experimentally, e.g. competence windows, pulsed signaling, and oscillatory dynamics due to the impracticality of executing. Automated liquid-handling systems can change that by enabling stable, repeatable temporal execution.
Exploring the design space
Cell fate and tissue morphology have very high-dimensional development search spaces. This comes from the interaction of multiple biochemical and mechanical variables that evolve over time. Contrasting this intuition or slow sequential tuning is currently used. Automation enables systematic, multi-parameter exploration of said search space. The goal of automation is to find non-linear responses and regime changes that are not obvious in manual experiments.
Guiding Experiments
Development proceeds through continuous feedback between tissue state and signaling environment, yet most experiments are designed in an open-end fashion, with data analysis and redesign being manual and in a discrete fashion. Automation should allow experiments to close the loop, where readouts inform the next steps of the experiment. The goal is to adaptively explore tissue formation.
Final Project Ideas
As explained in this week’s recitation, add 1-3 slides in your Node’s section of this slide deck with 3 ideas you have for an Individual Final Project. Be sure to put your name, city, and country on your slide!
slides are added
Use of Generative AI
Generative AI was used as a conceptual drafting aid during the development of this project. Specifically, it supported the structuring and refinement of complex ideas related to automated biological experimentation and experimental design in tissue engineering, as well as clarifying concepts around temporal control, high-dimensional experimentation, and reproducibility in laboratory automation. The AI was used to iteratively improve clarity of language and to explore alternative conceptual framings. No generative AI was used for the implementation or coding components of the project, and all substantive ideas, technical decisions, and final judgments were made by the author.
Week 4 HW: Protein Design I
Part A: Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming that approx 20-25% of the weight is protein, that leaves us with 100-125g protein in a 500 gram meat piece. The average amino acid molar mass is approx. 100 g/mol. With Avogadro’s number 6.022×1023 the number of molecules is between:
1 x 6.022×1023 = 6.022×1023
and
1.25 x 6.022×1023 = 7.5 ×1023 (approx)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because the DNA of the food doesn’t get copied into the host. Digestion breaks proteins into amino acids or small peptides. Then the human body rebuilds new (human proteins) using DNA instructions and its own cell machinery.
Why are there only 20 natural amino acids?
As with all biology it’s probably an energy equilibrium. Biological life build itself with the smallest amount of chemical diversity it could find, e.g. charged, polar, hydrophobic, aromatic, sulfur, etc, that would make accurate encoding possible.
Can you make other non-natural amino acids? Design some new amino acids.
Chemists have shown that they can make other non-natural amino acide. Some useful ones would be
Clickable amino acid: an alanine-like side chain with an azide or alkyne group (bio-orthogonal labeling).
Photo-crosslinker: phenylalanine-like side chain bearing a benzophenone (forms covalent links under UV to map contacts).
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids could have potentially made by
Atmospheric / energy chemistry: classic spark/UV experiments can generate amino acids from simple gases.
Hydrothermal / geochemical routes: mineral surfaces can catalyze formation and concentration.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
D-amino acids form the opposite handedness, so an α-helix made of D-amino acids is left-handed.
Can you discover additional helices in proteins?
Depends on the definition on the alpha-helices. One can define an alpha-helices, so that there are helices that don’t match that group.
There are examples in nature like the 310-helices.
Why are most molecular helices right-handed?
The Chirality of building blocks is relevant, as biology uses mostly L-amino acids, which makes the right-handed α-helix sterically favorable and the left-handed version strained.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-strands have “sticky edges”, the backbone has H-bond donors/acceptors that like to be satisfied.
Additionally β-sheets can often extend by adding another strand at an exposed edge.
Many β-strands present alternating side chains, creating flat, complementary surfaces that pack well.
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
As referenced in Week 2, I’m interested in the Protein Myostatin. It’s function is to downregulate new muscle growth. That makes it an attractive protein to inhibit for biohackers as well as clinicians treating patients with muscular dystrophy diseases.
Identify the amino acid sequence of your protein.
To answer these questions, I went to Uniprot, looking up the protein (Entry ID: “O14793, GDF8_HUMAN”), I copied the sequence from the subchapter “Sequence”.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Using the Colab notebook that was provided by the HTGAA TAs (see link in the HW announcement). I copied the protein sequence into the variable protein_sequence in the notebook. After running the code block I got the answer:
The length of the protein is: 375 aminoacids.The most common amino acid is: L, which appears 33 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
With a BLAST search, it was determined that the protein had 250 homologes (see Fig. 1)
Does your protein belong to any protein family?
Myostatin belongs to the Transforming Growth Factor - beta (TGF-β) protein family.
Identify the structure page of your protein in RCSB
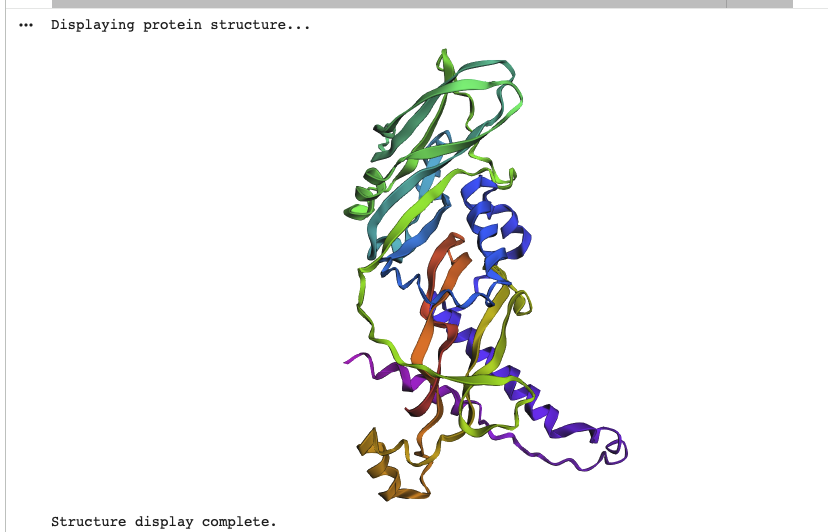
The protein I use to answer the following questions has the ID 5JI1 | pdb_00005ji1 in RCSB. This is not a protein from Homo Sapiens, but from mus musculus (a mouse). While techincally not the same protein, both proteins are very similar. The choice for the mouse protein is mainly, that there is no isolated myostatin protein from homo sapiens in RCSB.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
According to the overview page of the protein, it was deposited on the 21 April 2016 and released on the 22 March 2017.
The resolution of the structure is 2.25 Å, which is below the threshhold mentioned in the question text, therefore one can assume, that the resolution and therefore quality of the protein is good.
Are there any other molecules in the solved structure apart from protein?
As mentioned above the protein is isolated, therefore there are no other structures.
Does your protein belong to any structure classification family?
As mentioned myostatin is part of the TGF-β superfamily (growth factor, cystine-knot fold).
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
I tried to install pymol with homebrew, this failed repeatedly due to errors with the QT5 library, that manages desktop applications in python. Initializing a conda environment and installing pymol was successful though.
I read through the tutorial, though i decided to use the command line interface of pymol, as its better documented.
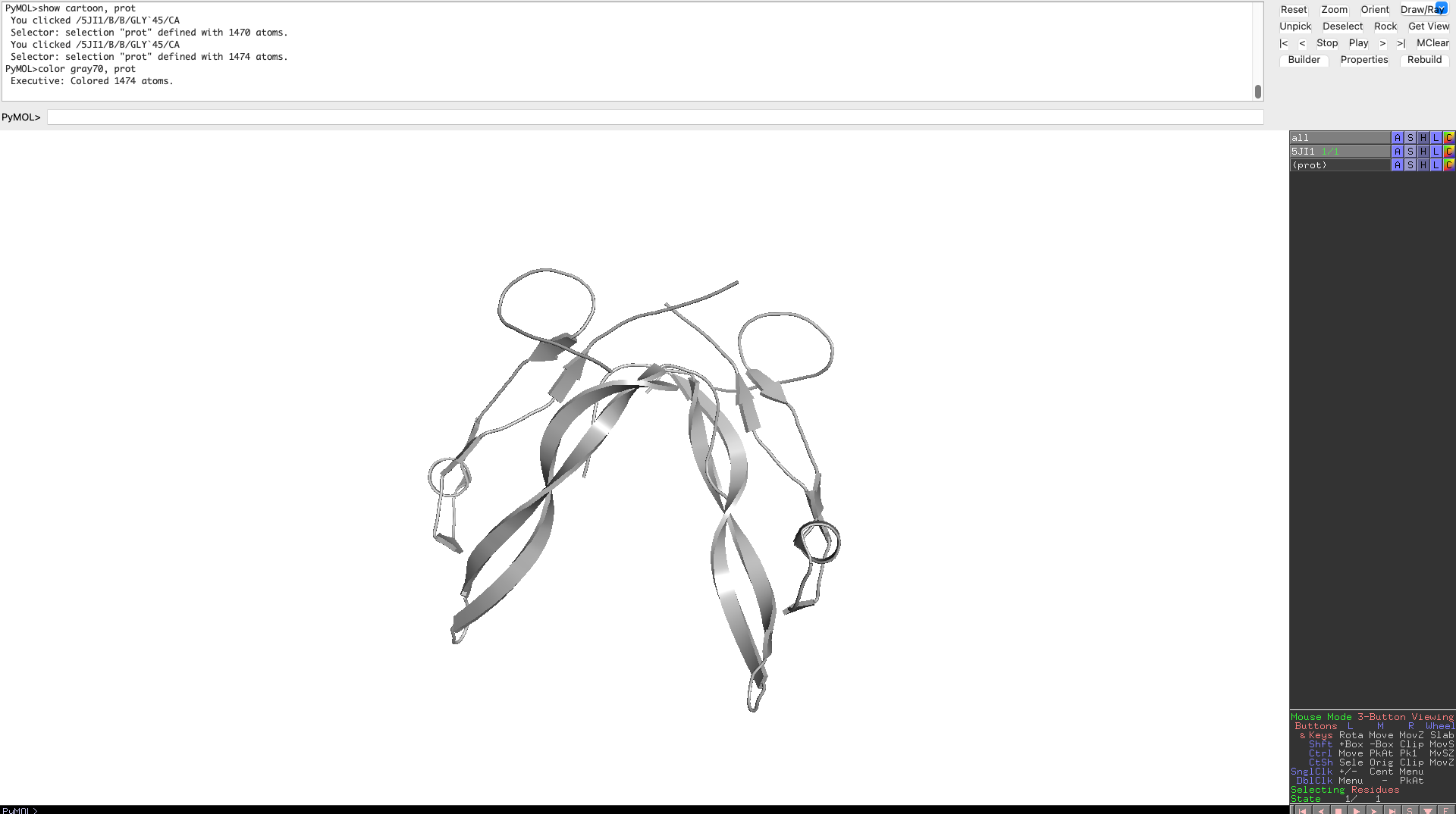
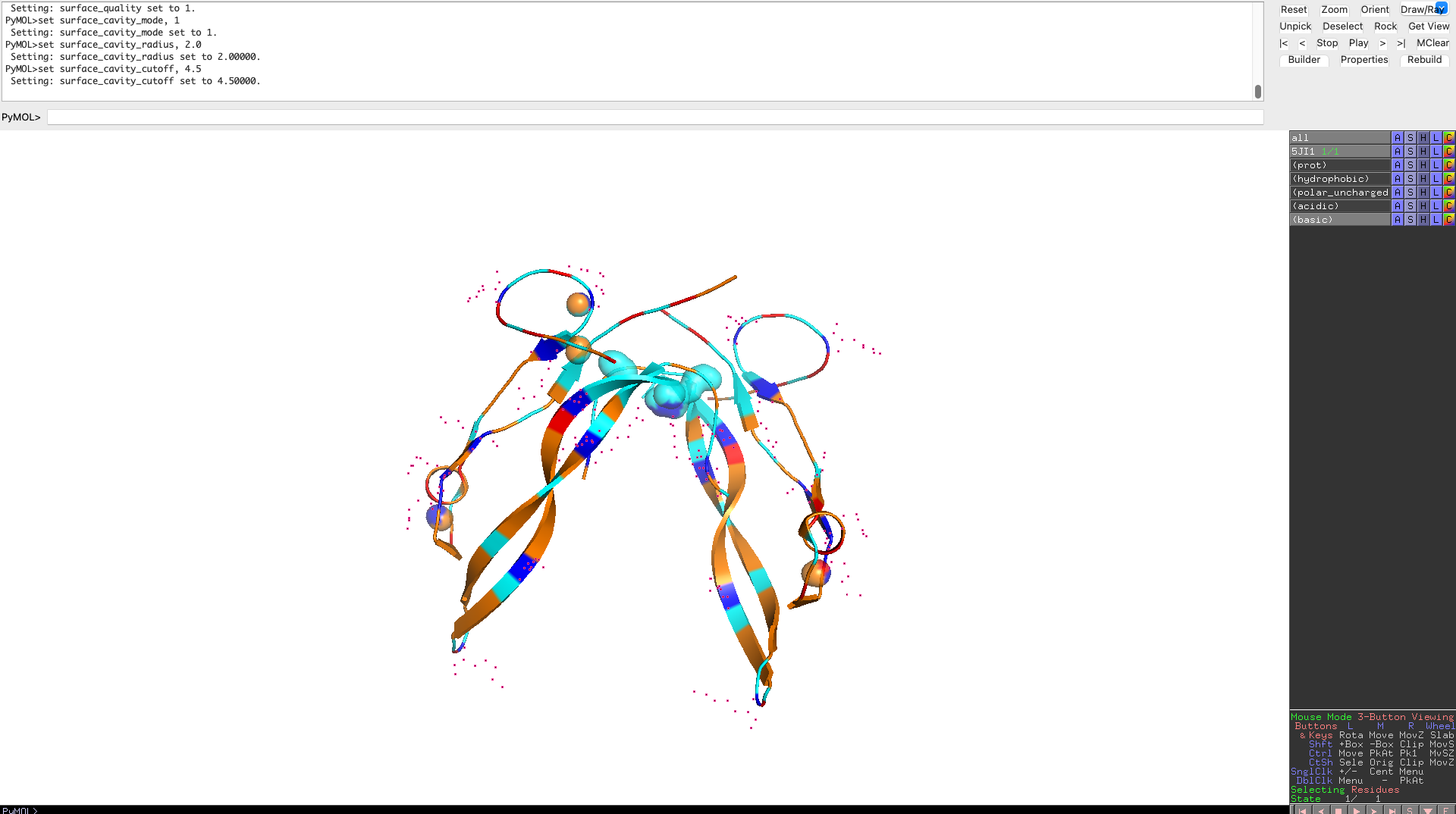
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
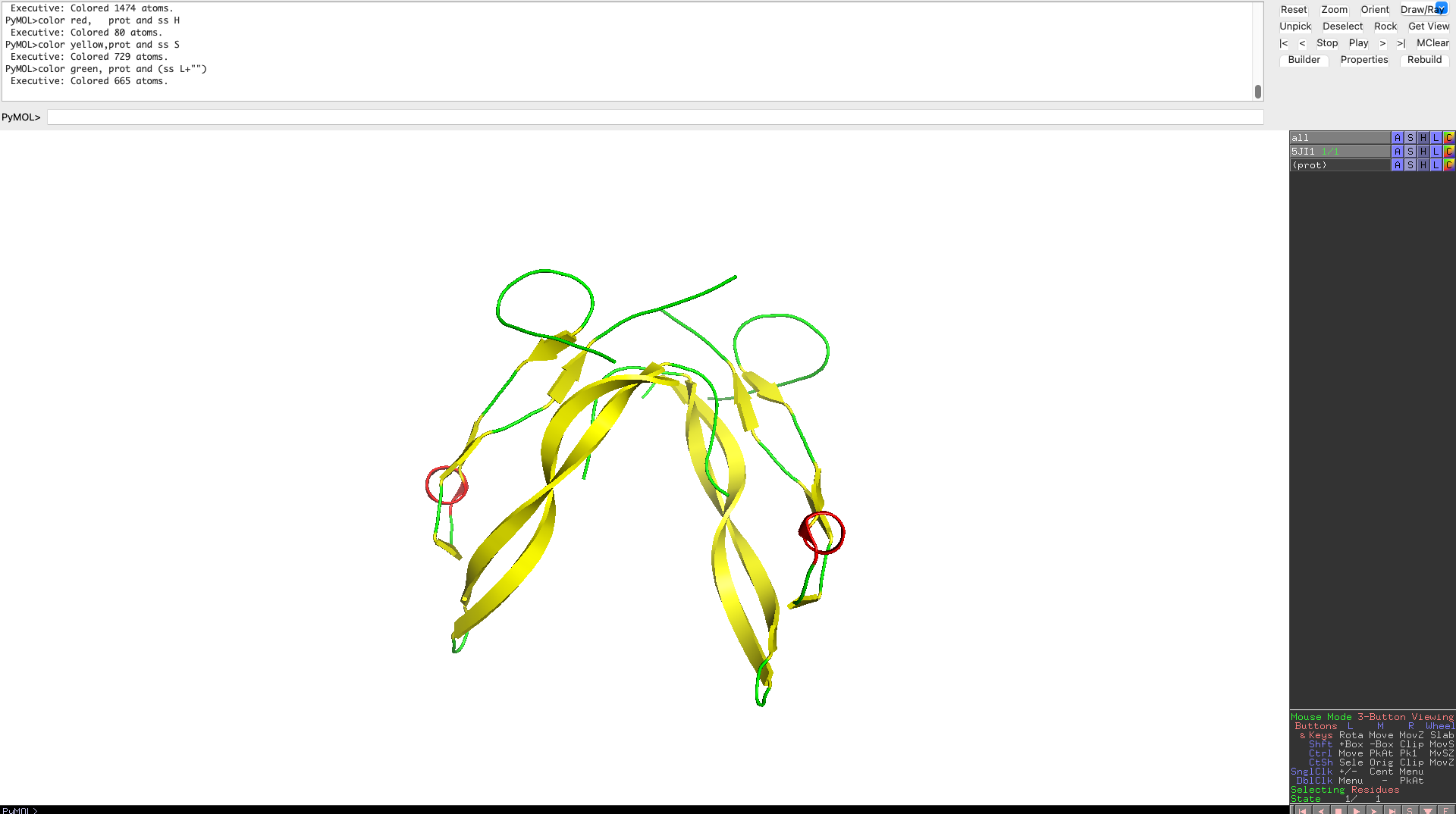
Color the protein by secondary structure. Does it have more helices or sheets?
As one can clearly see the protein has more beta sheets
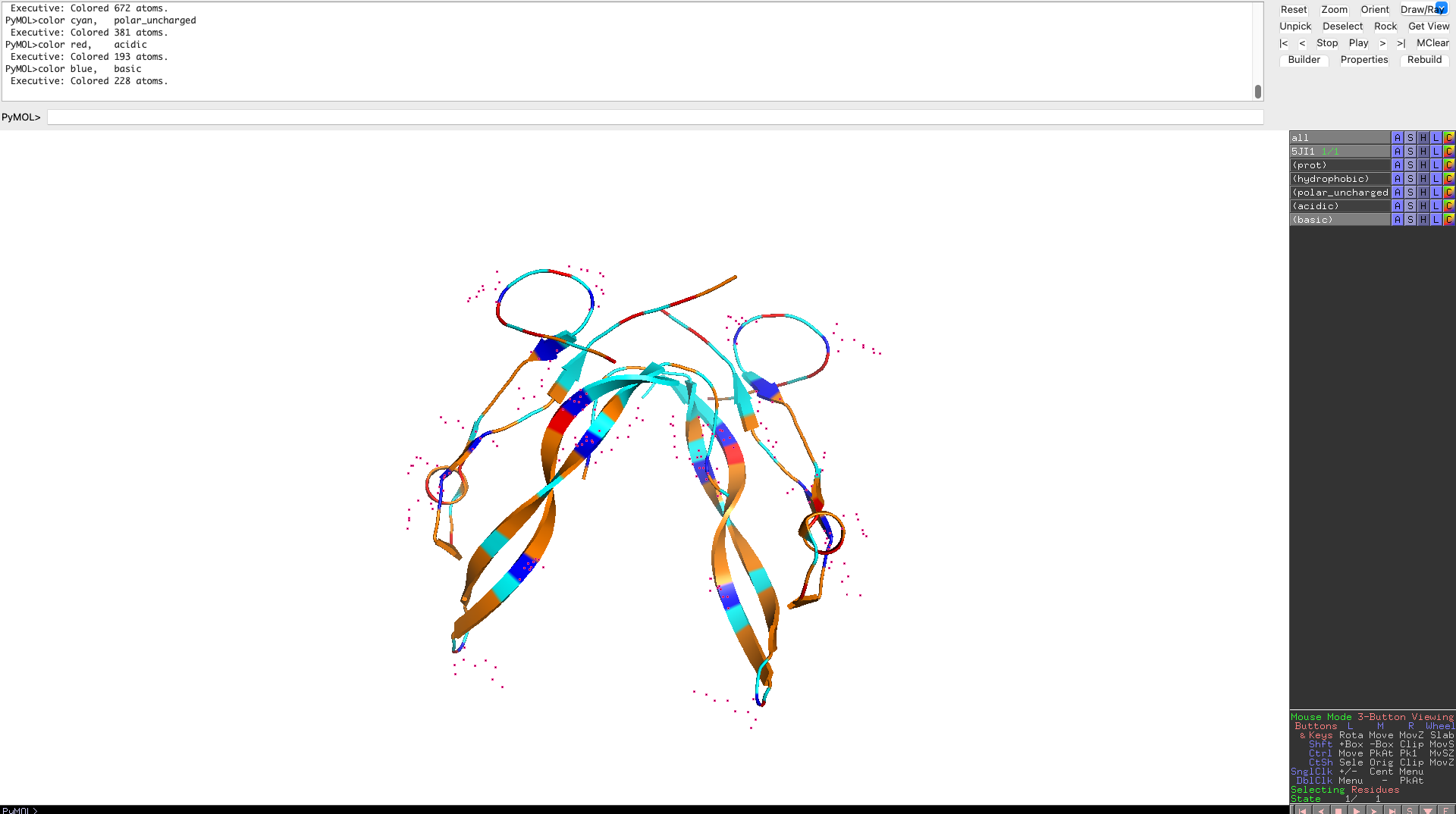
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic residues cluster in the interior of the protein, forming a stabilizing core, while hydrophilic and charged residues are predominantly surface-exposed. This distribution reflects a well-folded, soluble signaling protein whose surface properties are optimized for molecular recognition rather than catalysis.
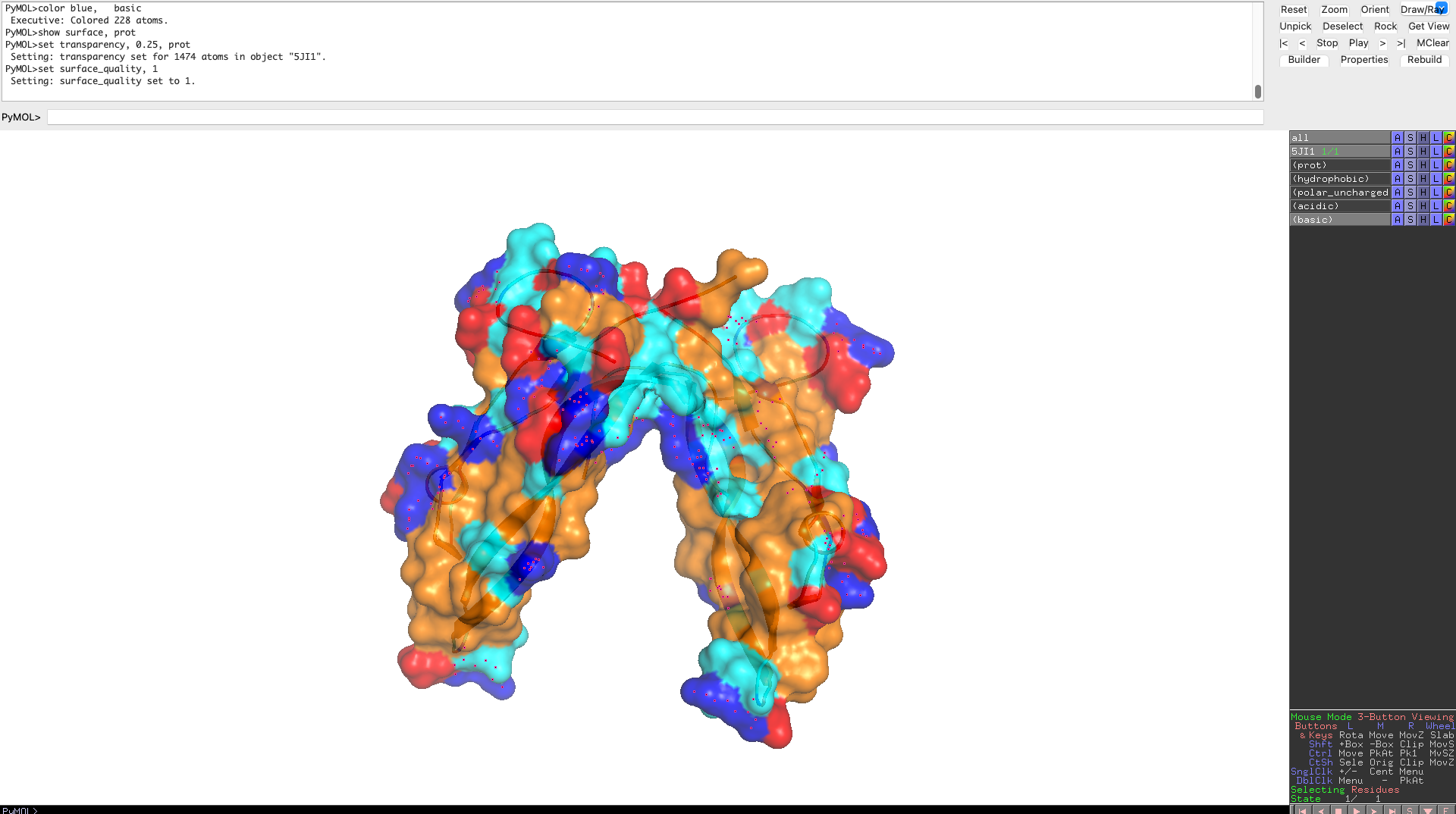
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
The protein surface lacks deep, well-defined binding pockets. Instead, it displays shallow surface depressions and grooves, characteristic of protein–protein interaction interfaces rather than enzyme active sites. This indicates that myostatin binds interaction partners through extended surface patches rather than classical binding pockets.
Part C: Using ML-Based Protein Design Tools
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
I set up a copy of the notebook and set it up with a Google T4 GPU.
Choose your favorite protein from the PDB.
Consistent with the rest of my homework, I chose the myostatin protein, to keep it consistent with Part B, i chose to continue to use the mus musculus sequence.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Deep Mutational Scans
I decided to use the smallest model “esm2_t6_8M_UR50D” to conserve gpu ressources and avoid hitting rate limits, as the protein in question is rather large. Still I hit the rate limit.
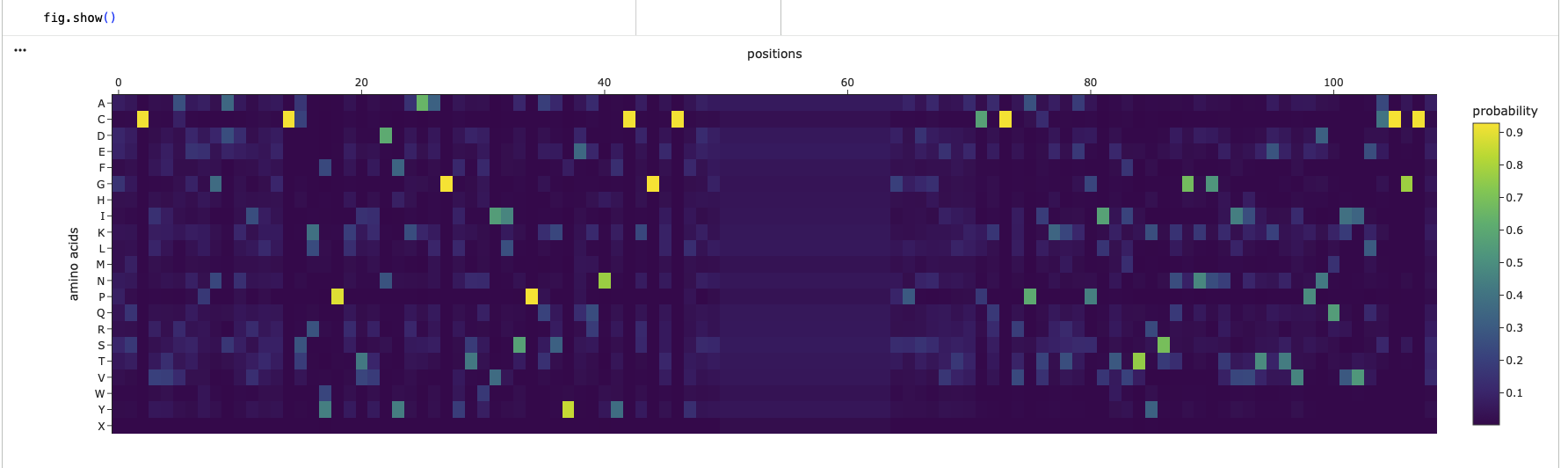
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
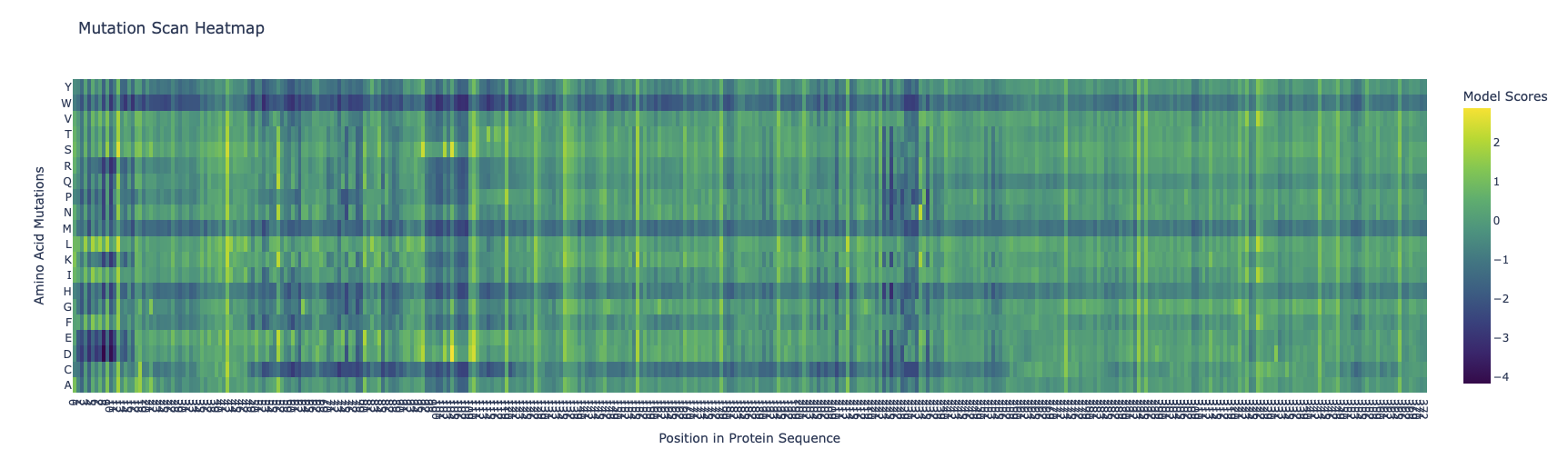
The protein has three super columns (7-13, 97-108, 225-235), with very low scores, indicating that they are very constrained, this could indicate a lynchipin in the protein, as the areas are structurally or functionally important.
A conserved cysteine in the mature TGF-β–like domain, mutated to Ser or Ala (Cys→Ser / Cys→Ala).
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
I struggled to find the data necessary ot make the comparison.
Latent Space Analysis
I downloaded the model and processed the batches:
Processing batches: 0%| | 0/15177 [00:00<?, ?it/s]sdpa attention does not support output_attentions=True. Please set your attention to eager if you want any of these features.
Processing batches: 100%|██████████| 15177/15177 [18:21<00:00, 13.77it/s]
Finished forwarding sequences through the LLM and collecting mean embeddings.
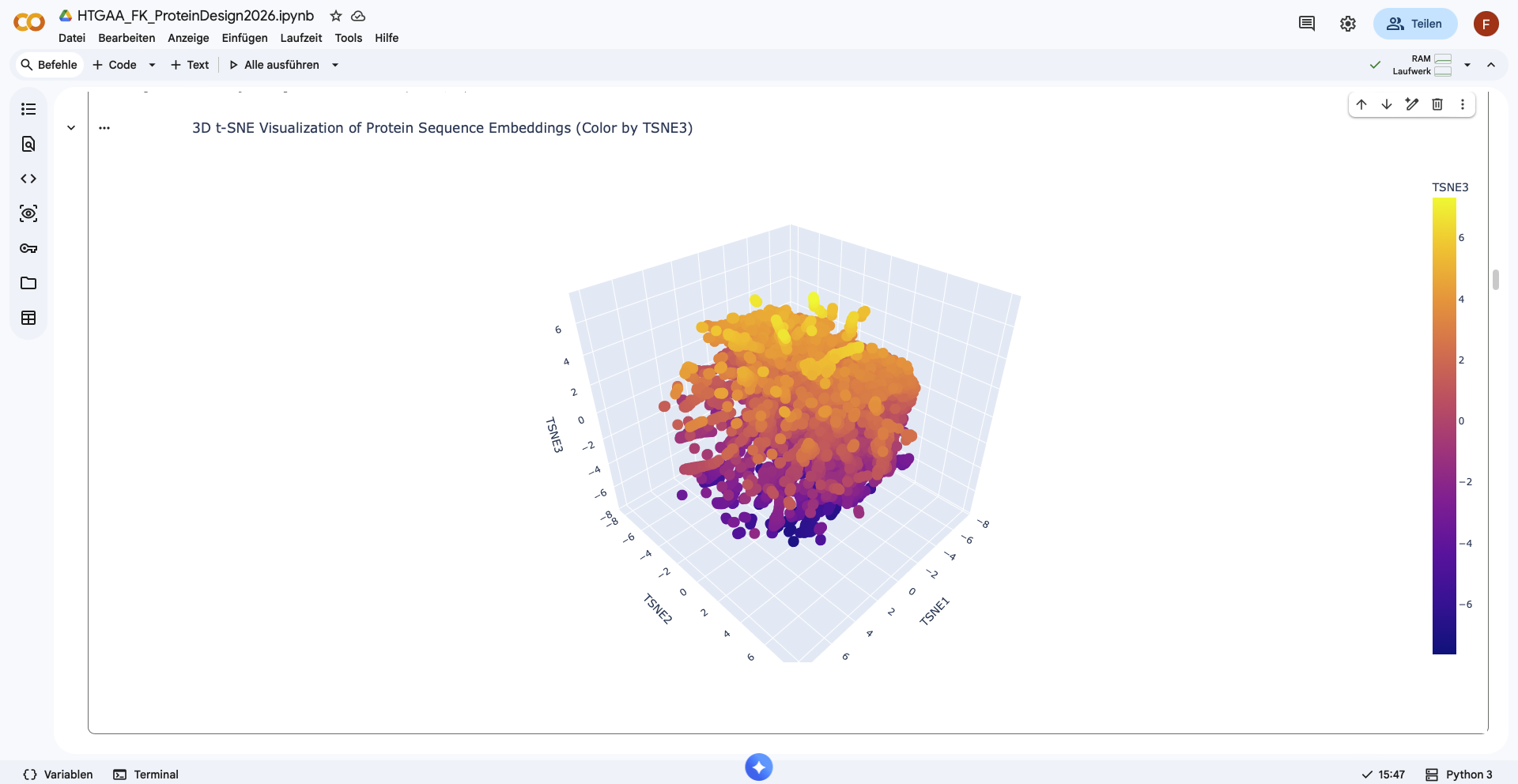
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Shape of embeddings array before 3D t-SNE: (15177, 320)
Shape of embeddings array before 3D t-SNE: (15177, 3)
Analyze the different formed neighborhoods: do they approximate similar proteins?
There form of the latent space looks like a sphere, with one half being densly filled, while the other one is more sparsely filled. Nevertheless no clear neighborhoods are distinguishable.
When looking at one nearest neighbor neighborhood, I still fail to see any pattern in the data, as the organisms are different, as well as the type of the protein, and the function.
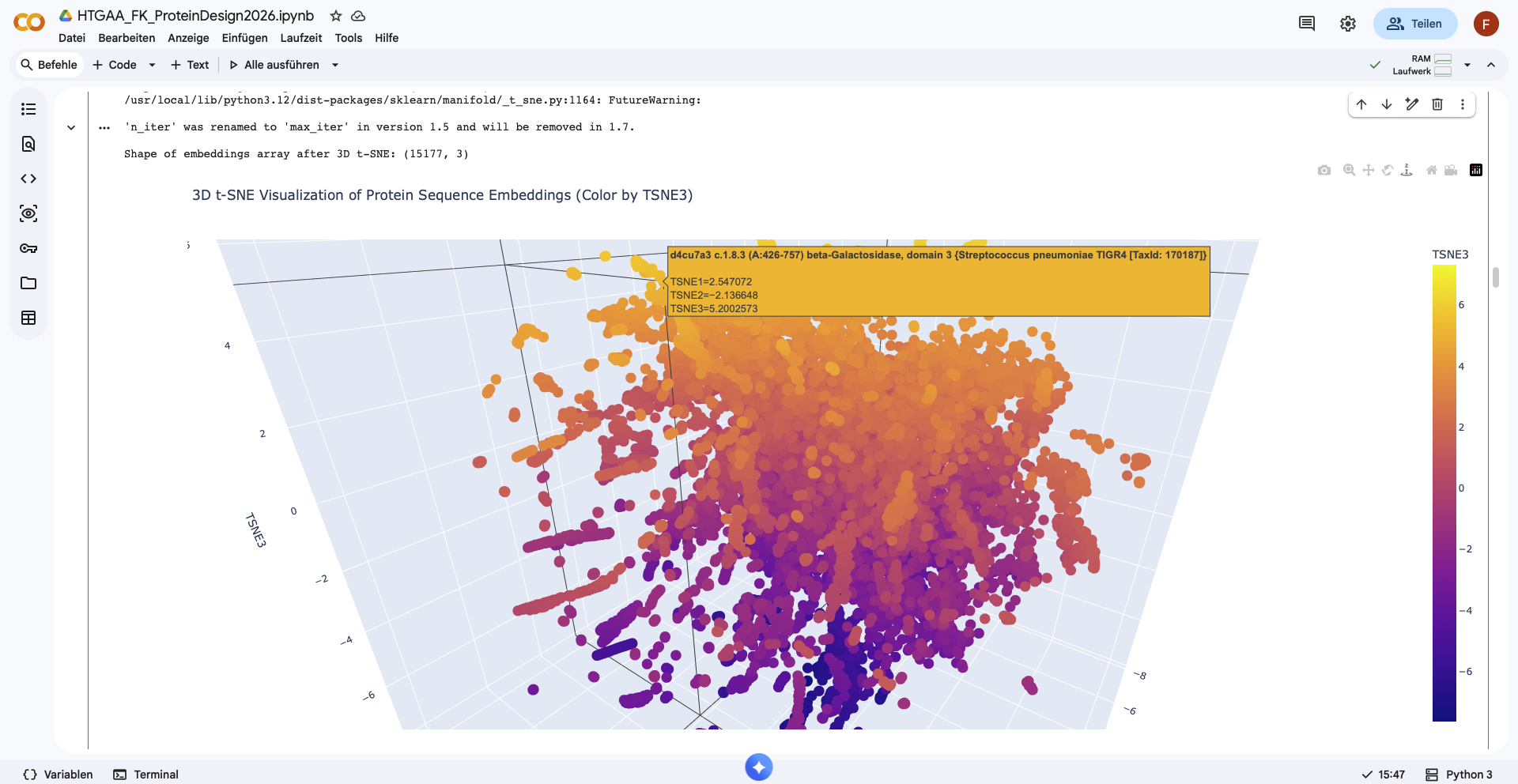
Place your protein in the resulting map and explain its position and similarity to its neighbors.
I was unable to place the myostatin in the resulting map.
C2. Protein Folding
Folding a protein
I used Version 1 of ESMFold.
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I named the job “myostatin_v1”, with 1 copy and 3 num_recycles. To visualize i used the colorsheme “rainbow”
While the overall structure can be described as V or Y shaped in both predicted and actual structure, both structures don’t match on closer look. The original is mostly beta-sheets and has a destinct V shape, while the predicted model is more Y shaped, has several alpha-helices and has a more complex shape to it.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I repeatedly tried to change the structure, though i hit ratelimits with the GPU. Furthermore, as the predicted structure doesn’t match the database myostatin, either trying mutations or changing large segments will likely further remove the predicted structure from it experimental data.
Proposed mutations:
Splitting the protein in 4 equal parts and mutating the sequence at that point
switching 20 aa for a randomly generated 20 aa section.
Inverting the aa sequence.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
I used the model “v_48_020”, the pdb 5JI1, the homomer designed_chain A, num_seqs=1, sampling_temp=0.1
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Looking at the two heatmaps, they aren’t very similar, as the predicted structure, has selectively high probabilites in random spots and else rather low probabilites. Its completely missing the 3 super clusters of low confidence, like the original one
Generating sequences…
New Sequence:GQNIVAGPGAAITECSLWPKTVDFKAAGYDWVISPKSYERNYCSGTCTSSXXXXXXXXXXXXXXGPDNVTEKRCVPTETAPITMTYSLGDGKIITETVPNQIVKACGCV
The predicted sequence isn’t similar to the originally procured sequence.
Input this sequence into ESMFold and compare the predicted structure to your original.
Trying to input the newly generated sequence, led to an error. While I cannot determine the reason for said error, my best guess is the large sequence of “X”.
Part D: Group Brainstorm on Bacteriophage Engineering
Due to later start of our Node, we had limited time to find groups and set up a meeting, therefore the drafts of our group are mainly individual, and not discussed
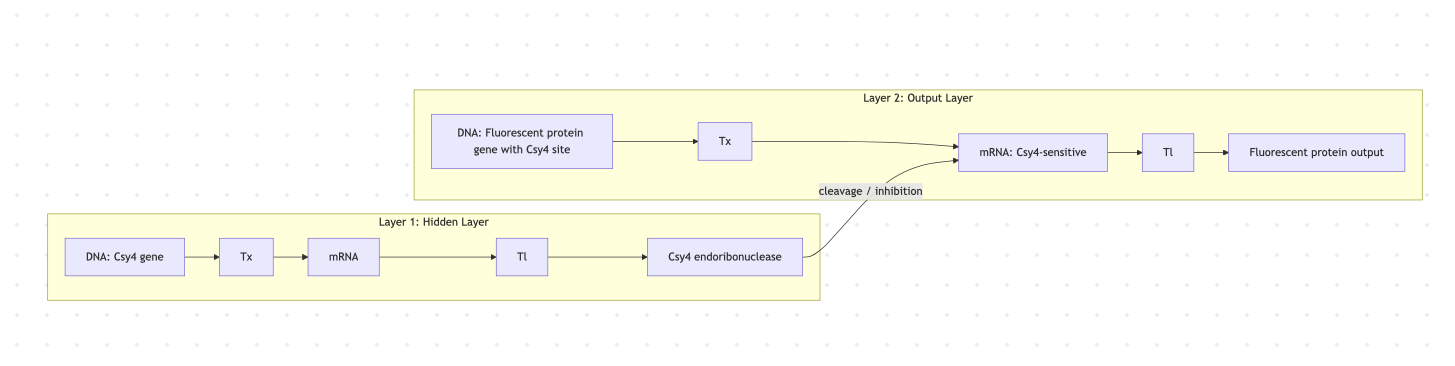
Goal
We target two complementary objectives: (A) Increased stability of the L protein, specifically engineering DnaJ-independent variants that fold correctly without host chaperone assistance; and (B) Higher toxicity / faster lysis, by optimizing the transmembrane oligomerization interface to accelerate pore formation. Goal A is prerequisite to Goal B: a stable, chaperone-independent L is resistant to the most documented E. coli escape mechanism (DnaJ P330Q mutation), and faster lysis narrows the window for resistance acquisition.
Scientific Rational
Three findings define our design space.
DnaJ binds the highly basic N-terminal domain (res. 1–36) of L and relieves a steric inhibition blocking target engagement; removing this domain eliminates DnaJ dependency and accelerates lysis (Chamakura, J Bacteriol 2017).
Near-saturating mutagenesis shows the LS motif (Leu48-Ser49) and flanking residues form a heterotypic interface with an unknown target; exquisitely conservative mutations matter (L44V = dead, L44I = functional) and all are recessive, pointing to a specific binding event rather than membrane disruption (Chamakura, Microbiology 2017).
MS2-L oligomerizes into 10+ mers in nanodisc membranes via its TM domain; cryo-EM shows large envelope lesions starting at the outer membrane (Mezhyrova et al., 2023).
Strategy: neutralize basic charges in Domain 1 so DnaJ is no longer required, while leaving Domains 2–4 (the lytic machinery) untouched.
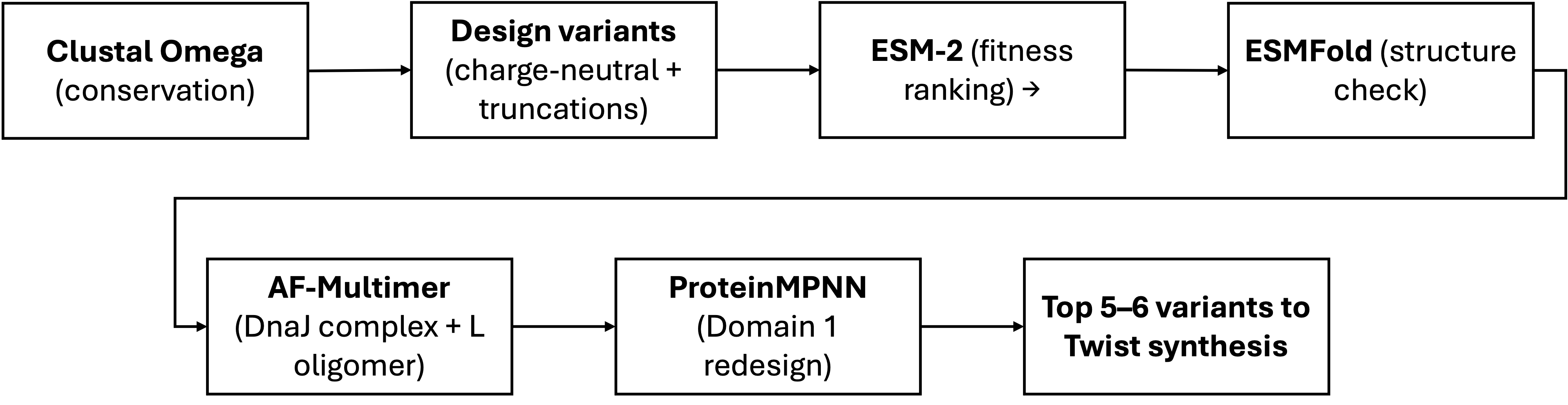
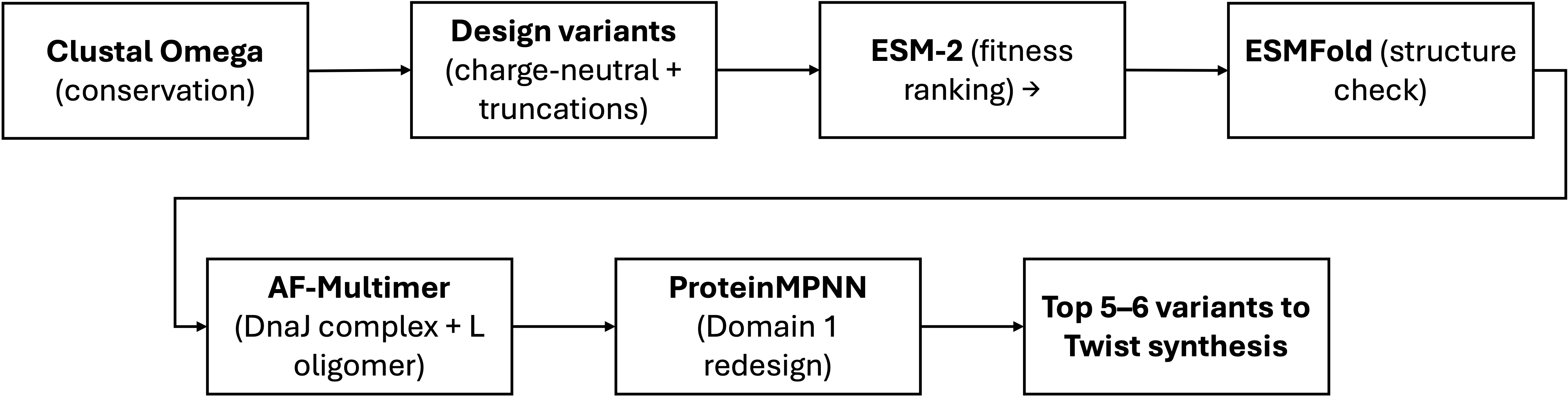
Computational Tools
Tool
Application
Why it helps
Clustal Omega
Align L homologs to identify which aminoacids are freely mutable
Reproduces and extends the LS-motif alignment from Chamakura (2017). Essential first step: tells us where NOT to mutate.
ESMFold
Predict 3D structure and each designed variant; verify the TM helix remains intact after mutations
Fast single-sequence predictor. For a 75 aa peptide with few homologs, much more practical than full AlphaFold for screening many candidates.
AlphaFold-Multimer
Model the L–DnaJ complex; confirm charge-neutralized variants show reduced interface confidence. Also model L–L homodimers to check TM packing.
Key validation for Goal A: if predicted L–DnaJ interface weakens for our variants, that supports DnaJ independence.
ProteinMPNN
Inverse folding: redesign Domain 1 (res. 1–36) to be uncharged while fitting the ESMFold-predicted backbone. Domains 2–4 fixed as hard constraints.
new sequence for existing fold with position-specific constraints. Generates diverse candidates we can then filter with ESM-2.
ESM
Zero-shot fitness scoring: rank all candidate variants by pseudo-log-likelihood as a sequence-level sanity check
Independent of structure prediction. Benchmarked first against known mutants — if it captures L biology, we use it to filter; if not, we rely on conservation alone.
Schematic
Pitfalls
We cannot model the most critical interaction (L with its unidentified host target) computationally.
ML models may not capture L biology, as L is a 75 aa phage toxin with very few homologs, far outside the training distribution of ESM-2 and AlphaFold
Use of Generative AI
Generative AI was used as a conceptual drafting aid during the development of this project. Specifically, it supported the structuring and refinement of complex ideas related to protein design and use of computational tools. It was instrumental in drafting the computational strategy of engineering the MS2 Lysis Protein L, as well as clarifying the scientific concepts in the related reading.
The AI was used to iteratively improve clarity of language and to explore alternative conceptual framings. All final judgments were made by the author. The link for the prompts and responses is attached in the repository.
Week 5 HW: Protein Design II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
The Protein can be found with this link
The Protein sequence is:
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Colab Notebook was copied to my personal drive. I applied for access, even though that doesn’t seem to be necessary anymore.
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.Record the perplexity scores that indicate PepMLM’s confidence in the binders.
I checked the single-sequence checkmark and pasted the protein sequence above. I gave it the job name “SOD1_fk_v1”.
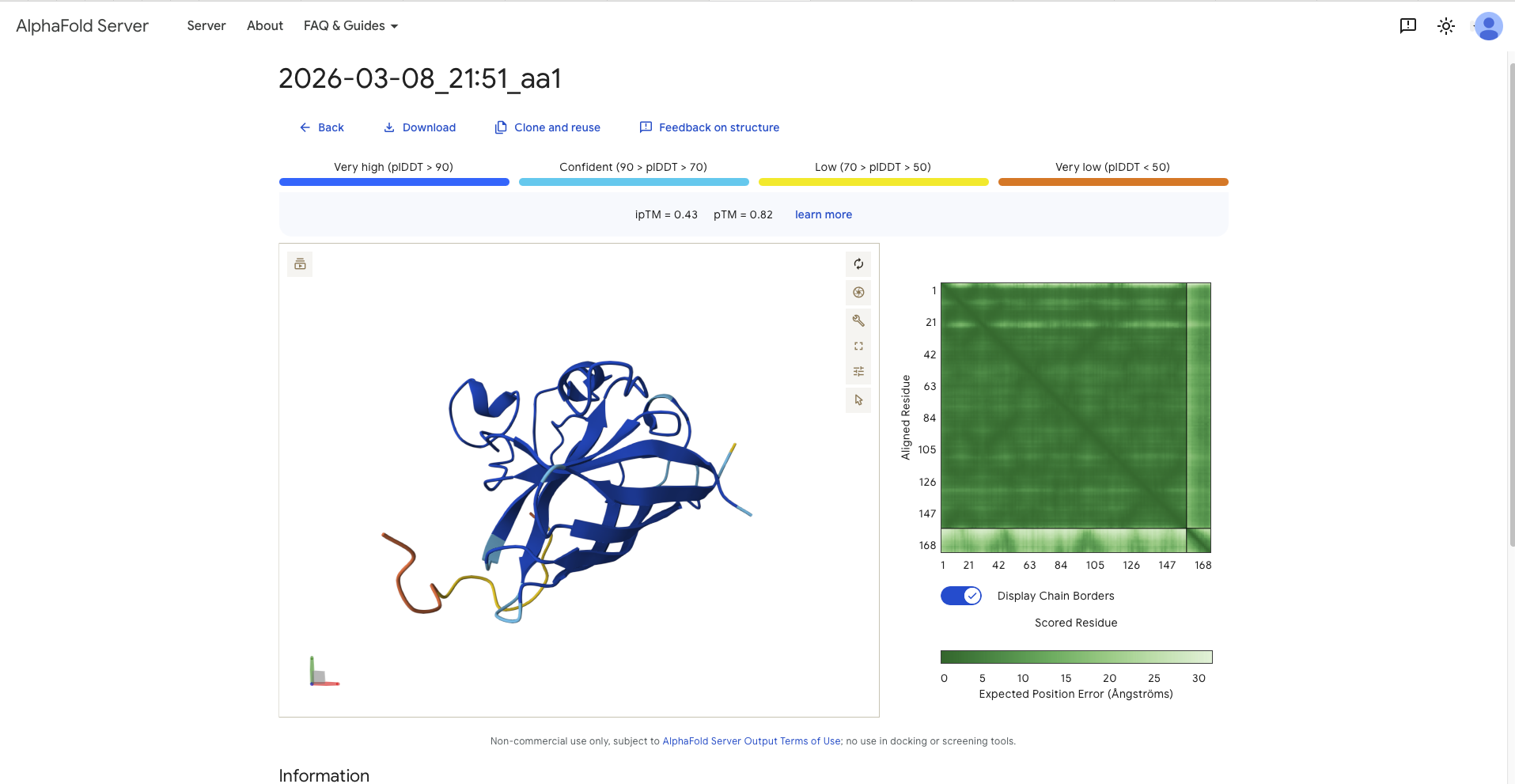
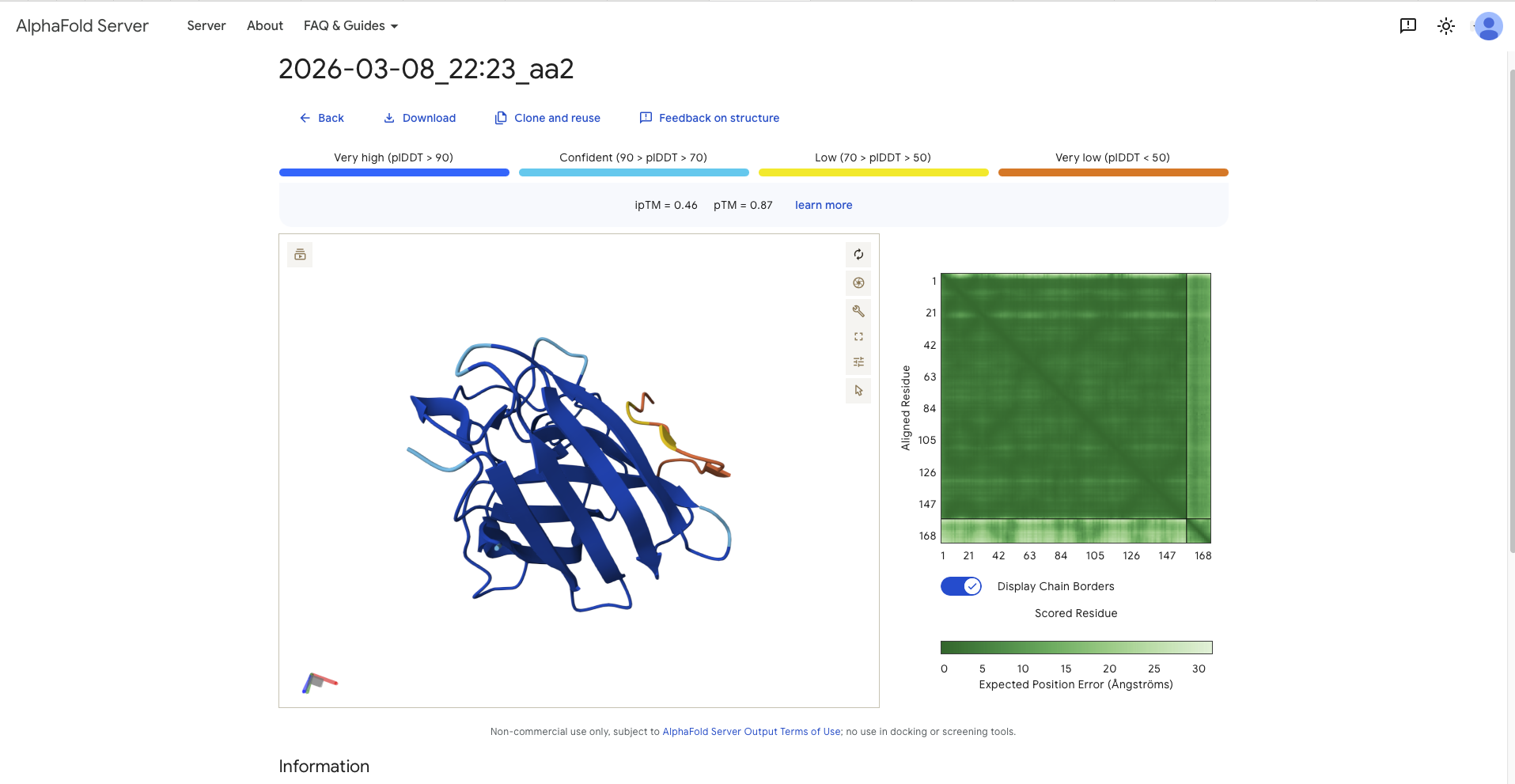
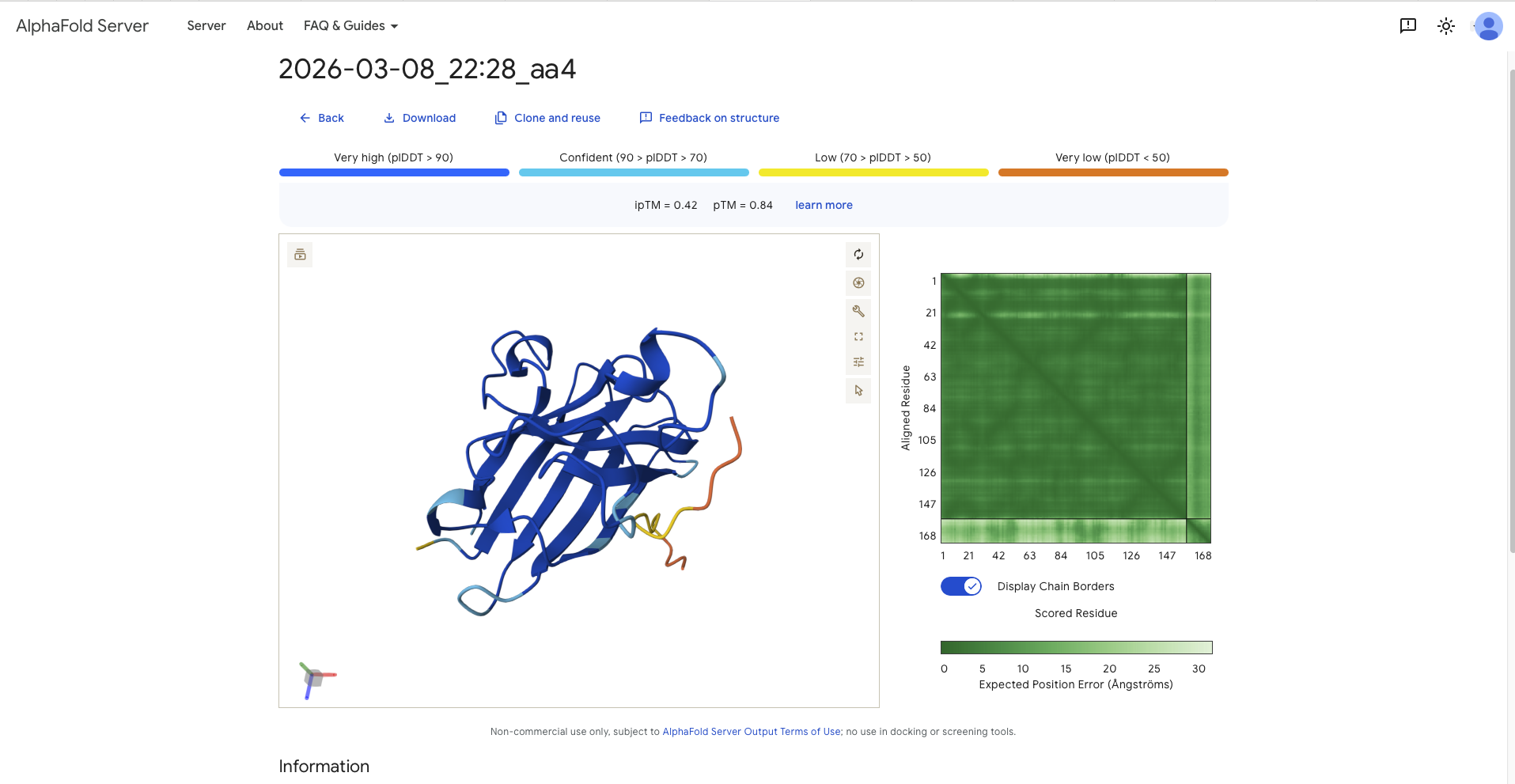
Afterwards I continued with the model and generated several sequences. This had to be done in different runs as most batches contained sequences with an “X”, which later would not be accepted by AlphaFold. In the end I decided to mix and match from different batches. These are the sequences (aa1 - aa4) with the reference sequence being named aa0. The following table recodes the sequence and the perplexity score.
ID
Sequence
Pseudo Perplexity
aa0
FLYRWLPSRRGG
-
aa1
AHYGVLAAAVKWRRK
15.4397
aa2
SRYDVYVGRVKARAK
18.3568
aa3
WRYDPVTGRYAAKKA
9.3430
aa4
SWVPVYTAVVKLKRK
20.8359
Part 2: Evaluate Binders with AlphaFold3
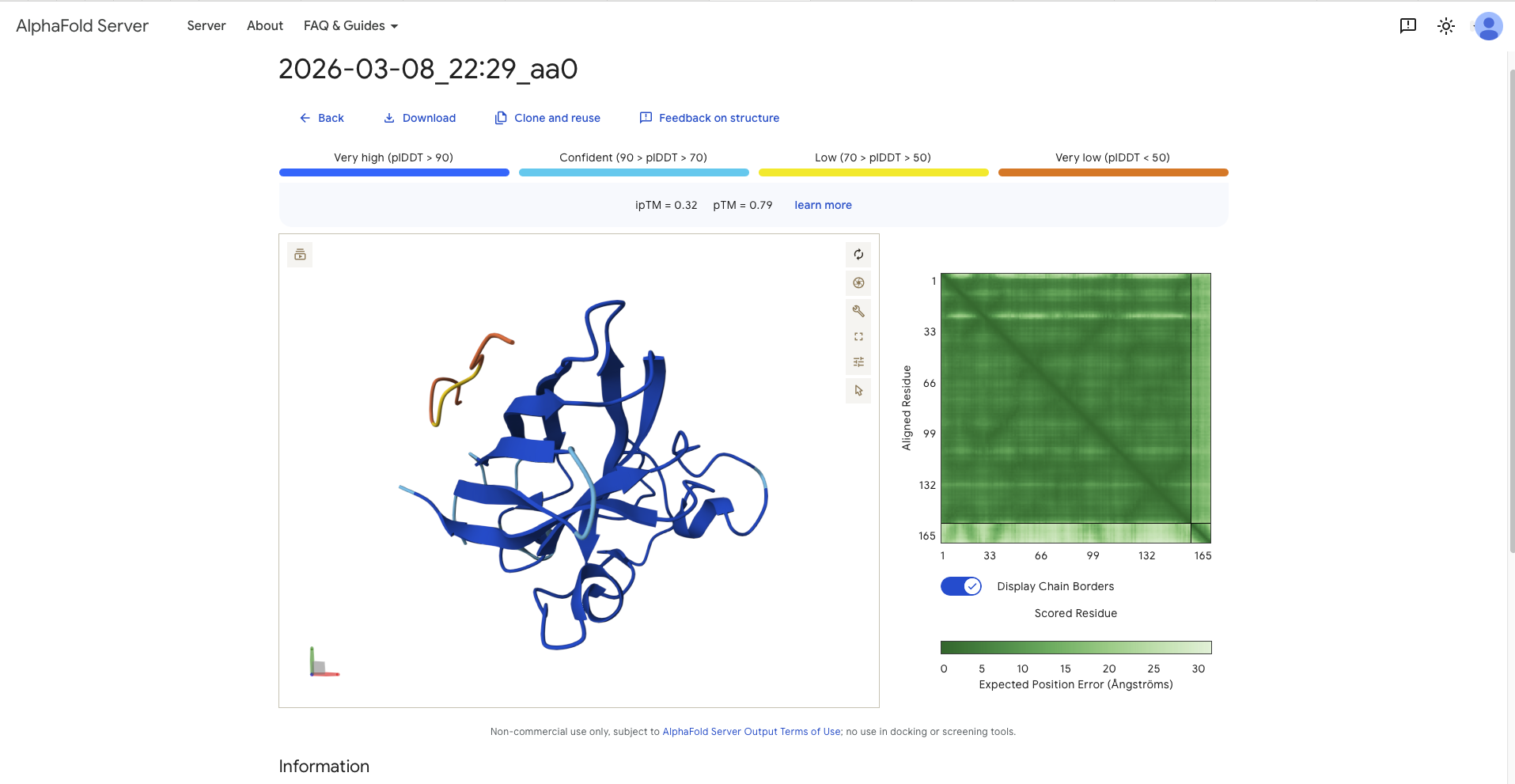
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
ID
Sequence
Pseudo Perplexity
ipTM
pTM
Bindung Site
Binding Format
aa0
FLYRWLPSRRGG
-
0.32
0.79
- In the same region as N-terminus, parallel to the beta barrel - in the same region as the dimer interface (the dimer interface is where N-Terminus and C-Terminus meet, or easier, the beginning and the end of the protein. It has the form of a strand
Surface BoundSurface Bound
aa1
AHYGVLAAAVKWRRK
15.4397
0.43
0.82
- On the opposite side of the protein from the N-terminus - wrapping around the beta barrel from the side and the top - on the opposite side of the protein from the N-terminus
Surface Bound
aa2
SRYDVYVGRVKARAK
18.3568
0.46
0.87
- around a 90 degree away from N-Terminus and therefore the dimer interface - paralell to the beta barrel, though it has the form of a C
Surface Bound
aa3
WRYDPVTGRYAAKKA
9.3430
0.23
0.84
- around a 90 degree away from N-Terminus and therefore the dimer interface - perpendicular to the beta barrel - it has the form of a C, two beta sheets on either side with the belly of the C pointing towards the protein
mostly surface bound, though more burried than the others
aa4
SWVPVYTAVVKLKRK
20.8359
0.42
0.84
- around a 120 degree away from N-Terminus and therefore the dimer interface - paralell to the beta barrel - shape is random, but wrapped into the protein in a 3D shape
mostly surface bound, though of all the generated the most burried than the others
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
All ipTM values exceed the refrence linker, except aa3, the, though all pTM values exceed the reference sequence. After discussion with TA all values above 0.8 constitute a good confidence score that the overall structure is correct.
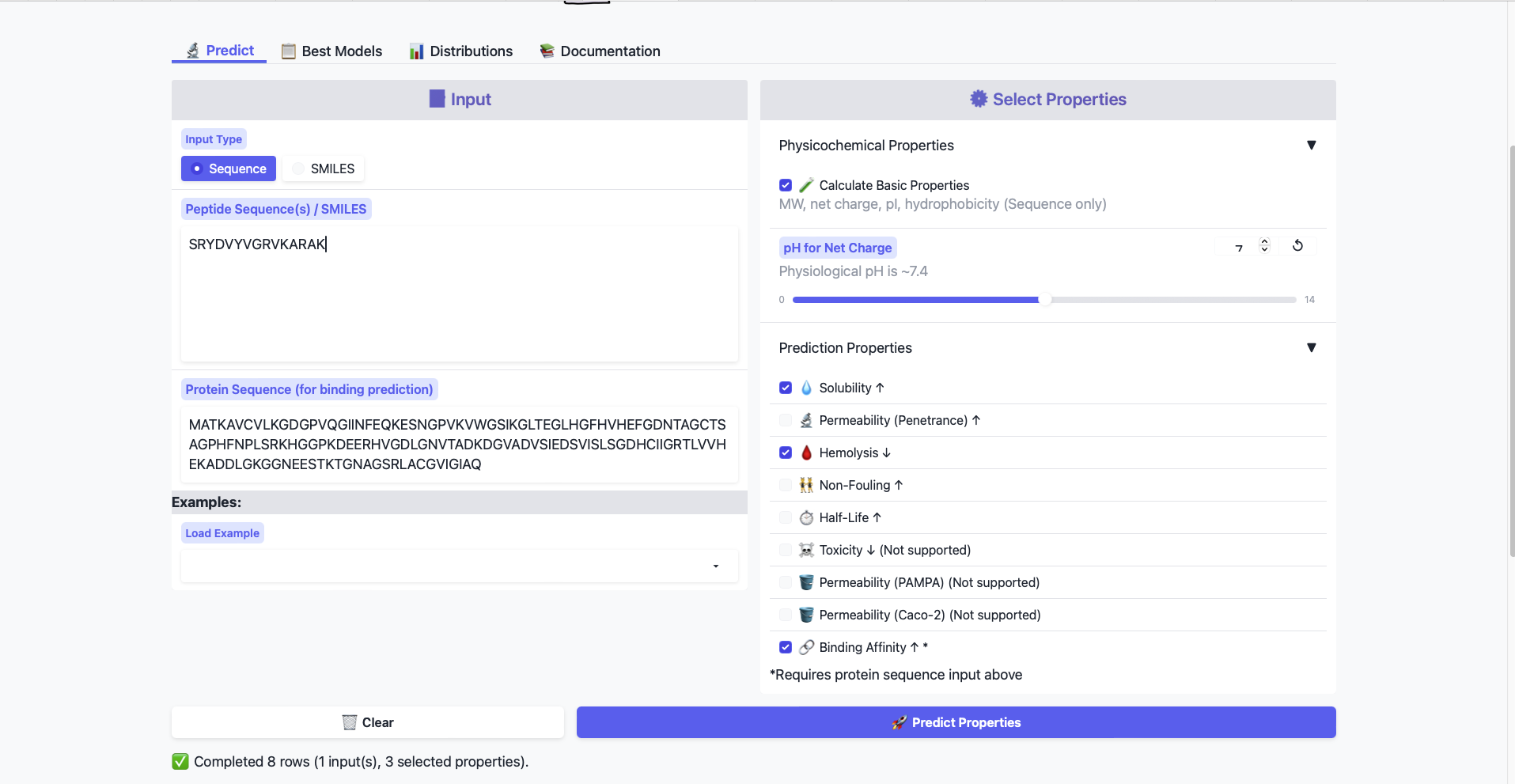
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see.
All peptides show weak binding affinity. This is somewhat expected from the AlphaFold data, as the generated Peptides all show only surface level binding.
Do peptides with higher ipTM also show stronger predicted affinity?
According to a quick trendline analysis, the relationship is negative. The affinity scores scatter around 6.27 and with a standard deviation of around 200.
Are any strong binders predicted to be hemolytic or poorly soluble?
My predictions didn’t produce any strong binders, therefore I cannot answer the question. All predicted peptides are non-hemolytic (values range between 22 and 77) and soluable
Which peptide best balances predicted binding and therapeutic properties?
As non of my peptides have strong binding, while they have good therapeutic properties, the sequence with the highest binding affinity is the best balance. Which is aa3, that does have the lowest ipTM score.
Choose one peptide you would advance and justify your decision briefly.
I’d choose aa3, as the sample is doesn’t produce a predicted sequence that has a strong binding affinity, while the therapeutic properties are solid, the peptide with the highest binding affinity is chosen.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card Make a copy and switch to a GPU runtime. In the notebook: Paste your A4V mutant SOD1 sequence. Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). Set peptide length to 12 amino acids. Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
Unfortunately I wasn’t able to complete this part as google didn’t allow for the use of A100 or L4 GPU. I got the tried with T4 GPUs but the first cell ran in a infinite loop till my GPU credits ran out.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
See above
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele) [optional for Committed Listeners]
Part 0: Sign-up to Boltz Lab
I signed up for access to the Boltz Lab, though as of writing the Homework i have not received any credentials
Part 1: Structural Predictions in the Sandbox
Part 2: Setting Up a BRD4 Design Project
Part 3: Running Your Virtual Screen
Part 4: Analysis and Discussion
Part C: Final Project: L-Protein Mutants
Option 1: Mutagenesis
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
I copied the Colab notebook and worked through it to the best of my ability. Any experimental work will be done in the BioPunk Node at a later time, as per discussion with Eliott Roth (Our Node Leader).
Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid. It can be positive or negative for the protein. We want to identify possible mutations that are “positive” If you run this notebook - you will see a .csv file in the sidebar. You can download it and look at it in the google sheets if that’s easier.
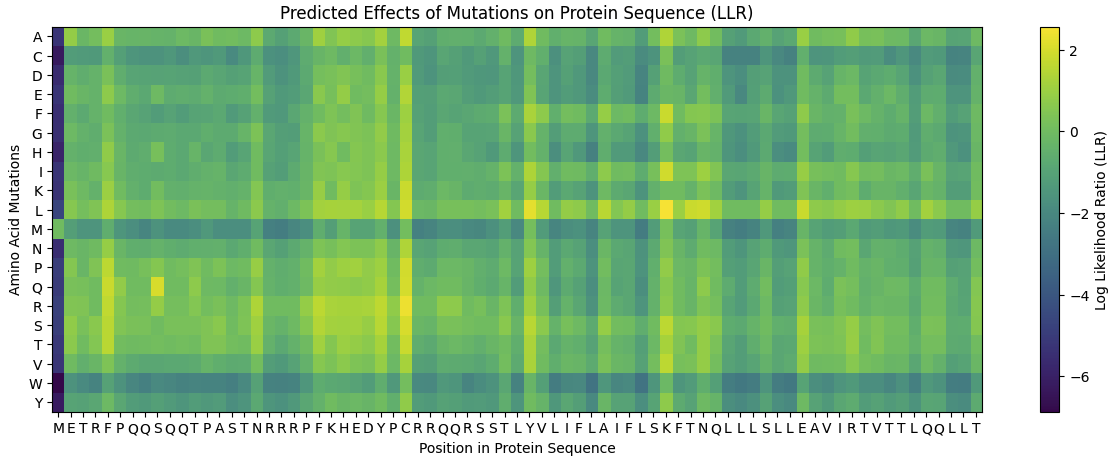
Running the Colab notebook, gave me several outputs, unfortunately they were quite badly documented therefore all information should be used with caution.
The first run analyses the entire protein sequence and produces an exhaustive list of potential mutation sites:
Model 1
Position
Wild_Type_AA
Mutation_AA
LLR_Sccore
50
K
L
2,561468
29
C
R
2,395427
39
Y
L
2,24178
29
C
S
2,04315
9
S
Q
2,014325
29
C
Q
1,997049
29
C
P
1,971029
29
C
L
1,960646
50
K
I
1,928801
53
N
L
1,864932
61
E
L
1,818098
52
T
L
1,813968
50
K
F
1,802069
29
C
T
1,797247
29
C
K
1,795878
5
F
Q
1,795244
5
F
R
1,659717
29
C
A
1,648656
27
Y
R
1,628061
22
F
R
1,602028
5
F
P
1,596891
50
K
V
1,594576
50
K
S
1,574557
5
F
T
1,559024
5
F
S
1,556417
45
A
L
1,539248
39
Y
S
1,517457
27
Y
S
1,497053
40
V
L
1,47763
27
Y
L
1,474637
22
F
S
1,423358
29
C
E
1,383281
39
Y
A
1,364999
29
C
N
1,362601
50
K
A
1,357795
29
C
I
1,344121
5
F
L
1,332615
17
N
R
1,323651
39
Y
I
1,320103
39
Y
T
1,302804
26
D
R
1,268762
29
C
H
1,246107
39
Y
F
1,245851
39
Y
V
1,24439
23
K
R
1,236555
25
E
R
1,22935
24
H
R
1,227779
50
K
T
1,222131
27
Y
Q
1,218851
27
Y
T
1,215567
Model 2
ID
Amino Acid
Position
LLR_Score
0
L
50
2,561468
1
L
39
2,24178
2
I
50
1,928801
3
L
53
1,864932
4
L
52
1,813968
5
F
50
1,802069
6
V
50
1,594576
7
S
50
1,574557
8
L
45
1,539248
9
S
39
1,517457
10
L
40
1,47763
11
A
39
1,364999
12
A
50
1,357795
13
I
39
1,320103
14
T
39
1,302804
15
F
39
1,245851
16
V
39
1,24439
17
T
50
1,222131
18
L
54
1,12086
19
R
39
1,064191
Model 3
Position
Wild_Type_AA
Mutation_AA
LLR_Score
50
K
L
2,561468
29
C
R
2,395427
39
Y
L
2,24178
29
C
S
2,04315
9
S
Q
2,014325
29
C
Q
1,997049
29
C
P
1,971029
29
C
L
1,960646
50
K
I
1,928801
53
N
L
1,864932
Model 4
Position
Wild_Type_AA
Mutation_AA
LLR_Score
50
K
L
2,561468
29
C
R
2,395427
29
C
R
2,395427
39
Y
L
2,24178
29
C
S
2,04315
29
C
S
2,04315
9
S
Q
2,014325
9
S
Q
2,014325
29
C
Q
1,997049
29
C
Q
1,997049
Here is the Heatmap generated from the Colab Notebook.
Use the experimental data here. This dataset contains information about mutants of the L-Protein and their effect on lysis in the lab - L-Protein Mutants
I copied the csv file from the website to the googlesheet and the Colab.
First check, does the experimental data correlate with the scores from the notebook in (b)? This should give you a clue on how well these language embeddings capture information about this protein sequence.
Let’s first look at the experimental data. Here the positions, mutations are displayed, together with a binary, whether the lysis has happened. Therefore one needs to look at the lysis = 1. These are 35 entries. Now one needs to compare, whether the positions are in the model generated mutation sites.
I’m unsure what the instructors mean with correlating the scores from the experimental data with the predicted data. The above mentioned workflow is a comparison of targets.
In a next step on can check, whether the identified targets have a positive LLR score.
Using information about the effect of protein mutations at these sites - both the scores and the experimental data in the drive, come up with 5 mutations for each student along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (refer to notes above on what amino acid positions these are) and 2 of them must be in the soluble region . Remember that you can also use the pBLAST to see which residues are conserved and not mutate them if you want to. One easy way to generate sequence mutations could be to look for residue positions and mutations that have a positive mutational effect either in the experimental or have a positive score from step 1. And pick a combination of those mutations.
ID
Model
Position
Wildtype
Mutation
1
1
61
E
L
2
1
50
K
F
3
2
53
N
L
4
2
52
L
5
3
53
N
L
You can utilize Af2_Multimer to generate a Multimeric Assembly; you can do this by making your query sequence as. We want to do this because - A running hypothesis for how this protein functions is that it assembles to make a perforation in the bacterial membrane.
No Colab was provided for that. If applicable the colab from option 2 could be utilized. This can be submitted at a later point.
Use of Generative AI
Generative AI was used as a conceptual drafting aid during the development of this project. Specifically, it supported the structuring and refinement of complex ideas related to protein design and use of computational tools. It was instrumental in explaining the methods used in the colab notebook to generate mutations of the lysis protein, as well as clarifying the scientific concepts in the related reading. The AI was used to iteratively improve clarity of language and to explore alternative conceptual framings. All final judgments were made by the author. The link for the prompts and responses is attached in the repository.
Week 6 HW: Genetic Circuits I
DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
I didn’t find anything pertaining the matter in the protocol itself, though a quick websearch (Link 1, Link 2), revealed that the PCR Master Mix contains 4 main ingreadients.
Phusion DNA Polymerase
Copying the Gene
Deoxynucleotide Triphosphates (dNTPs)
Building blocks (ATCG) used by the polymerase to synthesize the new strand of DNA
Reaction Buffer
Maintaining the ideal environment for the polymerase to function
Magnesium Chloride
Cofactor for the polymerase reaction
What are some factors that determine primer annealing temperature during PCR?
The annealing temperature Ta is determined by the melting temperature Tm, around 3-5°C lower. The melting temperature is determined by the primer length and the GC content.
Primer length and GC Content: The longer the primer length, the and the higher the GC content, the higher the annealing temperature can become. GC have three hydrogen bonds, that needs more energy (temperature) to break.
Salt and Ion Concentration: DNA backbone is negatively charged and two strand would repel each other. The salt provides cations to cancel the negative charge.
Primer Concentration: Similar logic as 1). The higher the concentration of the primer the higher the annealing temperature
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
In short PCR builds DNA up from scratch, while restriction digests cut existing DNA down.
PCR binds to single stranded DNA and complements the strand with free flowting dtNs, effectively “negative copying” the DNA. It needs little input DNA. The polymerase docks at custom primers. It can output billions of strands of DNA.
The PCR Protocol is thermocyclic and consists of denaturation (95°C), annealing (55-65°C) and extension (72°C). This process is then repeated 30-40 times.
Restriction Enzyme Digests cuts DNA fragments by enzymatic cleavage. It needs large amounts of clean DNA fragments. The enzyme binds to restriction sites and cuts them.
Protocol for restriction enzyme digest is isothermal. The main steps include mixing DNA, restriction enzyme and buffer, incubation at 37°C for 30 min, then inactivation of the enzyme (and the reation) through heat (around 65-85°C).
The two methods are very different, but from the description it gets clear when to pick each method.
PCR should be used when you have little samples, when you want to add sequences, like tags and the sequence is known (essential for primer design)
Restriction enzymes should be used when you are cloning (as you need sticky sites, checking the validity of a plasmid, or you’re working with unknown sequences.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson Assembly operates with homologous overlapping ends, and doesn’t require specific restriction sites or leave behind “scars” like traditional restriction cloning. Therefore the main requirement for a successful Gibson Assembly is the correct design of the 20-40bp long overlap.
How does the plasmid DNA enter the E. coli cells during transformation?
There are several transformation methods, like heat shock, electroporation or conjugation, etc. The lab went into detail with heat shock, therefore this will be the focus of the answer.
The cells and plasmids are suspended in a cold calcium chlorid solution (mostly in an ice bucket), then heated up in a waterbath or PCR machine (42°C) for 30-60 seconds.
Describe another assembly method in detail (such as Golden Gate Assembly)
I choose Golden Gate Assembly.
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a molecular cloning method that uses Type IIS restriction enzymes to assemble multiple DNA fragments into a single vector in one reaction. Type IIS enzymes (like BsaI or BbsI) cut at a specific distance away, leaving behind 4-base “overhangs” that are not resticted by the enzyme’s binding sequence, unlike traditional enzymes that cut within their recognition site. Plainly they have a recognition site, where they dock, but then instead of cutting in their recognition site, they reach over and cut a few basepairs away. By thoughtfully designing these overhangs, you can ensure that multiple fragments anneal only in a specific, predetermined order. Because the recognition sites are oriented to be “cut out” during the reaction, the final assembled product lacks the original enzyme sites, making the process irreversible . This “one-pot” reaction combines digestion and ligation in a single tube, allowing for the seamless assembly of multiple fragments simultaneously.
Model this assembly method with Benchling or Asimov Kernel!
Asimov Kernel
NOTICE 2026-03-17: As of the deadline, we have not received access to the Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
Week 7 HW: Genetic Circuits Part II
Part 1
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
The key advantage of intracellular analog neural networks (IANNs) over traditional genetic circuits lies in the shift from discrete logic to continuous computation. Classical genetic circuits are typically engineered as Boolean systems: inputs are interpreted as “on” or “off”. This abstraction is convenient for engineering and design, but it is fundamentally misaligned with how biology actually operates, where signals exist as continuously varying concentrations and reaction rates.
IANNs, in contrast, operate on these continuous biochemical signals directly. Instead of forcing a threshold-based decision, where a small fluctuation can flip the output entirely, they produce graded responses. In practice, this means a cell can scale its response to stimulus intensity rather than switching abruptly, which is far more consistent with natural regulatory systems.
A second major advantage is robustness as biological systems are inherently noisy. Boolean circuits are fragile under these conditions because they rely on precise thresholding. IANNs mitigate this by distributing computation across multiple interacting components. Much like artificial neural networks, they integrate signals through weighted interactions, which effectively averages out noise and reduces the likelihood of incorrect responses.
Closely related to this is the issue of scalability. As traditional genetic circuits grow in complexity, the number of required components increases rapidly, often becoming impractical to implement in living cells. IANNs compress complexity into tunable parameters such as binding affinities and interaction strengths. This allows them to represent much more complex input–output relationships with fewer physical parts, making them more feasible for real biological systems.
Another important distinction is the type of functions each approach can represent. Boolean circuits are naturally suited for discrete decision boundaries, but they struggle with complex, nonlinear mappings across many inputs. IANNs, on the other hand, can approximate arbitrary nonlinear functions. This enables cells to perform tasks that resemble pattern recognition, such as distinguishing subtle combinations of molecular signals that define a disease state or environmental condition.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Useful applications of an intracellular analog neural network (IANN) can be cell-based diagnostics and therapy of cancer, where a cell must interpret a complex molecular environment and make a precise decision about whether to release a therapeutic drug. This is a setting where Boolean genetic circuits struggle, because disease states are rarely defined by single markers or sharp thresholds; instead, they emerge from subtle, multidimensional patterns of signals,which are impractical to engineer with boolean logic gates.
Concept Engineering an immune cell that can distinguish between healthy tissue and tumor tissue by integrating multiple intracellular and extracellular signals. The cell evaluates a combination of cues, such as surface receptors, cytokine levels, metabolic indicators, and stress signals (pick peer reviewed literature for input adoption) and determines whether the overall pattern corresponds to a malignant state. If the pattern matches, the cell releases a therapeutic molecule; if not, it remains inactive.
Input
The inputs to the IANN would be continuous biochemical signals, for example:
Expression levels of tumor-associated antigens
Cytokine concentrations such as IL-6 or TGF-β in the microenvironment
Indicators of hypoxia or altered metabolism
Output The output is also continuous but can be coupled to thresholded downstream actions. For example:
Low output function → no response (cell remains inactive)
Intermediate output → mild response (e.g., secretion of signaling molecules to recruit immune cells)
High output → full activation (e.g., cytotoxic killing, release of therapeutic proteins, or induction of apoptosis in the target cell)
Advantages
This application benefits directly from the properties of IANNs. The system must interpret noisy, overlapping biological signals and map them to a decision boundary that is not easily expressible as a simple logical rule. An IANN can approximate this complex mapping, remain robust to fluctuations, and avoid false positives that would arise from single-marker detection.
Limitations
Two main limitations apply. First, there is the implementation issue: encoding precise weights and nonlinearities in biochemical systems is difficult, as binding affinities and expression levels are not infinitely tunable and are subject to drift over time. Second, training and calibration remain a major challenge; unlike digital circuits, the correct parameter set must often be found through iterative screening or evolution, which is experimentally intensive.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Part 2
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials usually based on mycelium (the filamentous network of fungi) are increasingly used as sustainable alternatives to plastics, foams, leather, and some construction materials.
Mycelium packaging materials
These are grown by letting mycelium bind loose biomass into a foam-like structure and are used for protective packaging (e.g., replacing Styrofoam). Their main advantage is that they are fully biodegradable, low-energy to produce, and utilize waste streams as feedstock. However, they suffer from lower mechanical consistency, reduced moisture resistance, and currently higher costs compared to petrochemical foams.
Mycelium-based construction materials
These include insulation panels, acoustic materials, and experimental bricks. They offer a very low carbon footprint, good thermal and sound insulation, and natural fire resistance. In contrast, they have much lower compressive strength than concrete or engineered wood, are sensitive to moisture, and face significant regulatory hurdles before widespread adoption in construction.
Mycelium leather and emerging fungal textiles
These materials are derived from fungal structures grown into dense mats or processed into fiber-like forms for use in fashion, upholstery, and potentially broader textile applications. They offer clear advantages, including avoiding animal use, significantly reducing environmental impact, and enabling tunable material properties during growth. Additionally, they hold promise as biodegradable alternatives to synthetic fibers produced through low-energy processes. However, they currently face several limitations: lower durability compared to high-quality leather, reliance on coatings to achieve desired performance characteristics, insufficient mechanical strength for many applications, and ongoing challenges in processing and scaling production with consistent quality.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi represent an underexploited chassis for synthetic biology. Unlike bacteria, they are eukaryotic, naturally adapted to degrade complex substrates, and already widely used in industry. This makes them particularly attractive for engineering applications that require secretion, material formation, or complex metabolic processing.
Enhancing material properties for fungal biomaterials
One compelling direction is to engineer fungi to produce mycelium-based materials with improved mechanical strength, water resistance, or functionality. This could involve modifying cell wall composition (e.g., chitin, glucans), introducing crosslinking proteins, or enabling secretion of hydrophobic compounds.
Programming fungi for advanced biomanufacturing
Fungi could be engineered to produce high-value compounds such as pharmaceuticals, enzymes, or specialty chemicals, especially those requiring post-translational modifications. Compared to bacteria, fungi (as eukaryotes) can correctly fold complex proteins, perform glycosylation, and secrete products efficiently into the medium.
Improving degradation of complex waste streams
Fungi are naturally capable of breaking down lignocellulose, plastics, and other recalcitrant materials. This enables applications in waste valorization and circular bioeconomy systems.
Developing functional living materials
Fungi could be programmed to create living materials that respond to environmental stimuli, such as self-healing construction materials or responsive insulation. This might involve integrating sensing circuits with growth or material production pathways.
Week 9 HW: Cell-Free Systems
Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis provides major advantages over traditional in vivo protein expression because the reaction occurs outside living cells, giving researchers direct control over reaction conditions and components. Variables such as DNA concentration, salts, energy substrates, cofactors, temperature, and additives can be adjusted independently without needing to maintain cell viability, allowing rapid optimization and faster experimental iteration.
Another key advantage is flexibility: proteins can be expressed immediately after adding DNA templates, without time-consuming cloning, transformation, or cell culturing steps. Cell-free systems also allow incorporation of non-natural amino acids, toxic proteins, or synthetic circuits that would otherwise harm or interfere with living cells.
Two important cases where cell-free expression is more beneficial than cell-based production are:
Expression of toxic proteins — proteins that damage membranes, inhibit metabolism, or kill cells can often still be produced efficiently in cell-free systems because there is no living host to maintain.
Rapid prototyping of genetic constructs or biosensors — cell-free systems enable fast testing of promoters, enzymes, fluorescent proteins, or metabolic pathways within hours rather than days, making them highly useful for synthetic biology and high-throughput design workflows.
Describe the main components of a cell-free expression system and explain the role of each component.
A Cell-free protein synthesis system contains several core components that work together to synthesize proteins outside living cells:
Cell lysate (extract): The lysate is the main biological machinery of the system and is typically prepared from organisms such as E. coli. It contains ribosomes, tRNAs, aminoacyl-tRNA synthetases, translation factors, metabolic enzymes, and often RNA polymerase, all of which are required for transcription and translation.
DNA template: The DNA plasmid or linear DNA contains the gene encoding the target protein under a promoter such as T7. This serves as the instruction set for producing mRNA and ultimately the protein of interest.
Amino acids: Free amino acids are supplied as the building blocks used by ribosomes during protein synthesis.
Energy source / energy regeneration system: Molecules such as phosphoenolpyruvate (PEP), 3-PGA, or ATP regeneration substrates provide the energy needed for transcription, translation, and other enzymatic reactions over the course of the reaction.
Nucleotides (NTPs): ATP, GTP, CTP, and UTP are needed for RNA synthesis during transcription and also contribute energy for translation processes.
Salts and buffering agents: Components such as magnesium glutamate, potassium glutamate, and HEPES buffer maintain optimal ionic strength and pH. Magnesium is especially critical for ribosome stability and enzyme activity.
Cofactors and supplements: Additional molecules such as NAD, CoA, folinic acid, spermidine, or tRNAs help support enzyme function, metabolic balance, and efficient translation.
RNA polymerase (if not already in lysate): In T7-based systems, T7 RNA polymerase transcribes the DNA template into mRNA. Some lysates already contain this enzyme, such as BL21(DE3)-derived extracts.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision is critical because cell-free protein synthesis consumes large amounts of ATP and GTP during transcription, translation, aminoacyl-tRNA charging, and protein folding, but unlike living cells the reaction cannot continuously regenerate energy through normal metabolism. Once ATP is depleted, protein synthesis slows or stops, so fluorescence output over a long incubation depends strongly on maintaining energy supply.
One method is to include an energy regeneration system, such as 3-phosphoglycerate (3-PGA) or phosphoenolpyruvate (PEP), together with the enzymes in the lysate that convert these substrates into ATP. This continuously replenishes ATP during the reaction, allowing longer protein production and higher final fluorescence over the 36-hour incubation.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems (commonly based on E. coli lysates) are fast, inexpensive, and produce very high protein yields. They are ideal for expressing simple bacterial proteins or rapidly prototyping genetic circuits because they have efficient transcription and translation machinery and are easy to optimize. However, they generally lack advanced post-translational modifications such as glycosylation and may struggle with complex eukaryotic proteins.
For a prokaryotic system, I would choose to produce sfGFP because it folds efficiently, matures rapidly, and does not require complex modifications. Its robustness makes it highly suitable for high-yield expression in E. coli-based cell-free reactions.
Eukaryotic cell-free systems (such as wheat germ, insect, or mammalian extracts) better support complex protein folding, disulfide bond formation, and post-translational modifications that are important for many human proteins. Although they are usually more expensive and produce lower yields, they are advantageous for expressing proteins that require native-like processing and functionality.
For a eukaryotic system, I would choose a human monoclonal antibody fragment or another glycosylated therapeutic protein because proper folding and post-translational modifications are critical for biological activity. A eukaryotic cell-free system would better reproduce the cellular environment needed for correct structure and function.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are difficult to express because their hydrophobic regions often aggregate or misfold without a membrane environment. To optimize expression in a Cell-free protein synthesis system, I would test reactions containing liposomes, nanodiscs, or mild detergents to support proper insertion and folding.
I would vary conditions such as Mg²⁺ concentration, temperature, DNA concentration, and chaperone addition while measuring both fluorescence and protein functionality. Lower temperatures and membrane mimics would likely improve soluble, functional protein yield by reducing aggregation and promoting correct folding.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Homework question from Kate Adamala
Pick a function and describe it.
antibiotic-sensing reporter vesicle
What would your synthetic cell do? What is the input and what is the output?
The synthetic cell detects the antibiotic tetracycline in the environment.
Input: tetracycline outside the vesicle.
Output: green fluorescence from sfGFP expression inside the vesicle.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, a bulk cell-free reaction could detect tetracycline, but encapsulation makes it more “cell-like” and allows compartmentalized sensing, which is useful for artificial-cell experiments.
Could this function be realized by genetically modified natural cell?
Yes. A bacterium could be engineered with a tetracycline-responsive promoter controlling GFP expression. However, living cells introduce growth, metabolism, toxicity, and regulatory complexity.
Describe the desired outcome of your synthetic cell operation.
When tetracycline is present, the synthetic cell produces GFP and becomes fluorescent. Without tetracycline, fluorescence remains low.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
The membrane would be a lipid vesicle, for example made from POPC and cholesterol. This creates a stable artificial membrane around the cell-free reaction.
What would you encapsulate inside? Enzymes, small molecules.
cell-free Tx/Tl mastermix
DNA encoding the reporter circuit
amino acids
NTPs
salts and buffer
ATP regeneration system
TetR regulatory protein or DNA encoding TetR
sfGFP reporter gene
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
An E. coli cell-free system is sufficient because the circuit can use a bacterial tetracycline-responsive promoter. A mammalian system is not needed unless using mammalian promoters such as Tet-ON.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Tetracycline is small and can diffuse across membranes to some extent, but permeability may be limited. To improve input access, I would include a membrane pore such as α-hemolysin so small molecules can enter the vesicle.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids:
POPC: main phospholipid for vesicle membrane
Cholesterol: improves membrane stability
Optional: PEG-lipid such as DSPE-PEG to reduce vesicle aggregation
hla from Staphylococcus aureus: encodes α-hemolysin pore, if membrane permeability needs improvement
How will you measure the function of your system?
I would measure GFP fluorescence over time using a plate reader or fluorescence microscope. The key comparison would be vesicles with tetracycline versus vesicles without tetracycline; successful function means tetracycline-treated vesicles show clearly higher fluorescence after incubation.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
A smart protective textile could use freeze-dried cell-free biosensors printed into fabric to detect harmful chemicals or pollutants and produce a visible color or fluorescence warning.
How will the idea work, in more detail? Write 3-4 sentences or more.
The textile would contain small printed patches of freeze-dried cell-free expression mix, DNA sensor circuits, and color-producing reporter proteins. When the fabric becomes wet from sweat, rain, or a small added water droplet, the cell-free system rehydrates and becomes active. If a target molecule, such as a toxic industrial chemical, pesticide, or air pollutant, is present, it triggers expression of a reporter such as GFP, mScarlet, or an enzyme that produces a visible pigment. The result would be a wearable, low-cost warning system for workers, soldiers, farmers, or people in polluted environments.
What societal challenge or market need will this address?
Many people are exposed to unsafe chemicals without immediate detection tools, especially in agriculture, factories, disaster zones, and low-resource settings. A sensor textile could provide fast, portable, and easy-to-read exposure information without needing electronics or laboratory equipment.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Because cell-free reactions need water, the sensor patches would be sealed in dry protective microcapsules or printed hydrogel spots that activate only when intentionally wetted or exposed to moisture. Stability could be improved by freeze-drying with protectants such as trehalose or sucrose. Since many cell-free sensors are single-use, the textile could include replaceable sensor patches, similar to disposable test strips integrated into reusable clothing.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-duration spaceflight exposes astronauts to radiation, microgravity, and closed habitats, all of which can increase cellular stress and DNA damage. Monitoring biological stress quickly and with limited equipment is important because astronauts cannot rely on full Earth-based laboratories during missions. This topic is significant for humanity because future Moon and Mars missions will require autonomous health monitoring. It is scientifically interesting because space conditions may change how DNA damage and repair pathways behave compared with Earth.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
p53 pathway stress response, represented by a synthetic DNA construct expressing fluorescent protein in response to DNA damage-related signaling.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The p53 pathway is strongly linked to DNA damage, radiation response, and cellular stress. In space, increased radiation exposure may cause DNA lesions that activate stress-response pathways. A simplified BioBits cell-free reporter cannot fully recreate a human cell, but it can model whether a designed genetic sensor circuit can convert a DNA damage-related input into a visible fluorescent output.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
One hypothesizes that a freeze-dried BioBits cell-free reaction can be used as a simple space-compatible biosensor for DNA damage-related stress signals. If a DNA damage-mimicking input or regulatory DNA sequence is added to the reaction, the system should produce a stronger fluorescent signal than the negative control. The reasoning is that cell-free systems are lightweight, shelf-stable, and do not require living cells, making them useful for space biology experiments where resources, storage, and safety are constrained. The research goal is to test whether a cell-free fluorescent reporter could serve as a prototype for astronaut health or environmental radiation monitoring.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would prepare BioBits cell-free reactions containing a fluorescent reporter construct and test three conditions: no input negative control, positive control with constitutive fluorescent protein expression, and experimental reaction with the DNA damage-responsive sensor design. Reactions would be rehydrated with water and incubated. Fluorescence would be measured using the P51 Molecular Fluorescence Viewer over time, for example at 0, 6, 12, and 24 hours. The main data would be fluorescence intensity and time to visible signal. A successful sensor would show higher fluorescence in the experimental condition than the negative control.
Part B: Individual Final Project
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1
Done ;)
Submit this Final Project selection form if you have not already.
Done ;)
Begin planning how you will write your final project documentation based on these guidelines
Done ;)
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.
Done ;)
Week 10 HW: Advanced Imaging and Measurements
Final Project
Measurement Plan
The project’s central question — do AI-guided designs outperform standard, random, and unguided foundation-model designs in cell-free expression? — requires measurements at three levels: the DNA (to confirm we test what we designed), the protein output (the primary readout feeding the surrogate), and the surrogate model itself (to know whether the loop is learning).
1. DNA Identity and Quality (Build verification)
Before any cell-free reaction, every linear construct from Twist is verified:
Sequence identity — Sanger sequencing of the variable region using a T7-anchored primer (~$5/reaction, Eurofins/Azenta), aligned to the design CSV. Run on all Grade-A candidates (e.g. construct #40) and on any anomalous downstream result.
DNA concentration and purity — NanoDrop (A260/A280, A260/A230), confirmed by Qubit dsDNA HS for top candidates. Target ≥10 ng/µL, A260/A280 ≈ 1.8–2.0.
DNA integrity — 1% agarose gel electrophoresis (GelRed, 100 V, 30 min) on a per-batch sample, or Agilent TapeStation for higher throughput. Single tight band at ~770 bp confirms full-length, non-degraded DNA.
2. Protein Expression Output (Test step — primary readout)
The headline measurement that drives the entire loop. Each construct is run in triplicate in 10 µL TX-TL reactions in a black, clear-bottom 96-well plate, sealed, and incubated at 30 °C in a kinetic-fluorescence plate reader (BioTek Synergy H1 or Tecan Spark) with 485/528 nm ex/em for sfGFP. Reads every 3 min for 8 h yield ~160 timepoints per well, from which four features are extracted:
Fluorescence AUC — the single number used to train the surrogate.
Maximum fluorescence (F_max) — proxy for total protein yield.
Time to signal (t_signal) — proxy for expression rate.
Replicate CV — the Aim-2 reproducibility metric.
A purified recombinant sfGFP standard curve on every plate converts RFU to nM, making the readout comparable across plates and rounds. For top hits, total protein is independently confirmed by BCA assay (Thermo Pierce, ~$100/kit) and SDS-PAGE with Coomassie (clean band at ~27 kDa). In later rounds, when sfGFP is replaced by C1-metabolism enzymes, intact-mass LC-MS (Waters BioAccord) catches truncation products that would otherwise masquerade as low expression.
3. Construct-Level Sequence Features (surrogate inputs)
Computed in silico on every designed construct and stored in the library CSV:
Three quantities tracked on the model between rounds:
Held-out R² and MAE on a 20% test split. Expected trajectory: ~0.3 (Round 1, n=47) → ~0.5–0.6 (Round 3, n=144).
Top-K enrichment — fraction of the surrogate’s predicted top-10 that fall in the experimental top-10 of the next round. The operationally relevant metric.
Uncertainty calibration — reliability diagrams of predicted std vs. observed error; essential for the exploration/exploitation balance.
The plate-reader fluorescence AUC is the single most important measurement — it is what the surrogate trains on and what the entire closed loop optimizes for. Every other measurement either verifies the DNA matches the design, catches artifacts that would corrupt the fluorescence signal, or assesses whether the loop itself is learning.
Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Calculating this from the exact sequence, then adjust for the GFP chromophore because that modification changes the intact mass. I’ll also flag the common ambiguity around N-terminal methionine processing.
So the expected intact mass for the mature fluorescent eGFP standard is approximately:
27,986.6 Da, or 27.987 kDa.
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine z for each adjacent pair of peaks (n, n+1) using: z = (m/z_n+1)/((m/z_n) - (m/z_n+1))
I selected two adjacent charge-state pairs from Figure 1:
The assignment asks us to use the adjacent charge-state approach on the intact eGFP LC-MS spectrum, using the labeled m/z peaks in Figure 1. For ESI protein spectra, adjacent peaks usually differ by one charge, and the molecular weight can be calculated from the relationship m/z=(MW+zH)/z, where H is the proton mass.
Pair 1: 903.7148 and 875.4421
z=875.4421 / 903.7148−875.4421 = 30.96 (31)
Waters Part II — Secondary/Tertiary structure
Waters Part III — Peptide Mapping - primary structure
The eGFP sequence contains:
3.1
analyzing the sequence in Benchling, we get to
20 Lysines (K)
6 Arginines (R)
3.2
Using the Expasy Tool PeptideMass according to the instructions i arrive at:
27 peptides
3.3
18 chromatographic peaks
3.4
No, as there are 27bpeptides predicted, but only 18 peaks counted
3.5
most abundant peptide peak at
m/z = 525.76712
isotopic spacing is 1/z
z = 2
Calculating the single charged Mass
MH+ = (525.767 × 2) − 1.0078 = 1050.53 Da
3.6
unsure, as none of the masses fit.
Waters Part IV — Oligomers
The goal is to identify KLH oligomeric species in the CDMS mass spectrum using the known masses of the KLH polypeptide subunits. The Homework gives the KLH subunit masses as 7FU = 340 kDa and 8FU = 400 kDa, and asks us to assign the oligomeric states on the spectrum.
Given subunit masses
Subunit
Mass
7FU
340 kDa
8FU
400 kDa
Calculations and peak assignments
Oligomeric species
Calculation
Theoretical mass
Approximate location on spectrum
7FU Decamer
(10 x 340 kDa)
3400 kDa = 3.4 MDa
Peak near 3.4 MDa
8FU Didecamer
(20 x 400kDa)
(8000 kDa) = 8.0 MDa
Major peak near 8.33 MDa
8FU 3-Decamer
(30 x 400 kDa)
(12000 kDa) = 12.0 MDa
Peak near 12.67 MDa
8FU 4-Decamer
(40 x 400 kDa)
(16000 kDa) = 16.0 MDa
Weak/broad signal near 16–17 MDa
Final answer
The KLH oligomeric species can be assigned as follows:
7FU Decamer: near 3.4 MDa
8FU Didecamer: near 8.0–8.33 MDa
8FU 3-Decamer: near 12.0–12.67 MDa
8FU 4-Decamer: near 16.0–17.0 MDa
The strongest species in the spectrum is the 8FU didecamer, observed as the largest peak around 8.33 MDa.
Waters Part V — Did I make GFP?
I didn’t receive any further documents.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Let me try to become a TA for How to biomanufacture almost anything
Make a note on your HTGAA webpages including:
what you liked about the project
The final bioart project looked crazy cool. I love seeing all my contributers
what about this collaborative art experiment could be made better for next year.
Focusing more on the protocol generation for Gingko Nebula. While the artwork is a cool and visually stimulating project, it would be cooler to understand the automated lab part.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
The lysate contains the cellular machinery required for transcription and translation, including ribosomes, tRNAs, translation factors, metabolic enzymes, and cofactors extracted from E. coli. The incorporated T7 RNA polymerase drives strong transcription from T7 promoters, enabling high protein production in the cell-free system.
Salts/Buffer
Potassium Glutamate
Potassium glutamate makes sure that the the major intracellular ionic environment and helps stabilize ribosomes and enzymatic activity are provided. Glutamate also mimics the natural cytoplasmic conditions of E. coli, improving protein synthesis efficiency.
HEPES-KOH pH 7.5
HEPES is a buffering agent that maintains a stable pH during the reaction. Stable pH is essential because transcription, translation, and energy metabolism enzymes are highly pH sensitive.
Magnesium Glutamate
HEPES is a buffering agent that maintains a stable pH during the reaction. Stable pH is essential because transcription, translation, and energy metabolism enzymes are highly pH sensitive.
Potassium phosphate monobasic
This phosphate salt contributes to phosphate buffering capacity and helps maintain ionic strength. It also participates in maintaining phosphate availability for metabolic reactions.
Potassium phosphate dibasic
The dibasic phosphate form works together with the monobasic form to create a phosphate buffer system. This stabilizes pH and supports phosphate-dependent enzymatic reactions during long incubations.
Energy / Nucleotide System
Ribose
Ribose acts as a precursor for nucleotide regeneration through endogenous salvage pathways present in the lysate. It supports sustained RNA synthesis over long reaction times.
Glucose
Glucose serves as a slow-release energy substrate that fuels glycolytic metabolism within the lysate. This enables continuous ATP regeneration during extended incubations.
AMP
AMP is a nucleotide precursor that can be enzymatically converted into ATP inside the lysate. This reduces the need to directly add expensive high-energy triphosphates.
CMP
CMP serves as a precursor for cytidine nucleotide regeneration. Cellular enzymes recycle it into higher-energy nucleotide forms needed for RNA synthesis.
GMP
Although listed as 0 µM in the formulation, GMP would normally function as a guanosine nucleotide precursor for RNA synthesis and energy metabolism.
UMP
UMP is a precursor for uridine nucleotides used in RNA synthesis. Lysate enzymes convert it into higher phosphorylated nucleotide forms as needed.
Guanine
Guanine can be salvaged by endogenous enzymes to generate GMP and eventually GTP. This provides a cheaper and potentially more stable alternative to directly supplying guanosine nucleotides.
Translation Mix (Amino Acids)
17 Amino Acid Mix
This mixture supplies most amino acids required for protein synthesis. Providing amino acids externally prevents depletion during translation and improves protein yield.
Tyrosine
Tyrosine is added separately because of its relatively low solubility and instability in standard amino acid mixes. Separate optimization allows higher effective concentrations without precipitation.
Cysteine
Cysteine is also supplied separately because it is chemically reactive and prone to oxidation. Independent addition improves stability and availability for protein synthesis.
Additives
Nicotinamide
Nicotinamide is a precursor for NAD⁺ biosynthesis and supports redox metabolism in the lysate. Sustaining cellular redox balance is particularly important during long-duration reactions.
Backfill
Nuclease Free Water
Nuclease-free water is used to bring the reaction to the desired final volume while preventing degradation of RNA and DNA templates by contaminating nucleases.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP system relies on directly supplying high-energy molecules such as phosphoenolpyruvate (PEP) and fully phosphorylated nucleoside triphosphates (ATP, GTP, CTP, UTP), enabling very rapid and high initial protein production. However, this approach is relatively expensive and less sustainable because energy substrates are consumed quickly and inhibitory byproducts can accumulate.
In contrast, the 20-hour NMP-ribose-glucose system uses lower-energy nucleotide precursors (AMP, CMP, UMP, guanine), along with ribose and glucose, allowing the endogenous metabolic enzymes in the lysate to slowly regenerate nucleotides and ATP over time. This creates a more metabolically self-sustaining and cost-effective reaction optimized for long-duration protein expression.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because the lysate contains nucleotide salvage enzymes that convert guanine into GMP, then GDP, and finally GTP through sequential phosphorylation reactions. The system therefore generates the required GTP internally rather than adding GMP or GTP directly to the reaction mixture.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
sfGFP is engineered for very efficient folding and rapid chromophore maturation, making it highly robust in cell-free systems even under partially stressful reaction conditions. Its strong folding stability usually gives high fluorescence output and reliable expression readouts.
mRFP1
mRFP1 has a relatively slower chromophore maturation process compared to GFP variants, so fluorescence can lag behind actual protein synthesis in cell-free reactions. Red fluorescent proteins also tend to have lower brightness and more complex folding pathways.
mKO2
mKO2 is known for fast maturation among orange fluorescent proteins, which improves early signal detection in cell-free systems. However, its fluorescence can be somewhat sensitive to pH changes, affecting readout consistency if reaction conditions fluctuate.
mTurquoise2
mTurquoise2 has very high quantum yield and brightness, making it excellent for sensitive fluorescence detection in low-expression cell-free reactions. Like many CFPs, proper chromophore formation depends strongly on correct folding and oxygen availability.
mScarlet_I
mScarlet-I was engineered for rapid maturation and exceptionally high brightness among red fluorescent proteins, which improves signal intensity in cell-free systems. Nevertheless, red chromophore formation still generally requires more maturation time than GFP-like proteins.
Electra2
Electra2 is a more functionally specialized fluorescent protein whose optical behavior depends on conformational or environmental changes, making its readout potentially more sensitive to membrane mimicry, folding efficiency, and reaction composition in cell-free systems. Its larger and more complex architecture may also reduce expression efficiency relative to simpler fluorescent proteins.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mScarlet-I, increasing the energy regeneration system and maintaining adequate magnesium/potassium salt balance in the cell-free mastermix will improve total fluorescence over 36 hours by sustaining translation longer and supporting proper folding/chromophore maturation.
Specifically, slightly increasing 3-PGA or PEP as the energy substrate, together with optimized Mg-glutamate/Mg²⁺ and K-glutamate, should extend protein production and help mScarlet-I reach its high-brightness mature state. The expected effect is higher final red fluorescence after 36 hours, because more protein is synthesized and more of it has time to mature into the fluorescent chromophore.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
I did not receive an email.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate10 μL of 2X Optimized Master Mix from above2 μL of assigned fluorescent protein DNA template2 μL of your custom reagent supplementsTotal: 20 μL reaction
I haven’t received any data on the experiment.
Week 12 HW: Building Genomes
Continue making progress this week on your Individual Final Project and on DNA orders (due Friday midnight ET).
Done ;)
Week 13 HW: AI, SynBio, and Scaling Health
Homework: Work on your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
Done ;)
Week 14 HW:Biodesign & Biofabrication
Homework: Finish your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
AI-guided closed-loop design of complex biological constructs using foundation model and (cell-free systems) SECTION 1: ABSTRACT Significance. Designing biological constructs that perform reliably remains a major bottleneck in synthetic biology. Enzymes involved in C1 metabolism, such as those mediating methane-to-methanol conversion, are notoriously difficult to express and optimize, yet methane and other C1 feedstocks represent an abundant but underutilized carbon source with applications ranging from sustainable chemical production to carbon sequestration. Today, plasmid design still relies heavily on historical use, standard parts, and intuition (“vibes”), with optimization tools that are scattered and focused primarily on the coding sequence rather than the surrounding regulatory architecture.
Part D: Group Brainstorm on Bacteriophage Engineering Due to later start of our Node, we had limited time to find groups and set up a meeting, therefore the drafts of our group are mainly individual, and not discussed
Goal We target two complementary objectives: (A) Increased stability of the L protein, specifically engineering DnaJ-independent variants that fold correctly without host chaperone assistance; and (B) Higher toxicity / faster lysis, by optimizing the transmembrane oligomerization interface to accelerate pore formation. Goal A is prerequisite to Goal B: a stable, chaperone-independent L is resistant to the most documented E. coli escape mechanism (DnaJ P330Q mutation), and faster lysis narrows the window for resistance acquisition.
Subsections of Projects
Individual Final Project
AI-guided closed-loop design of complex biological constructs using foundation model and (cell-free systems)
SECTION 1: ABSTRACT
Significance. Designing biological constructs that perform reliably remains a major bottleneck in synthetic biology. Enzymes involved in C1 metabolism, such as those mediating methane-to-methanol conversion, are notoriously difficult to express and optimize, yet methane and other C1 feedstocks represent an abundant but underutilized carbon source with applications ranging from sustainable chemical production to carbon sequestration. Today, plasmid design still relies heavily on historical use, standard parts, and intuition (“vibes”), with optimization tools that are scattered and focused primarily on the coding sequence rather than the surrounding regulatory architecture.
Broad Objective. This project develops a closed-loop, AI-guided workflow that combines genomic foundation models, lightweight predictive modeling, and cell-free expression experiments to accelerate the design of high-performing genetic constructs that can ultimately be deployed in living cells.
Hypothesis. Foundation model–generated and AI-selected constructs will outperform standard and random designs in protein expression, and an iterative Design–Build–Test–Learn (DBTL) loop will progressively enrich for high-performing variants without requiring retraining of the underlying foundation model.
Specific Aims. (1) Experimentally test whether Evo 2–generated and AI-selected constructs improve protein expression in cell-free systems relative to standard and random designs, using a fluorescence readout. (2) Extend the workflow beyond simple expression to capture robustness, stability over time, reproducibility, and more complex or fully generated regulatory regions, including those needed for challenging or non-native proteins. (3) Generalize the system into a biological design engine that transfers from cell-free prototyping to host organisms such as E. coli and non-model species, with C1 bioconversion enzymes as a target application.
Methods. Candidate libraries spanning standard parts, random variants, and Evo 2–generated sequences are produced in the variable regulatory region of a fixed construct template, pre-screened with sequence-quality heuristics (GC content, homopolymers, forbidden sites), and exported as CSV/FASTA for synthesis. Constructs are evaluated in an E. coli–based TX-TL cell-free system with fluorescence as the readout (substituted in this iteration by a generative-model-based simulator producing point estimates with uncertainty, due to lack of wet-lab access). Each [sequence, measurement] pair is encoded as a feature vector and used to train a Random Forest surrogate model that ranks the next round of candidates, closing the DBTL loop across three iterative rounds.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of my final project is to test whether foundation model–generated and AI-selected constructs can improve protein expression in cell-free systems by utilizing a closed-loop Design–Build–Test–Learn (DBTL) workflow that integrates Evo 2 (40b/1b) for sequence generation, heuristic and surrogate-model–based pre-screening, an E. coli–based TX-TL cell-free expression system for validation, and a Random Forest regression surrogate model for ranking and informing subsequent rounds.
A library of constructs — comprising standard designs, random variants, and Evo 2–generated sequences in the variable regulatory region of a fixed construct template — will be generated through the project’s AI-Guided Construct Designer web application, pre-screened against quality heuristics (GC content, homopolymer stretches, forbidden restriction sites), and exported as CSV/FASTA files ready for ordering from DNA synthesis providers such as Twist Bioscience. Constructs will be expressed in a TX-TL cell-free system following the linear-DNA rapid-prototyping protocol of Sun et al. (2014) and evaluated by fluorescence readout. Each [sequence, measurement] pair will be converted into a feature vector and used to fit a RandomForestRegressor, whose predictions will then guide candidate ranking in the next iteration of the loop. Three sequential DBTL rounds will be performed, and performance will be compared head-to-head across standard, random, foundation model–generated, and AI-guided designs. Because wet-lab access was not available during this course, the experimental measurement step is substituted in the current iteration by a generative-model-based simulator that returns fluorescence point estimates with associated uncertainty; the workflow is otherwise fully end-to-end and wet-lab-ready.
Aim 2: Development Aim
Following a successful Aim 1, the next step is to extend the system beyond optimizing simple expression toward more robust and realistic construct optimization. This means (i) running the closed loop with real wet-lab data — a validation campaign is already discussed with the Biopunk Labs cohort, replacing the surrogate fluorescence simulator with measured TX-TL output — and (ii) incorporating additional performance metrics such as expression stability over time and reproducibility across replicate batches and reaction conditions. The design space will be expanded from substituting a fixed variable region to generating more complex or fully de novo regulatory architectures (promoter, 5’ UTR, RBS, terminator) as a single jointly optimized block. On the modeling side, the hand-crafted feature vector inside the surrogate grader will be replaced with learned embeddings drawn directly from the foundation model, so that the ranker reasons over sequence representations rather than tabulated descriptors — an approach that was prototyped during the course with mixed results and deserves a more thorough treatment with larger, real-data training sets. This stage also enables a deeper evaluation of how well foundation model–generated sequences translate into reliable experimental performance, particularly for challenging or non-native proteins that resist standard design heuristics.
Aim 3: Visionary Aim
The long-term vision is to develop a generalizable, self-improving biological design engine that optimizes genetic constructs across biological contexts and host organisms — bridging cell-free prototyping with reliable in vivo deployment in E. coli as well as non-model chassis. Realized fully, such a system would challenge the prevailing paradigm in synthetic biology, in which plasmid design still leans on historical use, standard parts, and intuition, with optimization tools scattered across the workflow and focused mainly on the coding sequence rather than the regulatory architecture surrounding it. By tightly coupling generative foundation models with rapid experimental feedback in a continuously learning loop, the engine would replace artisanal construct engineering with a data-driven workflow where computational models and experimental systems continuously inform each other, compounding in capability with every round. The most immediate impact lies in industrial biotechnology — particularly the design of enzymes and pathways for C1 bioconversion, where reliable expression of complex methane- and methanol-handling enzymes remains a key barrier to using methane and other abundant C1 feedstocks for sustainable chemical production and carbon sequestration. More broadly, this approach would lower the activation energy for engineering any difficult-to-express protein, opening up enzyme classes, pathways, and host organisms that are currently out of reach for standard cloning workflows.
SECTION 3: BACKGROUND
Background and Literature Context
Synthetic biology has matured into a discipline capable of constructing increasingly complex genetic systems, yet a persistent bottleneck remains: the reliable design of constructs whose expression behavior can be predicted a priori. Despite decades of accumulated parts catalogs (e.g., the iGEM Registry, Anderson promoter series, standardized RBS collections), most plasmid design still proceeds by combining well-characterized “standard parts” with intuition drawn from historical use — an approach that breaks down precisely where it is most needed: for non-native, structurally complex, or metabolically demanding proteins. Existing optimization tools tend to be scattered across the workflow and focus narrowly on the coding sequence (codon optimization, rare-codon avoidance) rather than the regulatory architecture (promoter, 5’ UTR, RBS, spacer regions) that frequently dominates expression variance. Cell-free TX-TL systems, as pioneered for rapid linear-DNA prototyping by Sun et al. (2014), offer a fast, low-overhead readout for testing many constructs in parallel, and recent reviews of cell-free synthetic biology (Yurchenko et al., 2024) emphasize the growing role of mechanism-based and data-driven modeling in this space. In parallel, genomic foundation models — most notably Evo 2 (Brixi et al., 2026), trained on genomes spanning all domains of life — have demonstrated the ability to generate and score biological sequences at a scale and quality that was inaccessible only a few years ago. What is still missing is a tight, automated loop that couples these generative models to experimental measurement and uses the resulting data to steer the next round of designs.
Summarization of two peer-reviewed research citations relevant to your research.
Genome modelling and design across all domains of life with Evo 2
Brixi, G., Durrant, M.G., Ku, J. et al. Genome modelling and design across all domains of life with Evo 2. Nature 652, 1349–1361 (2026). https://doi.org/10.1038/s41586-026-10176-5
Evo 2 is a large-scale biological foundation model trained on 9 trillion DNA base pairs from the OpenGenome2 dataset, spanning all domains of life (bacteria, archaea, eukarya, and bacteriophage) with a context window of up to 1 million nucleotides at single-nucleotide resolution. The model uses a novel StripedHyena 2 architecture that combines convolutional and attention mechanisms, enabling efficient training and inference on both short and long genomic sequences. Evo 2 demonstrates strong zero-shot prediction capabilities for mutational effects across proteins, RNAs, and organismal fitness, including accurate pathogenicity prediction for human clinical variants in both coding and noncoding regions. Through mechanistic interpretability analysis using sparse autoencoders, the model reveals learned biological features such as exon-intron boundaries, transcription factor binding motifs, protein secondary structures, and prophage genomic regions. As a generative model, Evo 2 can produce organelle-scale, prokaryotic genome-scale, and eukaryotic chromosome-scale sequences with greater naturalness than previous methods. The researchers also demonstrate experimental validation of designed mammalian chromatin accessibility patterns by coupling Evo 2 with inference-time guidance using predictive models like Enformer and Borzoi. The model, training code, inference code, and OpenGenome2 dataset are fully open-sourced to accelerate biological research, with safety measures including exclusion of eukaryotic-infecting viral sequences to mitigate dual-use risks.
Protocols for Implementing an Escherichia coli Based TX-TL Cell-Free Expression System for Synthetic Biology
Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, Noireaux V. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J Vis Exp. 2013 Sep 16;(79):e50762. doi: 10.3791/50762. PMID: 24084388; PMCID: PMC3960857.
This academic paper describes a detailed five-day protocol for preparing and executing an endogenous Escherichia coli-based transcription-translation (TX-TL) cell-free expression system. Unlike traditional systems that rely heavily on specialized T7 bacteriophage RNA polymerase transcription, this platform preserves endogenous E. coli molecular mechanisms to more accurately mimic in vivo cellular dynamics. The protocol details the bulk preparation of the crucial reagents, including crude cell extract via cost-effective bead-beating lysis, an amino acid solution, and a 3-phosphoglyceric acid (3-PGA) energy solution. It also includes an optimization phase where magnesium glutamate, potassium glutamate, and DTT are strictly calibrated to achieve maximum protein expression yields. Once these storage-stable reagents are created, setting up individual 10-microliter reactions takes less than 8 hours from preparation to final data collection. The system achieves high protein yields comparable to T7-driven commercial alternatives, but at a 98% reduction in material costs. Ultimately, the authors position this adaptable platform as a versatile “biomolecular breadboard” optimized for synthetic biology prototyping, exploring biological circuit assembly, and analyzing regulatory mechanics.
Novelty and Innovation
This project is novel in three connected ways. First, rather than retraining or fine-tuning a foundation model — which is computationally prohibitive and data-hungry — it pairs an off-the-shelf frontier generator (Evo 2) with a lightweight, task-specific surrogate model (Random Forest regressor) that learns from a handful of experimental data points and is used to rank candidates for the next round; the foundation model supplies sequence diversity while the surrogate supplies experiment-informed selection pressure. Second, it operationalizes the full Design–Build–Test–Learn cycle as a single end-to-end web application (the AI-Guided Construct Designer), spanning library generation, heuristic and model-based pre-screening, CSV/FASTA export ready for DNA synthesis vendors such as Twist, and ingestion of measurement data back into the surrogate — replacing the scattered, ad hoc tooling that currently dominates plasmid design. Third, the approach treats the regulatory variable region of a construct as the primary design target rather than the coding sequence, which inverts the usual emphasis of codon-optimization-centric tools and aligns the design space with what most often limits expression in practice. Taken together, these choices challenge the prevailing “standard parts plus intuition” paradigm and offer a route to data-driven construct engineering that compounds in capability with every round of experiments.
Why This Project Matters
The pressing real-world problem addressed here is the unreliable expression of difficult biological constructs — a barrier that gates progress across industrial biotechnology, therapeutic protein production, and metabolic engineering. This matters acutely for C1 bioconversion: methane and other one-carbon feedstocks are among the most abundant and underutilized carbon sources on the planet, and the enzymes that act on them (methane monooxygenases, methanol dehydrogenases, formate-handling enzymes) are notoriously hard to express in heterologous hosts. Unlocking reliable expression of these enzymes would open routes to converting fugitive methane emissions and waste C1 streams into useful chemicals, advancing both sustainable chemical production and carbon sequestration — outcomes with direct climate and public-health relevance. Beyond C1 metabolism, the same workflow lowers the activation energy for engineering any hard-to-express protein, which would broadly accelerate enzyme discovery, pathway engineering, and biomanufacturing. At the field level, a successful demonstration would shift construct design from an artisanal, parts-library-driven craft toward a continuously learning, model-in-the-loop engineering practice — analogous to the shift that machine learning has already produced in protein structure prediction and small-molecule design. Finally, by making the workflow available as an open-source web application with seamless export to DNA synthesis vendors, the project lowers the technical floor for non-specialists, distributed-biology communities (such as Biopunk Labs and iGEM teams), and resource-limited laboratories to participate in cutting-edge construct design.
Ethical Implications
The ethical considerations of this project cluster around three principles. Non-maleficence is most immediately relevant because the tool generates novel DNA sequences and exports them in formats designed to be sent directly to commercial DNA synthesis providers; without appropriate screening, such a workflow could in principle be misused to design sequences with biosecurity concerns, even if that is far from the intended C1-bioconversion use case. Beneficence and justice are at stake in how the technology is distributed: a self-improving design engine could either concentrate capability in a small number of well-resourced labs or, conversely, democratize access to high-quality construct design — the choice depends on licensing, openness, and documentation. Responsibility applies to scientific claims: surrogate models trained on small numbers of cell-free measurements can produce confidently wrong predictions, and exporting “AI-ranked” libraries risks giving users a false sense of certainty about constructs that have never been measured. There are also dual-use considerations specific to generative genomic models, environmental considerations tied to deploying engineered organisms downstream, and authorship/data-provenance questions for any sequences that derive from foundation-model outputs trained on publicly available genomes.
Several concrete measures should be taken to ensure the project is conducted and deployed ethically. Proposed actions: (i) integrate sequence screening against established biosecurity databases (e.g., IGSC-style hazard screening) into the export step, so that any library leaving the tool has passed a hazard check; (ii) display calibrated uncertainty alongside every surrogate-model prediction in the UI, rather than presenting rankings as deterministic; (iii) release the workflow under an open-source license with clear documentation of training data, model versions, and known failure modes; (iv) restrict, at the application layer, generation targeted at known pathogen toxin genes or other flagged sequence classes; and (v) for any future wet-lab campaigns (such as the planned Biopunk Labs validation), follow institutional biosafety review and contain all work to non-pathogenic, well-characterized chassis. Potential unintended consequences include over-reliance on the surrogate ranker leading to systematically biased libraries that drift away from the experimental ground truth, false negatives that discard genuinely useful designs, and the broader risk of “vibe-engineering” in which users trust the model output without retaining biological intuition. What could be wrong: the central assumption that foundation-model-generated regulatory sequences translate from cell-free to in vivo performance has not been fully validated; the surrogate’s feature representation may miss the actual determinants of expression; and the simulator-based substitution used during this course is itself an approximation that may not reflect true wet-lab variance. Alternatives to address these risks include keeping a human-in-the-loop review step for all libraries above a certain size, restricting initial deployments to well-characterized model organisms, using ensemble or Bayesian surrogates that more honestly report uncertainty, and committing to publish negative results and failure modes alongside successes so that the community can calibrate trust in the tool. Taken together, these measures aim to capture the public-health and sustainability upside of better construct design — particularly for C1 bioconversion and climate-relevant biotechnology — while keeping the dual-use surface and the epistemic risks of AI-guided biology under active, transparent management.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Detailed Experimental Plan with Timeline
The plan below assumes a 12-week timeline that mirrors what would have been executed in a fully wet-lab-enabled version of this project, with the software workflow (already built during the course) feeding directly into the bench work. As the final project timeline allows for only 4 weeks of work, only one cycle of the DBTL cycle is discribed in this section.
An additional caveat is that the major time uncertainty is the ordering of the DNA constructs. For simplicity it was assumed that it will take 1 eek to deliver the constructs. As Sun et al. (2014) described, one wetlab experiemnt cycle takes five days, therefore the wetlab experiment is synonymous for the protocol that is described above.
Task 1 — Define the construct template and target enzyme (Week 1). Fix the construct backbone as a linear DNA template suitable for E. coli TX-TL: T7 promoter — variable regulatory region (~30 bp, including 5’ UTR/RBS/spacer) — coding sequence (CDS) — T7 terminator. The variable region is the design target; the CDS is held constant. For Round 1 the CDS is a fluorescent reporter (deGFP or sfGFP) to give a clean fluorescence readout; later rounds substitute a representative C1-metabolism enzyme (e.g., a soluble methanol dehydrogenase fragment) fused to a reporter for downstream relevance.
Task 2 — Round 1 library design (Week 1). Use the AI-Guided Construct Designer to generate 50 candidate sequences in the variable region: ~10 standard parts (canonical RBS/promoter combinations from the Registry of Standard Biological Parts), ~10 random variants as a negative-control baseline, and ~30 Evo 2–generated sequences (a mix of 1b and 40b model outputs). Pre-screen all candidates with sequence-quality heuristics (GC content within 35–65%, no homopolymer runs >5 nt, no forbidden restriction sites in the cloning context). Export ranked CSV and FASTA files.
Task 3 — Build: order linear DNA (Weeks 2–4). Submit a Twist Bioscience order for the 50 linear-DNA constructs as a gene-fragment library. Twist’s eBlocks or Gene Fragments format is well-suited to TX-TL prototyping because no plasmid cloning is needed; linear DNA can be used directly per Sun et al. (2014).
Task 4 — Test: Round 1 cell-free expression (Week 3). Run all 50 constructs in triplicate in an E. coli TX-TL reaction (10 µL reactions, 96-well plate format) and read fluorescence kinetically at 30 °C over 8 hours in a plate reader. Compute the area under the curve (AUC) of the fluorescence trace as the per-construct readout. Include positive (well-characterized strong RBS) and negative (no-DNA) controls on every plate. Expected result: Evo 2–generated constructs should produce a wider distribution of AUC values than the random baseline, with the top quartile matching or exceeding the standard-parts controls; this is consistent with the simulator-based Round 1 distribution observed during the course (best AUC ~101k).
Task 5 — Learn: train surrogate model (Weeks 3-4). Convert each [sequence, AUC] pair into a feature vector (k-mer frequencies, GC content, predicted secondary structure ΔG, RBS-calculator score, etc.) and fit a RandomForestRegressor on the Round 1 data. Hold out 20% for validation and report R²/MAE. Expected result: R² ≈ 0.3–0.5 with only ~47 training points, sufficient to enrich top-quartile candidates in the next round but not for confident point predictions.
Task 6 — Round 2 library design, build, test, learn (Week 4). Generate a second library of 50 candidates, this time ranking Evo 2 outputs with the trained surrogate rather than only with heuristics. Order via Twist, run cell-free reactions, retrain the surrogate on the combined Round 1 + Round 2 data (~96 points). Expected result: the median AUC of the AI-guided library should rise relative to Round 1, even if the single best construct does not (course simulation showed median improvement with a slightly lower top value, ~89k — this is the classic exploitation-vs-exploration tradeoff and informs Round 3).
This is outside the scope of HTGAA Final Project, for clarity it is included, to appreachiate the circularity and iteration of the DBTL cycle.
Task 7 — Round 3 library design, build, test, learn (Weeks 5-12). Inject an explicit exploration component (e.g., 30% of Round 3 candidates sampled from high-uncertainty regions of the surrogate’s prediction space). Run cell-free reactions, retrain surrogate on all three rounds (~144 points). Expected result: recovery of high-value designs and improved median performance, mirroring the Round 3 simulator output (best AUC ~95k with tighter distribution).
Task 8 — Comparative analysis and reporting (Week 12). Statistically compare AUC distributions across the four design categories (standard, random, foundation model–generated unguided, AI-guided) using a Kruskal-Wallis test followed by pairwise Mann-Whitney with Bonferroni correction. Test the hypothesis that AI-guided > foundation-model-unguided > standard ≥ random. Deposit all sequences, measurements, and trained surrogate weights in an open repository.
Workflow Figure
The DBTL workflow figure illustrates this loop end-to-end: Design (Evo 2 library generation with heuristic and surrogate pre-screening) → Build (CSV/FASTA export to Twist) → Test (TX-TL cell-free expression with fluorescence AUC readout) → Learn (RandomForest surrogate training and ranking for the next round).
Techniques Checklist
Lab Safety
Bioethical Considerations
DNA Editing
DNA Construct Design
Databases (e.g., GenBank, NCBI)
Lab Automation
Creating Code for Laboratory Automation
Designing a Twist Order
Protein Design
Models and Notebooks
Databases
Bioproduction
Registry of Standard Biological Parts
Cell-Free Systems
Cell Free Reactions
Expanded Techniques
Inputing the linear DNA construct. The DBTL cycle begins by inputting your full genetic sequence into the web tool, then identifying and removing the genetic sequence in question and replacing it with the [VARIABLE] placeholder to designate the region for modification. Next, you configure the generation parameters by specifying the number of variants to generate, the region length, and selecting the foundation model that will power the inference. You then decide whether to apply a model-based grader to evaluate the generated variants, and if enabled, adjust the grader’s parameters to customize how variants are scored and ranked. Finally, you execute the inference to generate and evaluate the genetic variants according to your configured settings.
Cell Free Reactions. The Test step of every DBTL round uses an E. coli–based TX-TL system following the linear-DNA rapid-prototyping protocol of Sun et al. (2014), which avoids the multi-day overhead of plasmid cloning and transformation between rounds and is the single biggest enabler of the three-rounds-in-twelve-weeks cadence. Each construct is run in triplicate in 10 µL reactions in a 96-well plate, with fluorescence read kinetically at 30 °C over 8 hours and AUC used as the per-construct readout to capture both expression rate and total yield. Positive controls (well-characterized strong RBS) and negative controls (no-DNA reactions) are included on every plate to anchor cross-round comparisons. Once C1 enzymes replace the fluorescent reporter in later experiments, the same cell-free format will support reporter-fusion or coupled-enzyme assays without needing to redesign the workflow.
This could unfortunately not be validated in the lab, as i didn’t have access to a wetlab. It is purely based on the Sun et al. (2014) and other standard auxiliary protocols
Industry Council Companies Associated with This Project
Twist Bioscience — primary DNA synthesis vendor for all three rounds of linear-DNA libraries; the export format of the AI-Guided Construct Designer is built around Twist’s gene-fragment ordering workflow.
Ginkgo Bioworks — relevant for the Aim 2/3 transition from cell-free prototyping to higher-throughput in vivo validation and scale-up.
Cultivarium — relevant for Aim 3’s extension to non-model host organisms, where the AI-guided design workflow would need to be retargeted to chassis beyond E. coli.
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
What Aspect of the Final Project Was Validated
The aspect validated in this work is the end-to-end software workflow of the closed-loop Design–Build–Test–Learn (DBTL) cycle, specifically the closed-loop behavior of the system across three sequential rounds: foundation-model–driven library generation in the variable regulatory region, heuristic and surrogate-model–based ranking of candidates, simulator-based fluorescence “estimation” with calibrated uncertainty (substituting for wet-lab cell-free expression that was not accessible), and surrogate-model retraining to inform the next round of designs. Because no wet-lab access was available during the course, this is a purely computational validation; its purpose is to demonstrate that the loop holds together mechanically, that the surrogate ranker improves with accumulated data, and that the export pipeline produces wet-lab-ready libraries — making the workflow ready to be plugged directly into a TX-TL setup as soon as bench access is available (discussed with the Biopunk Labs cohort).
To completely validate the project the simluator-based fluorescence “estimation” needs to be replaced with the proper wetlab experiment (see Section 4. Expanded Techniques)
Detailed Validation Protocol
Construct template definition. A fixed linear-DNA template was used in the AI-Guided Construct Designer: T7 promoter — variable regulatory region — fluorescent reporter CDS — T7 terminator. The variable region (~30 bp) was the design target.
Round 1 library generation. Fifty candidate sequences were generated in the variable region by querying Evo 2 (40b and 1b model endpoints) and combining with a small number of standard parts and random variants as baselines.
Pre-screening with heuristics. Each candidate was filtered against quality heuristics: GC content (35–65%), no homopolymer runs >5 nt, no forbidden restriction sites in the cloning context. Candidates failing hard filters were rejected; the rest were ranked.
Surrogate (“test”) measurement substitution. Because no wet-lab access was available, a generative-model-based simulator was used to assign each candidate a fluorescence AUC point estimate together with an uncertainty estimate. Prompts to the simulator encoded the same heuristics used in pre-screening, so the simulator returned biologically plausible (rather than uniformly random) values.
Surrogate ranker training. Each [sequence, simulated AUC] pair was converted into a feature vector (k-mer frequencies, GC content, sequence-quality descriptors, etc) and used to fit a RandomForestRegressor. Random Forest was chosen over deeper models because of strong performance on small tabular datasets and built-in feature-importance interpretability.
Round 2. Fifty new candidates were generated by Evo 2, re-ranked using the trained surrogate (rather than only heuristics), and “measured” by the simulator. The surrogate was retrained on the combined Round 1 + Round 2 data.
Round 3. Another fifty candidates were generated, re-ranked with the now-twice-trained surrogate, and “measured.” The surrogate was retrained on all three rounds.
Export validation. At every round, the library was exported as both CSV (with per-construct metadata: ID, sequence, predicted AUC, uncertainty, category) and FASTA (in a format directly uploadable to Twist Bioscience’s gene-fragment ordering interface).
Cross-round analysis. Best AUC, number of surviving candidates, and full AUC distributions were tracked across rounds to characterize loop behavior.
Synthetic Biology Techniques Used in Validation
This validation drew on several techniques from the course: DNA construct design was central — the project’s web application designs the variable regulatory region of a TX-TL-compatible linear-DNA construct, holding the T7 promoter, reporter CDS, and terminator fixed while varying the regulatory region as the optimization target. Designing a Twist order was practiced end-to-end: every library exports as a CSV/FASTA pair formatted to Twist Bioscience’s gene-fragment specification, including manufacturability pre-checks (GC content, homopolymers, repeats) so the output can be submitted without much effort. (Twist does similar checks in their webinterface, given that rejected variants arent included, this accelerates cycletime) Creating code for laboratory automation was applied throughout: the entire DBTL loop was implemented as a single web application that automates library generation, pre-screening, “measurement,” and surrogate retraining, removing manual handoffs between steps. Finally, databases and the Registry of Standard Biological Parts were used as the source of standard-part baselines (canonical RBS/promoter combinations) that anchor the comparison against random and Evo 2–generated designs, and bioethical considerations were applied throughout — including the recommendation that any library leaving the tool should pass biosecurity screening (e.g., via SecureDNA) before being submitted to a synthesis provider.
Data and Analysis
Three sequential rounds of the DBTL loop were executed in simulation, with the following summary results:
Round
Generated
Surviving
Discarded
Best AUC
Surrogate Training N
1
50
47
3
~101,425
47
2
50
49
1
~89,451
96
3
50
49
1
~95,790
144
Analysis. The validation produces three findings worth noting. First, the loop closes mechanically: each round successfully generated, filtered, “measured,” and trained on data, and the surrogate’s training set grew monotonically from 47 → 96 → 144 points, demonstrating that the data pipeline is intact end-to-end. Second, the best-AUC trajectory across rounds (101k → 89k → 96k) is non-monotonic, which is informative rather than a failure: it reflects the classic exploitation-vs-exploration tradeoff. Round 1 candidates are heuristic-ranked from a broad Evo 2 distribution and occasionally contain a strong outlier; Round 2 is re-ranked by a surrogate trained on only 47 points, which over-exploits a still-noisy estimate of the fitness landscape and slightly drops the top value; Round 3, with a more confident surrogate over 96 points, partially recovers. It is important to keep in mind that the AUC data is estimated, not experimentally validated, I did the analysis as is from the data I got, though I need to mention, that these estimations are error-prone, and therefore no experimental value can be derived from themThird, the surrogate-predicted AUC distributions (the green panels in slide 5) tighten across rounds while the simulated-fluorescence distributions (blue panels) shift in median, consistent with the ranker learning to identify the bulk of mid-to-high performers even when the single best construct is harder to recover. The headline takeaway is that the workflow is wet-lab-ready: the same loop, with the simulator swapped for a TX-TL reaction, can be run directly.
Unexpected Challenges and Mitigation Strategies
Several unexpected challenges arose, almost all on the software-engineering rather than the biological side. First, a significant fraction of the overall project time was consumed by getting the 40b Evo 2 model API endpoint to work reliably — the difficulty was not in the biology but in API authentication, rate limits, and response formatting, which is a useful warning about how much of “AI-guided synthetic biology” is in practice plumbing rather than science. Second, the originally planned approach of replacing the surrogate’s hand-crafted feature vector with embeddings drawn directly from the foundation model (so that the ranker would reason over learned sequence representations rather than tabulated descriptors) was attempted but produced mixed results, likely due to the small number of training points; the feature-vector RandomForest was retained as the more reliable choice for this scale of data, with embedding-based grading flagged as a development-aim follow-up. Third, the absence of wet-lab access (the project was executed from Germany while the node is in San Francisco, with the final month spent in China with limited resource access) forced the substitution of a simulator for the Test step; while this validates the loop’s mechanics, it does not validate the assumption that Evo 2–generated regulatory sequences actually translate to high cell-free expression — that assumption is the central scientific risk and can only be settled by the planned wet-lab campaign with the Biopunk Labs cohort. Fourth, the as-not-a-software-engineer reality that LLM-based coding agents are helpful but cannot yet produce enterprise-grade web applications via “vibe coding” alone meant a meaningful share of effort went into hardening the application — a limitation worth flagging because it generalizes: AI-guided biology workflows will only be as good as the (currently unglamorous) software infrastructure underneath them. Mitigation strategies for the remaining risks include running the planned wet-lab validation, swapping in real measurement data, retrying the embedding-based grader at larger N, and broadening the host-organism support to make the engine generalizable beyond E. coli TX-TL.
SECTION 6: ADDITIONAL INFORMATION
References
Brixi, G., Durrant, M.G., Ku, J., et al. (2026). Genome modelling and design across all domains of life with Evo 2. Nature.
Sun, Z.Z., Yeung, E., Hayes, C.A., Noireaux, V., and Murray, R.M. (2014). Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synthetic Biology, 3(6), 387–397.
Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, Noireaux V. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J Vis Exp. 2013 Sep 16;(79):e50762. doi: 10.3791/50762. PMID: 24084388; PMCID: PMC3960857.
Yurchenko, A., Özkul, G., van Riel, N.A., van Hest, J.C., and de Greef, T.F. (2024). Mechanism-based and data-driven modeling in cell-free synthetic biology. Chemical Communications, 60(51), 6466–6475.
HTGAA 2025 Course Materials, Lecture and Recitation 1 (plasmid design); Wet-lab and cell-free systems recitations.
Anthropic. Claude API (model: claude-opus-4-5) — used as the “Test” simulator and as the model-based grader in place of wet-lab measurement.
NVIDIA / Arc Institute. Evo 2 model API endpoints (1b and 40b parameter variants) — used for generative sequence design in the variable regulatory region.
Supply List and Budget
The budget below covers the 12-week wet-lab continuation of the project — i.e., what would be needed to run three real DBTL rounds (3 × 50 constructs) and validate the workflow with the Biopunk Labs cohort. Software/compute costs already incurred during this course are listed separately at the bottom. All prices are approximate, in USD, list-price as of 2026 (community-lab pricing may differ). When not possible to find an appropriate quote, I used a GenAI Websearch to find an number.
DNA Synthesis (Build step) — the largest cost item
Twist Bioscience Gene Fragments (eBlocks) — 3 rounds × 50 constructs × ~770 bp each = 150 fragments
Per-fragment cost at this length: ~$30–50
Subtotal: ~$5,000–7,500
Optional: full sequence verification (Sanger) for top hits per round — $5/reaction × 30 reactions = **$150**
Alternative: NEB PURExpress In Vitro Protein Synthesis Kit (~$450 / 10 rxn) — more expensive per reaction but better defined for difficult proteins; flagged as backup
Linear DNA stabilizer (GamS protein) — recommended for linear-template TX-TL per Sun et al. (2014), ~$200
Reporter Controls and Reference Parts
Positive control plasmid (well-characterized strong RBS + sfGFP) from Addgene — $75/plasmid × 2 = **$150**
Standard part references from the Registry of Standard Biological Parts — typically free with iGEM affiliation, otherwise $50/part × 3 = **$150**
Recombinant sfGFP / deGFP purified protein (for fluorescence standard curve) — ~$300
Equipment (assumed available at Biopunk Labs / community lab; listed for completeness)
Plate reader with fluorescence and kinetic readout capability (e.g., BioTek Synergy, Tecan Spark) — available
–80 °C and –20 °C freezers for TX-TL kit storage — available
Microcentrifuge, vortex, ice bucket, P2/P10/P20/P200/P1000 pipettes — available
Thermal cycler (optional, for any PCR verification) — available
Biosafety cabinet (BSL-1 work) — available
Software, Compute, and Cloud (per-project, 12 weeks)
Evo 2 API access (NVIDIA NIM / Arc Institute endpoints, 40b and 1b) — usage-based, estimated for ~300 generation calls and 10 retraining iterations: **$300–500**
Claude API access (for model-based grader and simulator substitution) — usage-based, estimated: ~$150–250
Web application hosting (Streamlit/Vercel/Hugging Face Spaces tier) — ~$20/month × 3 = $60
Storage / database (Supabase or equivalent for construct library and round-by-round data) — ~$25/month × 3 = $75
Compute for surrogate model training (small RandomForest, runs locally on a laptop) — ~$0
Biosecurity and Compliance
SecureDNA / IGSC-style sequence screening before each Twist order — typically bundled into vendor screening, but a dedicated screening service subscription if independently run: ~$200
Institutional Biosafety Committee (IBC) review fees at a community lab — ~$100 (variable)
Personnel and Travel (optional, often unfunded for community-lab projects)
Travel for in-person wet-lab work (Germany ↔ San Francisco for the Biopunk Labs validation campaign) — ~$1,500 (one trip, mid-project)
Budget Summary
Category
Estimated Cost
DNA synthesis (Twist gene fragments, 150 fragments)
If the goal is to validate only Round 1 with the top-tier candidates (e.g., the 10 Grade-A constructs plus controls):
Twist gene fragments: 12 × $40 = $480
TX-TL kit: 2 kits = $800
Consumables, controls, plates: $400
Software/API: $200
Minimum viable total: ~$1,900
This minimum-viable path is the one currently being scoped with the Biopunk Labs cohort for the first real-world validation of construct #40 and the top Grade-A candidates from the Round-1 library.
Group Final Project
Part D: Group Brainstorm on Bacteriophage Engineering
Due to later start of our Node, we had limited time to find groups and set up a meeting, therefore the drafts of our group are mainly individual, and not discussed
Goal
We target two complementary objectives: (A) Increased stability of the L protein, specifically engineering DnaJ-independent variants that fold correctly without host chaperone assistance; and (B) Higher toxicity / faster lysis, by optimizing the transmembrane oligomerization interface to accelerate pore formation. Goal A is prerequisite to Goal B: a stable, chaperone-independent L is resistant to the most documented E. coli escape mechanism (DnaJ P330Q mutation), and faster lysis narrows the window for resistance acquisition.
Scientific Rational
Three findings define our design space.
DnaJ binds the highly basic N-terminal domain (res. 1–36) of L and relieves a steric inhibition blocking target engagement; removing this domain eliminates DnaJ dependency and accelerates lysis (Chamakura, J Bacteriol 2017).
Near-saturating mutagenesis shows the LS motif (Leu48-Ser49) and flanking residues form a heterotypic interface with an unknown target; exquisitely conservative mutations matter (L44V = dead, L44I = functional) and all are recessive, pointing to a specific binding event rather than membrane disruption (Chamakura, Microbiology 2017).
MS2-L oligomerizes into 10+ mers in nanodisc membranes via its TM domain; cryo-EM shows large envelope lesions starting at the outer membrane (Mezhyrova et al., 2023).
Strategy: neutralize basic charges in Domain 1 so DnaJ is no longer required, while leaving Domains 2–4 (the lytic machinery) untouched.
Computational Tools
Tool
Application
Why it helps
Clustal Omega
Align L homologs to identify which aminoacids are freely mutable
Reproduces and extends the LS-motif alignment from Chamakura (2017). Essential first step: tells us where NOT to mutate.
ESMFold
Predict 3D structure and each designed variant; verify the TM helix remains intact after mutations
Fast single-sequence predictor. For a 75 aa peptide with few homologs, much more practical than full AlphaFold for screening many candidates.
AlphaFold-Multimer
Model the L–DnaJ complex; confirm charge-neutralized variants show reduced interface confidence. Also model L–L homodimers to check TM packing.
Key validation for Goal A: if predicted L–DnaJ interface weakens for our variants, that supports DnaJ independence.
ProteinMPNN
Inverse folding: redesign Domain 1 (res. 1–36) to be uncharged while fitting the ESMFold-predicted backbone. Domains 2–4 fixed as hard constraints.
new sequence for existing fold with position-specific constraints. Generates diverse candidates we can then filter with ESM-2.
ESM
Zero-shot fitness scoring: rank all candidate variants by pseudo-log-likelihood as a sequence-level sanity check
Independent of structure prediction. Benchmarked first against known mutants — if it captures L biology, we use it to filter; if not, we rely on conservation alone.
Schematic
Pitfalls
We cannot model the most critical interaction (L with its unidentified host target) computationally.
ML models may not capture L biology, as L is a 75 aa phage toxin with very few homologs, far outside the training distribution of ESM-2 and AlphaFold