Individual Final Project

AI-guided closed-loop design of complex biological constructs using foundation model and (cell-free systems)
SECTION 1: ABSTRACT
Significance. Designing biological constructs that perform reliably remains a major bottleneck in synthetic biology. Enzymes involved in C1 metabolism, such as those mediating methane-to-methanol conversion, are notoriously difficult to express and optimize, yet methane and other C1 feedstocks represent an abundant but underutilized carbon source with applications ranging from sustainable chemical production to carbon sequestration. Today, plasmid design still relies heavily on historical use, standard parts, and intuition (“vibes”), with optimization tools that are scattered and focused primarily on the coding sequence rather than the surrounding regulatory architecture.
Broad Objective. This project develops a closed-loop, AI-guided workflow that combines genomic foundation models, lightweight predictive modeling, and cell-free expression experiments to accelerate the design of high-performing genetic constructs that can ultimately be deployed in living cells.
Hypothesis. Foundation model–generated and AI-selected constructs will outperform standard and random designs in protein expression, and an iterative Design–Build–Test–Learn (DBTL) loop will progressively enrich for high-performing variants without requiring retraining of the underlying foundation model.
Specific Aims. (1) Experimentally test whether Evo 2–generated and AI-selected constructs improve protein expression in cell-free systems relative to standard and random designs, using a fluorescence readout. (2) Extend the workflow beyond simple expression to capture robustness, stability over time, reproducibility, and more complex or fully generated regulatory regions, including those needed for challenging or non-native proteins. (3) Generalize the system into a biological design engine that transfers from cell-free prototyping to host organisms such as E. coli and non-model species, with C1 bioconversion enzymes as a target application.
Methods. Candidate libraries spanning standard parts, random variants, and Evo 2–generated sequences are produced in the variable regulatory region of a fixed construct template, pre-screened with sequence-quality heuristics (GC content, homopolymers, forbidden sites), and exported as CSV/FASTA for synthesis. Constructs are evaluated in an E. coli–based TX-TL cell-free system with fluorescence as the readout (substituted in this iteration by a generative-model-based simulator producing point estimates with uncertainty, due to lack of wet-lab access). Each [sequence, measurement] pair is encoded as a feature vector and used to train a Random Forest surrogate model that ranks the next round of candidates, closing the DBTL loop across three iterative rounds.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of my final project is to test whether foundation model–generated and AI-selected constructs can improve protein expression in cell-free systems by utilizing a closed-loop Design–Build–Test–Learn (DBTL) workflow that integrates Evo 2 (40b/1b) for sequence generation, heuristic and surrogate-model–based pre-screening, an E. coli–based TX-TL cell-free expression system for validation, and a Random Forest regression surrogate model for ranking and informing subsequent rounds.
A library of constructs — comprising standard designs, random variants, and Evo 2–generated sequences in the variable regulatory region of a fixed construct template — will be generated through the project’s AI-Guided Construct Designer web application, pre-screened against quality heuristics (GC content, homopolymer stretches, forbidden restriction sites), and exported as CSV/FASTA files ready for ordering from DNA synthesis providers such as Twist Bioscience. Constructs will be expressed in a TX-TL cell-free system following the linear-DNA rapid-prototyping protocol of Sun et al. (2014) and evaluated by fluorescence readout. Each [sequence, measurement] pair will be converted into a feature vector and used to fit a RandomForestRegressor, whose predictions will then guide candidate ranking in the next iteration of the loop. Three sequential DBTL rounds will be performed, and performance will be compared head-to-head across standard, random, foundation model–generated, and AI-guided designs. Because wet-lab access was not available during this course, the experimental measurement step is substituted in the current iteration by a generative-model-based simulator that returns fluorescence point estimates with associated uncertainty; the workflow is otherwise fully end-to-end and wet-lab-ready.
Aim 2: Development Aim
Following a successful Aim 1, the next step is to extend the system beyond optimizing simple expression toward more robust and realistic construct optimization. This means (i) running the closed loop with real wet-lab data — a validation campaign is already discussed with the Biopunk Labs cohort, replacing the surrogate fluorescence simulator with measured TX-TL output — and (ii) incorporating additional performance metrics such as expression stability over time and reproducibility across replicate batches and reaction conditions. The design space will be expanded from substituting a fixed variable region to generating more complex or fully de novo regulatory architectures (promoter, 5’ UTR, RBS, terminator) as a single jointly optimized block. On the modeling side, the hand-crafted feature vector inside the surrogate grader will be replaced with learned embeddings drawn directly from the foundation model, so that the ranker reasons over sequence representations rather than tabulated descriptors — an approach that was prototyped during the course with mixed results and deserves a more thorough treatment with larger, real-data training sets. This stage also enables a deeper evaluation of how well foundation model–generated sequences translate into reliable experimental performance, particularly for challenging or non-native proteins that resist standard design heuristics.
Aim 3: Visionary Aim
The long-term vision is to develop a generalizable, self-improving biological design engine that optimizes genetic constructs across biological contexts and host organisms — bridging cell-free prototyping with reliable in vivo deployment in E. coli as well as non-model chassis. Realized fully, such a system would challenge the prevailing paradigm in synthetic biology, in which plasmid design still leans on historical use, standard parts, and intuition, with optimization tools scattered across the workflow and focused mainly on the coding sequence rather than the regulatory architecture surrounding it. By tightly coupling generative foundation models with rapid experimental feedback in a continuously learning loop, the engine would replace artisanal construct engineering with a data-driven workflow where computational models and experimental systems continuously inform each other, compounding in capability with every round. The most immediate impact lies in industrial biotechnology — particularly the design of enzymes and pathways for C1 bioconversion, where reliable expression of complex methane- and methanol-handling enzymes remains a key barrier to using methane and other abundant C1 feedstocks for sustainable chemical production and carbon sequestration. More broadly, this approach would lower the activation energy for engineering any difficult-to-express protein, opening up enzyme classes, pathways, and host organisms that are currently out of reach for standard cloning workflows.
SECTION 3: BACKGROUND
Background and Literature Context
Synthetic biology has matured into a discipline capable of constructing increasingly complex genetic systems, yet a persistent bottleneck remains: the reliable design of constructs whose expression behavior can be predicted a priori. Despite decades of accumulated parts catalogs (e.g., the iGEM Registry, Anderson promoter series, standardized RBS collections), most plasmid design still proceeds by combining well-characterized “standard parts” with intuition drawn from historical use — an approach that breaks down precisely where it is most needed: for non-native, structurally complex, or metabolically demanding proteins. Existing optimization tools tend to be scattered across the workflow and focus narrowly on the coding sequence (codon optimization, rare-codon avoidance) rather than the regulatory architecture (promoter, 5’ UTR, RBS, spacer regions) that frequently dominates expression variance. Cell-free TX-TL systems, as pioneered for rapid linear-DNA prototyping by Sun et al. (2014), offer a fast, low-overhead readout for testing many constructs in parallel, and recent reviews of cell-free synthetic biology (Yurchenko et al., 2024) emphasize the growing role of mechanism-based and data-driven modeling in this space. In parallel, genomic foundation models — most notably Evo 2 (Brixi et al., 2026), trained on genomes spanning all domains of life — have demonstrated the ability to generate and score biological sequences at a scale and quality that was inaccessible only a few years ago. What is still missing is a tight, automated loop that couples these generative models to experimental measurement and uses the resulting data to steer the next round of designs.
Summarization of two peer-reviewed research citations relevant to your research.
Genome modelling and design across all domains of life with Evo 2
Brixi, G., Durrant, M.G., Ku, J. et al. Genome modelling and design across all domains of life with Evo 2. Nature 652, 1349–1361 (2026). https://doi.org/10.1038/s41586-026-10176-5
Evo 2 is a large-scale biological foundation model trained on 9 trillion DNA base pairs from the OpenGenome2 dataset, spanning all domains of life (bacteria, archaea, eukarya, and bacteriophage) with a context window of up to 1 million nucleotides at single-nucleotide resolution. The model uses a novel StripedHyena 2 architecture that combines convolutional and attention mechanisms, enabling efficient training and inference on both short and long genomic sequences. Evo 2 demonstrates strong zero-shot prediction capabilities for mutational effects across proteins, RNAs, and organismal fitness, including accurate pathogenicity prediction for human clinical variants in both coding and noncoding regions. Through mechanistic interpretability analysis using sparse autoencoders, the model reveals learned biological features such as exon-intron boundaries, transcription factor binding motifs, protein secondary structures, and prophage genomic regions. As a generative model, Evo 2 can produce organelle-scale, prokaryotic genome-scale, and eukaryotic chromosome-scale sequences with greater naturalness than previous methods. The researchers also demonstrate experimental validation of designed mammalian chromatin accessibility patterns by coupling Evo 2 with inference-time guidance using predictive models like Enformer and Borzoi. The model, training code, inference code, and OpenGenome2 dataset are fully open-sourced to accelerate biological research, with safety measures including exclusion of eukaryotic-infecting viral sequences to mitigate dual-use risks.
Protocols for Implementing an Escherichia coli Based TX-TL Cell-Free Expression System for Synthetic Biology
Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, Noireaux V. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J Vis Exp. 2013 Sep 16;(79):e50762. doi: 10.3791/50762. PMID: 24084388; PMCID: PMC3960857.
This academic paper describes a detailed five-day protocol for preparing and executing an endogenous Escherichia coli-based transcription-translation (TX-TL) cell-free expression system. Unlike traditional systems that rely heavily on specialized T7 bacteriophage RNA polymerase transcription, this platform preserves endogenous E. coli molecular mechanisms to more accurately mimic in vivo cellular dynamics. The protocol details the bulk preparation of the crucial reagents, including crude cell extract via cost-effective bead-beating lysis, an amino acid solution, and a 3-phosphoglyceric acid (3-PGA) energy solution. It also includes an optimization phase where magnesium glutamate, potassium glutamate, and DTT are strictly calibrated to achieve maximum protein expression yields. Once these storage-stable reagents are created, setting up individual 10-microliter reactions takes less than 8 hours from preparation to final data collection. The system achieves high protein yields comparable to T7-driven commercial alternatives, but at a 98% reduction in material costs. Ultimately, the authors position this adaptable platform as a versatile “biomolecular breadboard” optimized for synthetic biology prototyping, exploring biological circuit assembly, and analyzing regulatory mechanics.
Novelty and Innovation
This project is novel in three connected ways. First, rather than retraining or fine-tuning a foundation model — which is computationally prohibitive and data-hungry — it pairs an off-the-shelf frontier generator (Evo 2) with a lightweight, task-specific surrogate model (Random Forest regressor) that learns from a handful of experimental data points and is used to rank candidates for the next round; the foundation model supplies sequence diversity while the surrogate supplies experiment-informed selection pressure. Second, it operationalizes the full Design–Build–Test–Learn cycle as a single end-to-end web application (the AI-Guided Construct Designer), spanning library generation, heuristic and model-based pre-screening, CSV/FASTA export ready for DNA synthesis vendors such as Twist, and ingestion of measurement data back into the surrogate — replacing the scattered, ad hoc tooling that currently dominates plasmid design. Third, the approach treats the regulatory variable region of a construct as the primary design target rather than the coding sequence, which inverts the usual emphasis of codon-optimization-centric tools and aligns the design space with what most often limits expression in practice. Taken together, these choices challenge the prevailing “standard parts plus intuition” paradigm and offer a route to data-driven construct engineering that compounds in capability with every round of experiments.
Why This Project Matters
The pressing real-world problem addressed here is the unreliable expression of difficult biological constructs — a barrier that gates progress across industrial biotechnology, therapeutic protein production, and metabolic engineering. This matters acutely for C1 bioconversion: methane and other one-carbon feedstocks are among the most abundant and underutilized carbon sources on the planet, and the enzymes that act on them (methane monooxygenases, methanol dehydrogenases, formate-handling enzymes) are notoriously hard to express in heterologous hosts. Unlocking reliable expression of these enzymes would open routes to converting fugitive methane emissions and waste C1 streams into useful chemicals, advancing both sustainable chemical production and carbon sequestration — outcomes with direct climate and public-health relevance. Beyond C1 metabolism, the same workflow lowers the activation energy for engineering any hard-to-express protein, which would broadly accelerate enzyme discovery, pathway engineering, and biomanufacturing. At the field level, a successful demonstration would shift construct design from an artisanal, parts-library-driven craft toward a continuously learning, model-in-the-loop engineering practice — analogous to the shift that machine learning has already produced in protein structure prediction and small-molecule design. Finally, by making the workflow available as an open-source web application with seamless export to DNA synthesis vendors, the project lowers the technical floor for non-specialists, distributed-biology communities (such as Biopunk Labs and iGEM teams), and resource-limited laboratories to participate in cutting-edge construct design.
Ethical Implications
The ethical considerations of this project cluster around three principles. Non-maleficence is most immediately relevant because the tool generates novel DNA sequences and exports them in formats designed to be sent directly to commercial DNA synthesis providers; without appropriate screening, such a workflow could in principle be misused to design sequences with biosecurity concerns, even if that is far from the intended C1-bioconversion use case. Beneficence and justice are at stake in how the technology is distributed: a self-improving design engine could either concentrate capability in a small number of well-resourced labs or, conversely, democratize access to high-quality construct design — the choice depends on licensing, openness, and documentation. Responsibility applies to scientific claims: surrogate models trained on small numbers of cell-free measurements can produce confidently wrong predictions, and exporting “AI-ranked” libraries risks giving users a false sense of certainty about constructs that have never been measured. There are also dual-use considerations specific to generative genomic models, environmental considerations tied to deploying engineered organisms downstream, and authorship/data-provenance questions for any sequences that derive from foundation-model outputs trained on publicly available genomes.
Several concrete measures should be taken to ensure the project is conducted and deployed ethically. Proposed actions: (i) integrate sequence screening against established biosecurity databases (e.g., IGSC-style hazard screening) into the export step, so that any library leaving the tool has passed a hazard check; (ii) display calibrated uncertainty alongside every surrogate-model prediction in the UI, rather than presenting rankings as deterministic; (iii) release the workflow under an open-source license with clear documentation of training data, model versions, and known failure modes; (iv) restrict, at the application layer, generation targeted at known pathogen toxin genes or other flagged sequence classes; and (v) for any future wet-lab campaigns (such as the planned Biopunk Labs validation), follow institutional biosafety review and contain all work to non-pathogenic, well-characterized chassis. Potential unintended consequences include over-reliance on the surrogate ranker leading to systematically biased libraries that drift away from the experimental ground truth, false negatives that discard genuinely useful designs, and the broader risk of “vibe-engineering” in which users trust the model output without retaining biological intuition. What could be wrong: the central assumption that foundation-model-generated regulatory sequences translate from cell-free to in vivo performance has not been fully validated; the surrogate’s feature representation may miss the actual determinants of expression; and the simulator-based substitution used during this course is itself an approximation that may not reflect true wet-lab variance. Alternatives to address these risks include keeping a human-in-the-loop review step for all libraries above a certain size, restricting initial deployments to well-characterized model organisms, using ensemble or Bayesian surrogates that more honestly report uncertainty, and committing to publish negative results and failure modes alongside successes so that the community can calibrate trust in the tool. Taken together, these measures aim to capture the public-health and sustainability upside of better construct design — particularly for C1 bioconversion and climate-relevant biotechnology — while keeping the dual-use surface and the epistemic risks of AI-guided biology under active, transparent management.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Detailed Experimental Plan with Timeline
The plan below assumes a 12-week timeline that mirrors what would have been executed in a fully wet-lab-enabled version of this project, with the software workflow (already built during the course) feeding directly into the bench work. As the final project timeline allows for only 4 weeks of work, only one cycle of the DBTL cycle is discribed in this section. An additional caveat is that the major time uncertainty is the ordering of the DNA constructs. For simplicity it was assumed that it will take 1 eek to deliver the constructs. As Sun et al. (2014) described, one wetlab experiemnt cycle takes five days, therefore the wetlab experiment is synonymous for the protocol that is described above.
Task 1 — Define the construct template and target enzyme (Week 1). Fix the construct backbone as a linear DNA template suitable for E. coli TX-TL: T7 promoter — variable regulatory region (~30 bp, including 5’ UTR/RBS/spacer) — coding sequence (CDS) — T7 terminator. The variable region is the design target; the CDS is held constant. For Round 1 the CDS is a fluorescent reporter (deGFP or sfGFP) to give a clean fluorescence readout; later rounds substitute a representative C1-metabolism enzyme (e.g., a soluble methanol dehydrogenase fragment) fused to a reporter for downstream relevance.
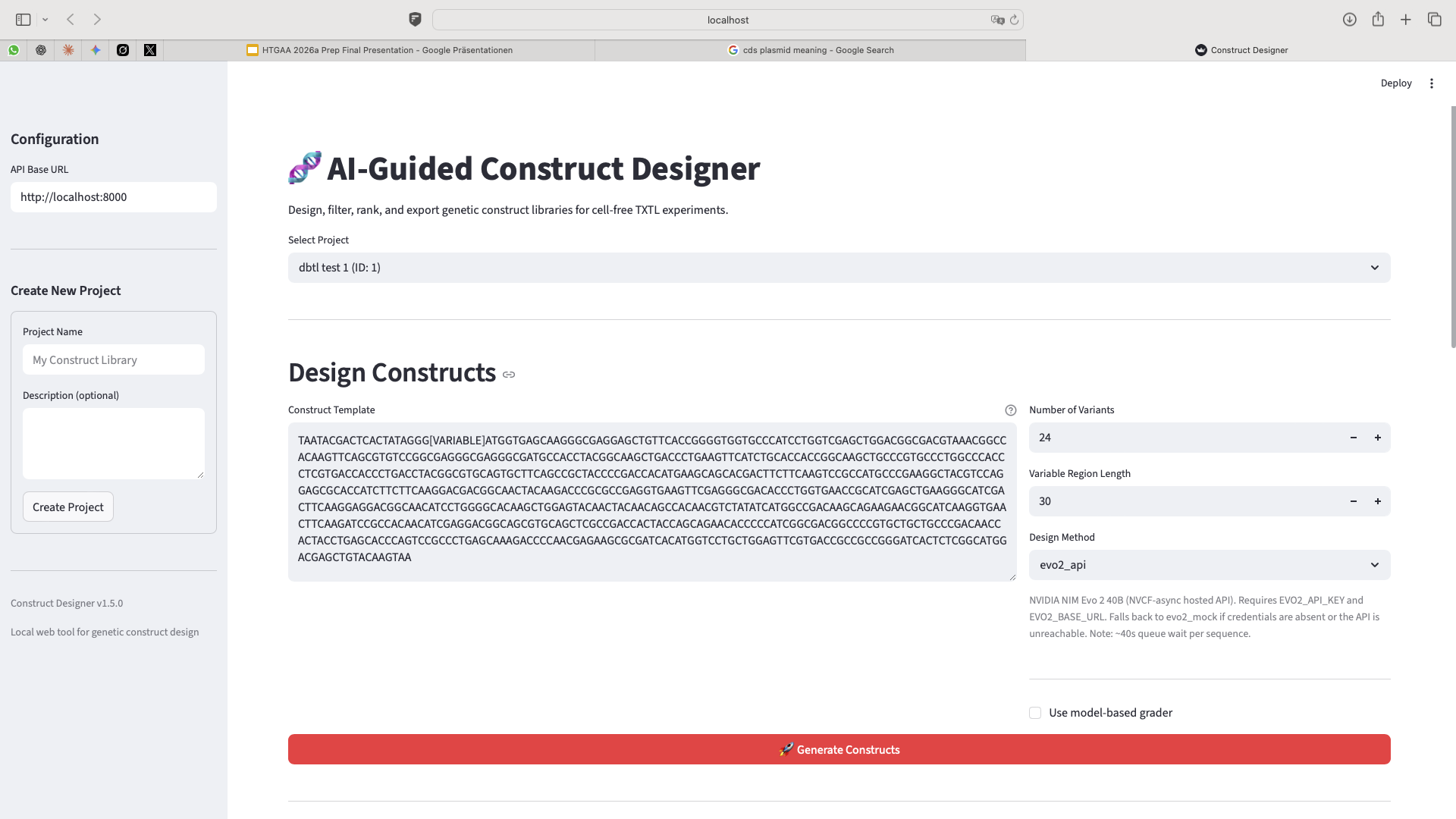
Task 2 — Round 1 library design (Week 1). Use the AI-Guided Construct Designer to generate 50 candidate sequences in the variable region: ~10 standard parts (canonical RBS/promoter combinations from the Registry of Standard Biological Parts), ~10 random variants as a negative-control baseline, and ~30 Evo 2–generated sequences (a mix of 1b and 40b model outputs). Pre-screen all candidates with sequence-quality heuristics (GC content within 35–65%, no homopolymer runs >5 nt, no forbidden restriction sites in the cloning context). Export ranked CSV and FASTA files.
Task 3 — Build: order linear DNA (Weeks 2–4). Submit a Twist Bioscience order for the 50 linear-DNA constructs as a gene-fragment library. Twist’s eBlocks or Gene Fragments format is well-suited to TX-TL prototyping because no plasmid cloning is needed; linear DNA can be used directly per Sun et al. (2014).
Task 4 — Test: Round 1 cell-free expression (Week 3). Run all 50 constructs in triplicate in an E. coli TX-TL reaction (10 µL reactions, 96-well plate format) and read fluorescence kinetically at 30 °C over 8 hours in a plate reader. Compute the area under the curve (AUC) of the fluorescence trace as the per-construct readout. Include positive (well-characterized strong RBS) and negative (no-DNA) controls on every plate. Expected result: Evo 2–generated constructs should produce a wider distribution of AUC values than the random baseline, with the top quartile matching or exceeding the standard-parts controls; this is consistent with the simulator-based Round 1 distribution observed during the course (best AUC ~101k).
Task 5 — Learn: train surrogate model (Weeks 3-4). Convert each [sequence, AUC] pair into a feature vector (k-mer frequencies, GC content, predicted secondary structure ΔG, RBS-calculator score, etc.) and fit a RandomForestRegressor on the Round 1 data. Hold out 20% for validation and report R²/MAE. Expected result: R² ≈ 0.3–0.5 with only ~47 training points, sufficient to enrich top-quartile candidates in the next round but not for confident point predictions.
Task 6 — Round 2 library design, build, test, learn (Week 4). Generate a second library of 50 candidates, this time ranking Evo 2 outputs with the trained surrogate rather than only with heuristics. Order via Twist, run cell-free reactions, retrain the surrogate on the combined Round 1 + Round 2 data (~96 points). Expected result: the median AUC of the AI-guided library should rise relative to Round 1, even if the single best construct does not (course simulation showed median improvement with a slightly lower top value, ~89k — this is the classic exploitation-vs-exploration tradeoff and informs Round 3).
This is outside the scope of HTGAA Final Project, for clarity it is included, to appreachiate the circularity and iteration of the DBTL cycle.
Task 7 — Round 3 library design, build, test, learn (Weeks 5-12). Inject an explicit exploration component (e.g., 30% of Round 3 candidates sampled from high-uncertainty regions of the surrogate’s prediction space). Run cell-free reactions, retrain surrogate on all three rounds (~144 points). Expected result: recovery of high-value designs and improved median performance, mirroring the Round 3 simulator output (best AUC ~95k with tighter distribution).
Task 8 — Comparative analysis and reporting (Week 12). Statistically compare AUC distributions across the four design categories (standard, random, foundation model–generated unguided, AI-guided) using a Kruskal-Wallis test followed by pairwise Mann-Whitney with Bonferroni correction. Test the hypothesis that AI-guided > foundation-model-unguided > standard ≥ random. Deposit all sequences, measurements, and trained surrogate weights in an open repository.
Workflow Figure
The DBTL workflow figure illustrates this loop end-to-end: Design (Evo 2 library generation with heuristic and surrogate pre-screening) → Build (CSV/FASTA export to Twist) → Test (TX-TL cell-free expression with fluorescence AUC readout) → Learn (RandomForest surrogate training and ranking for the next round).
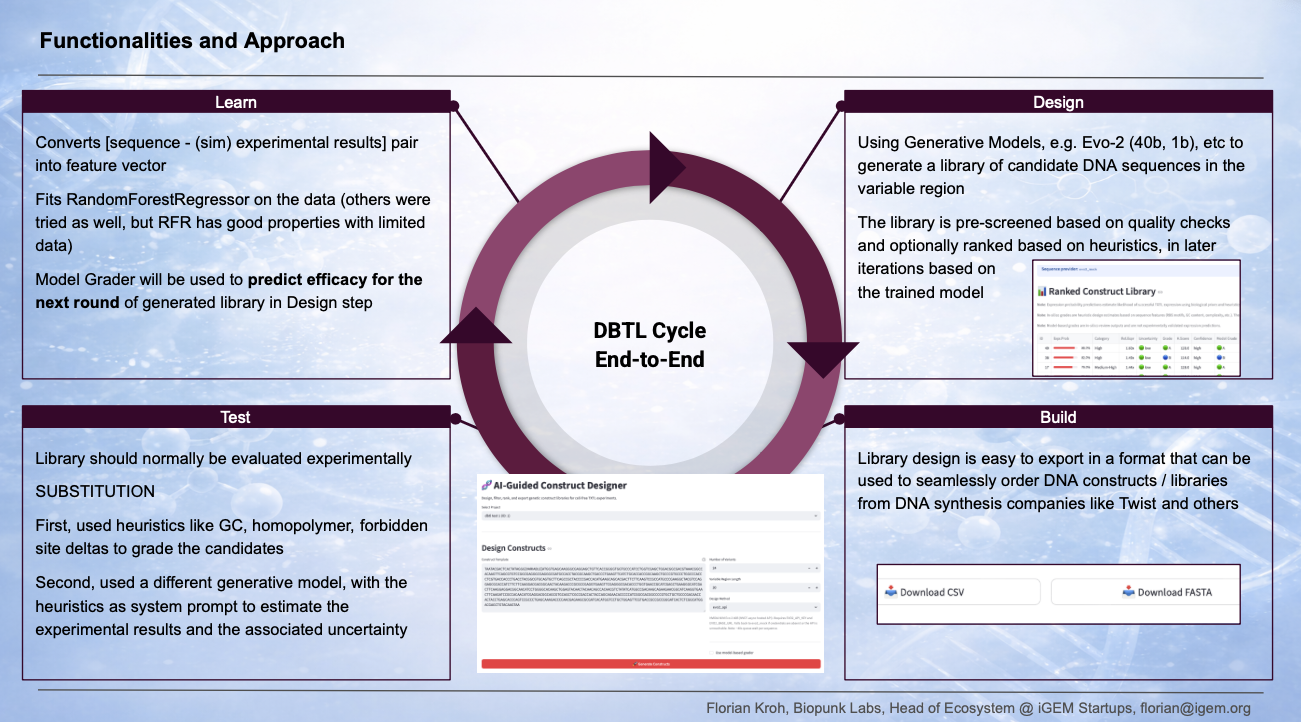
Techniques Checklist
Lab Safety
- Bioethical Considerations
DNA Editing
- DNA Construct Design
- Databases (e.g., GenBank, NCBI)
Lab Automation
- Creating Code for Laboratory Automation
- Designing a Twist Order
Protein Design
- Models and Notebooks
- Databases
Bioproduction
- Registry of Standard Biological Parts
Cell-Free Systems
- Cell Free Reactions
Expanded Techniques
Inputing the linear DNA construct. The DBTL cycle begins by inputting your full genetic sequence into the web tool, then identifying and removing the genetic sequence in question and replacing it with the [VARIABLE] placeholder to designate the region for modification. Next, you configure the generation parameters by specifying the number of variants to generate, the region length, and selecting the foundation model that will power the inference. You then decide whether to apply a model-based grader to evaluate the generated variants, and if enabled, adjust the grader’s parameters to customize how variants are scored and ranked. Finally, you execute the inference to generate and evaluate the genetic variants according to your configured settings.
Cell Free Reactions. The Test step of every DBTL round uses an E. coli–based TX-TL system following the linear-DNA rapid-prototyping protocol of Sun et al. (2014), which avoids the multi-day overhead of plasmid cloning and transformation between rounds and is the single biggest enabler of the three-rounds-in-twelve-weeks cadence. Each construct is run in triplicate in 10 µL reactions in a 96-well plate, with fluorescence read kinetically at 30 °C over 8 hours and AUC used as the per-construct readout to capture both expression rate and total yield. Positive controls (well-characterized strong RBS) and negative controls (no-DNA reactions) are included on every plate to anchor cross-round comparisons. Once C1 enzymes replace the fluorescent reporter in later experiments, the same cell-free format will support reporter-fusion or coupled-enzyme assays without needing to redesign the workflow. This could unfortunately not be validated in the lab, as i didn’t have access to a wetlab. It is purely based on the Sun et al. (2014) and other standard auxiliary protocols
Industry Council Companies Associated with This Project
- Twist Bioscience — primary DNA synthesis vendor for all three rounds of linear-DNA libraries; the export format of the AI-Guided Construct Designer is built around Twist’s gene-fragment ordering workflow.
- Ginkgo Bioworks — relevant for the Aim 2/3 transition from cell-free prototyping to higher-throughput in vivo validation and scale-up.
- Cultivarium — relevant for Aim 3’s extension to non-model host organisms, where the AI-guided design workflow would need to be retargeted to chassis beyond E. coli.
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
What Aspect of the Final Project Was Validated
The aspect validated in this work is the end-to-end software workflow of the closed-loop Design–Build–Test–Learn (DBTL) cycle, specifically the closed-loop behavior of the system across three sequential rounds: foundation-model–driven library generation in the variable regulatory region, heuristic and surrogate-model–based ranking of candidates, simulator-based fluorescence “estimation” with calibrated uncertainty (substituting for wet-lab cell-free expression that was not accessible), and surrogate-model retraining to inform the next round of designs. Because no wet-lab access was available during the course, this is a purely computational validation; its purpose is to demonstrate that the loop holds together mechanically, that the surrogate ranker improves with accumulated data, and that the export pipeline produces wet-lab-ready libraries — making the workflow ready to be plugged directly into a TX-TL setup as soon as bench access is available (discussed with the Biopunk Labs cohort). To completely validate the project the simluator-based fluorescence “estimation” needs to be replaced with the proper wetlab experiment (see Section 4. Expanded Techniques)
Detailed Validation Protocol
- Construct template definition. A fixed linear-DNA template was used in the AI-Guided Construct Designer: T7 promoter — variable regulatory region — fluorescent reporter CDS — T7 terminator. The variable region (~30 bp) was the design target.
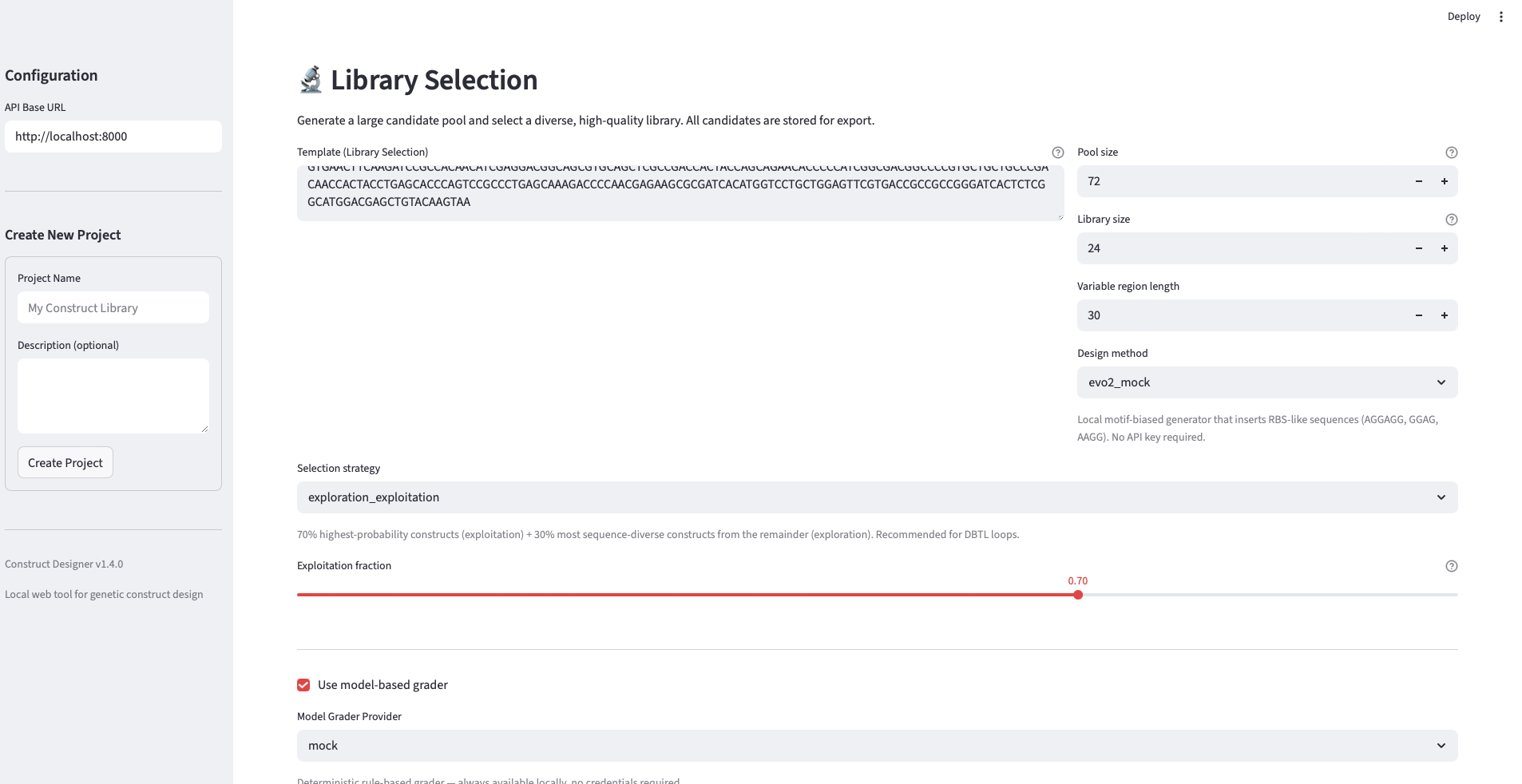
- Round 1 library generation. Fifty candidate sequences were generated in the variable region by querying Evo 2 (40b and 1b model endpoints) and combining with a small number of standard parts and random variants as baselines.
- Pre-screening with heuristics. Each candidate was filtered against quality heuristics: GC content (35–65%), no homopolymer runs >5 nt, no forbidden restriction sites in the cloning context. Candidates failing hard filters were rejected; the rest were ranked.
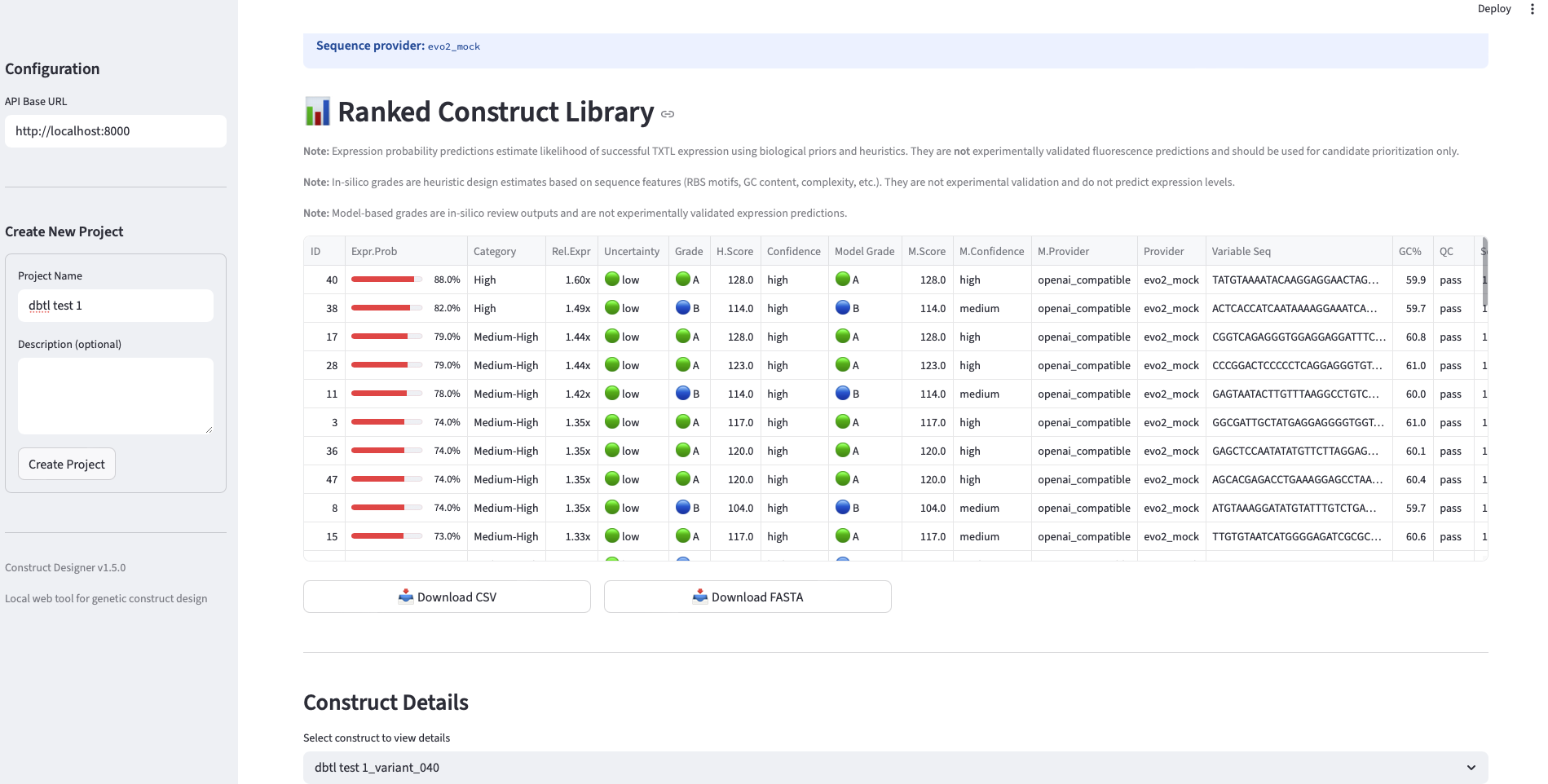
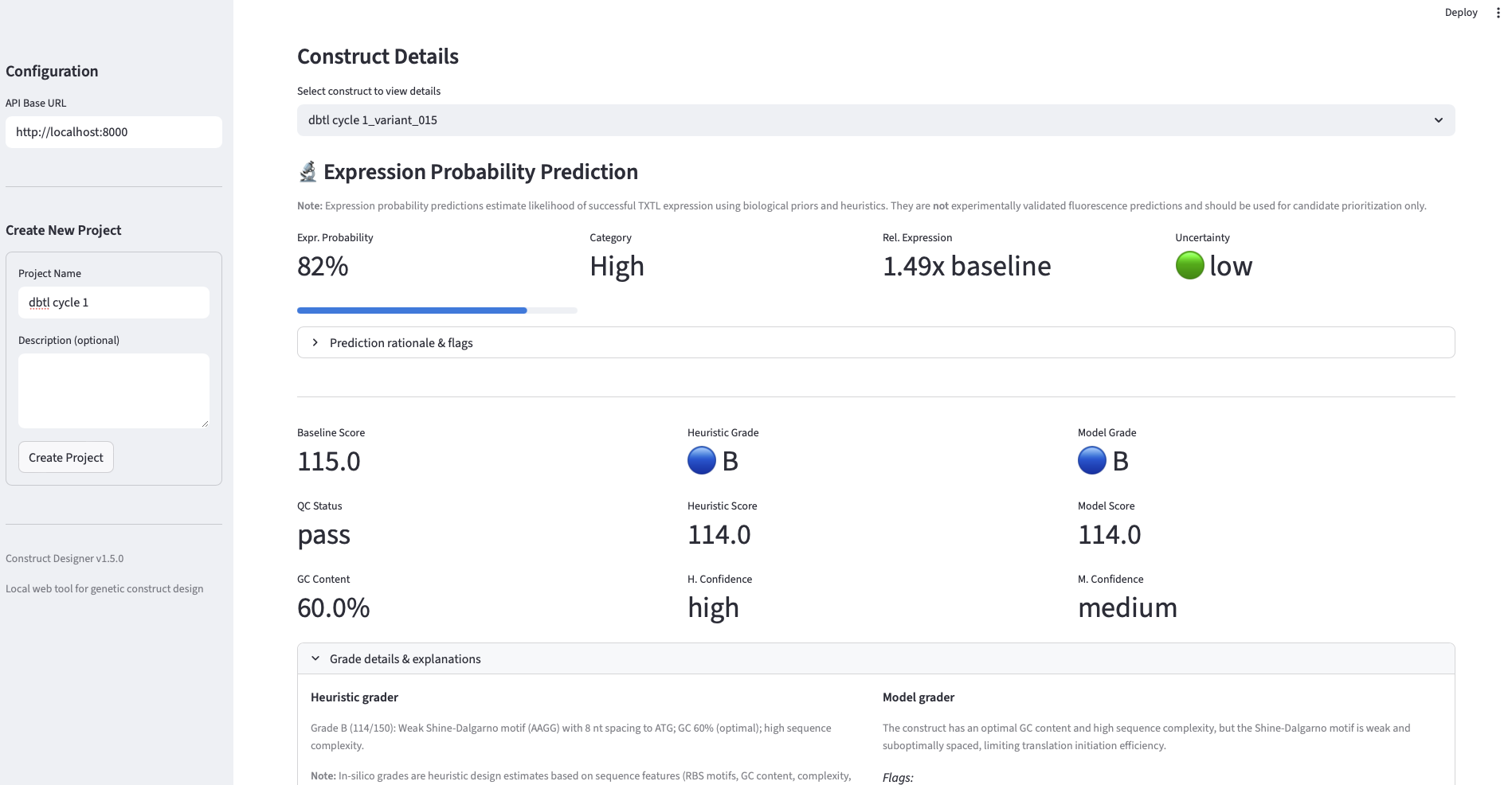
- Surrogate (“test”) measurement substitution. Because no wet-lab access was available, a generative-model-based simulator was used to assign each candidate a fluorescence AUC point estimate together with an uncertainty estimate. Prompts to the simulator encoded the same heuristics used in pre-screening, so the simulator returned biologically plausible (rather than uniformly random) values.
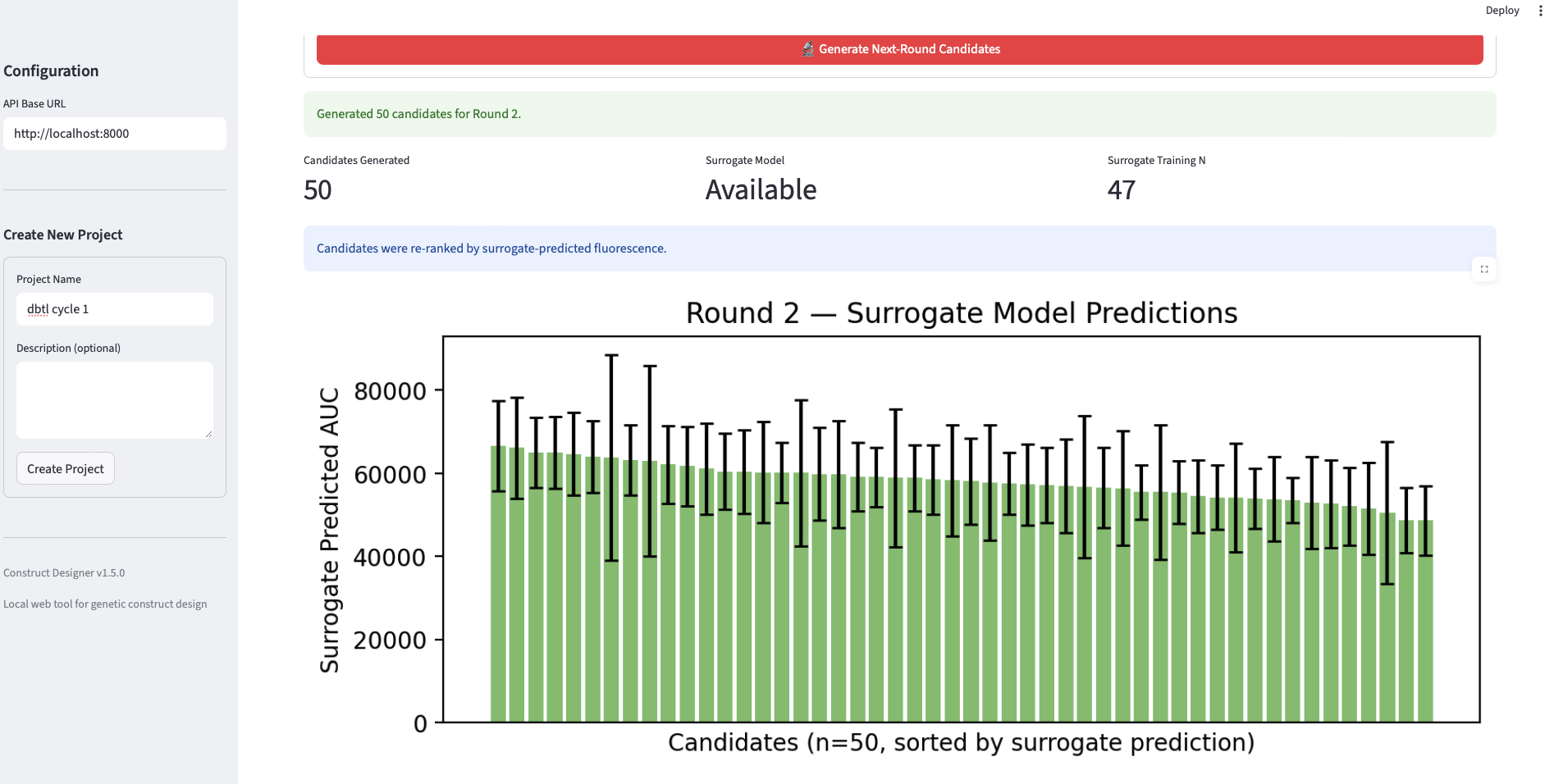
- Surrogate ranker training. Each [sequence, simulated AUC] pair was converted into a feature vector (k-mer frequencies, GC content, sequence-quality descriptors, etc) and used to fit a RandomForestRegressor. Random Forest was chosen over deeper models because of strong performance on small tabular datasets and built-in feature-importance interpretability.
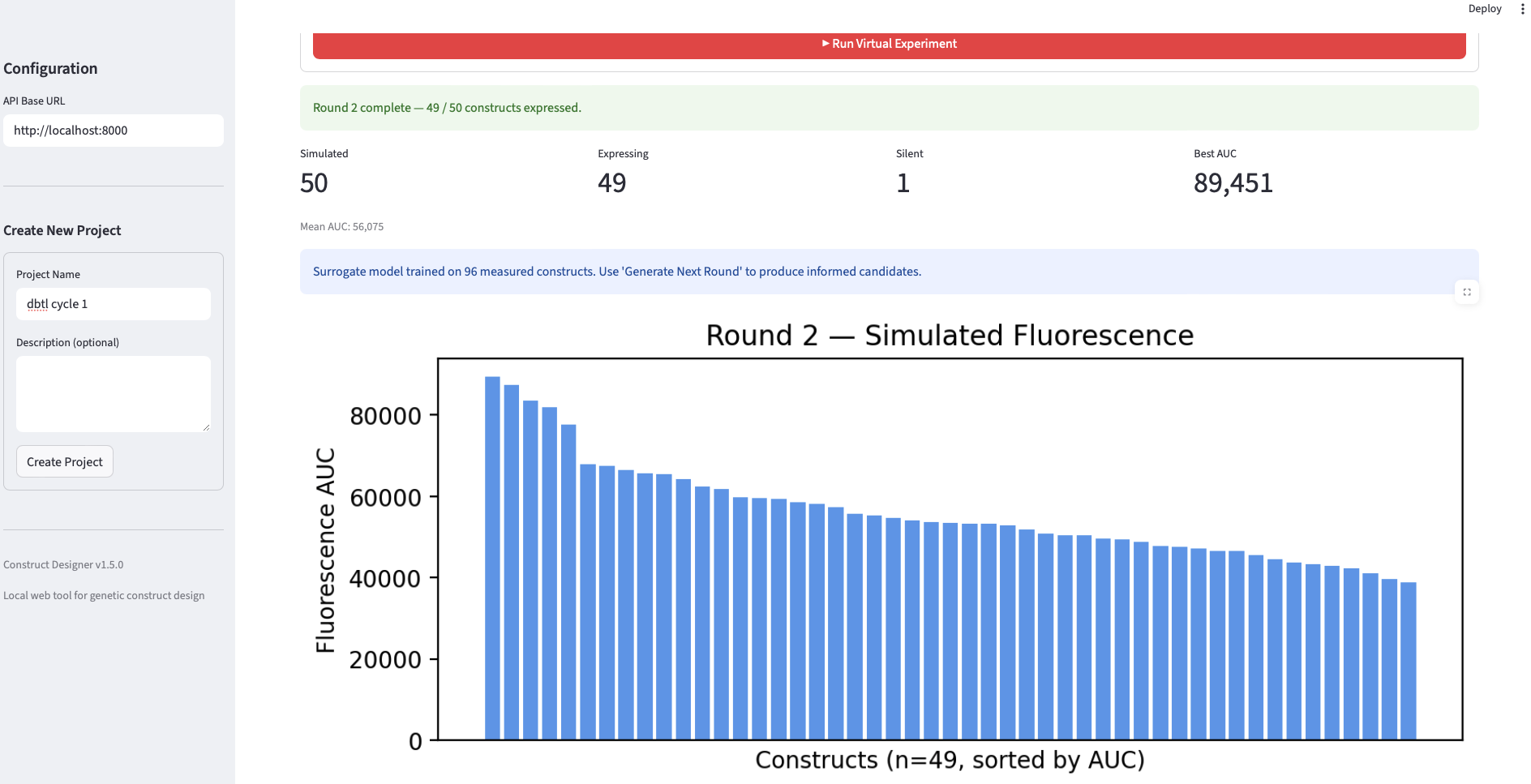
- Round 2. Fifty new candidates were generated by Evo 2, re-ranked using the trained surrogate (rather than only heuristics), and “measured” by the simulator. The surrogate was retrained on the combined Round 1 + Round 2 data.
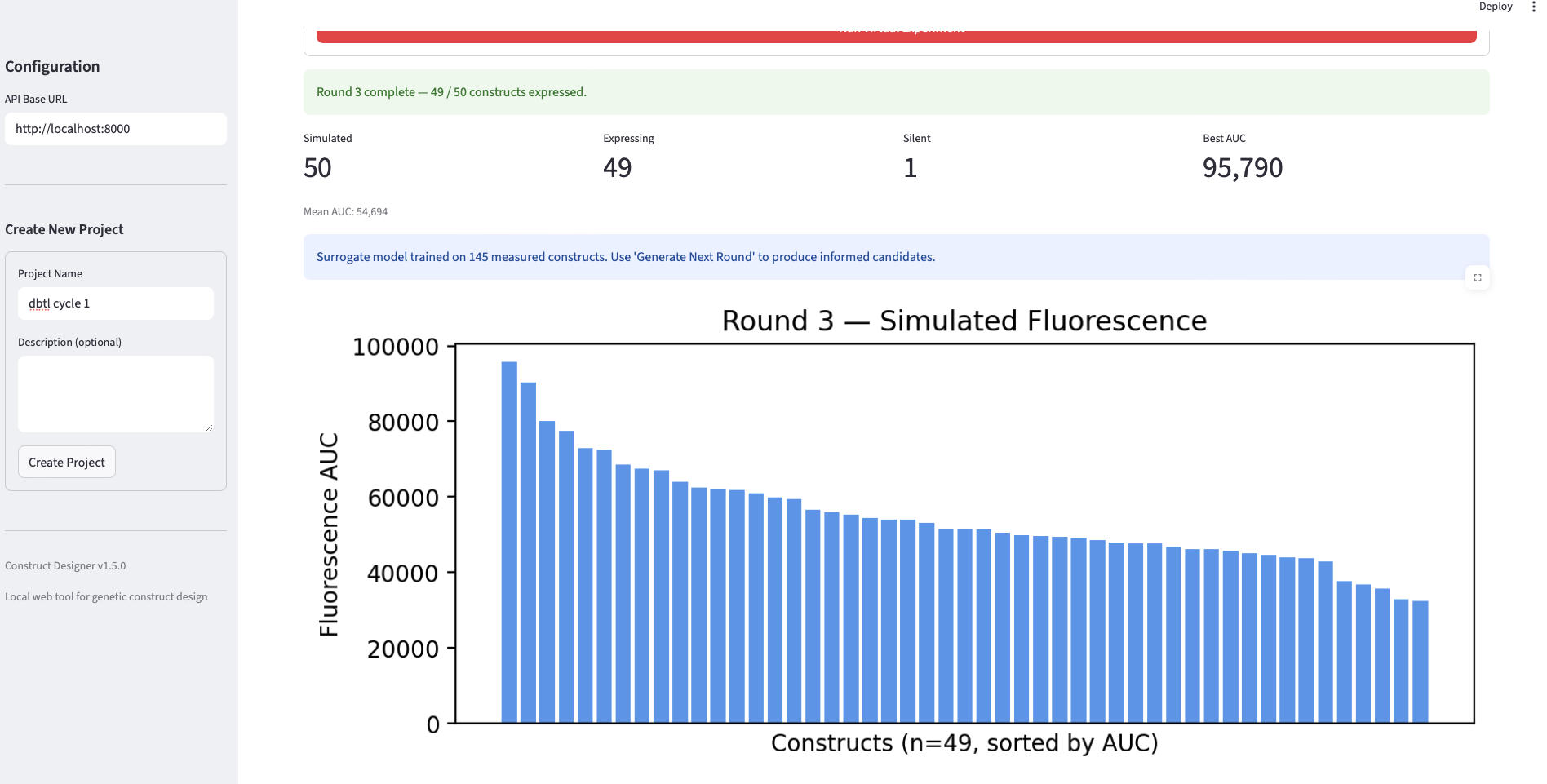
- Round 3. Another fifty candidates were generated, re-ranked with the now-twice-trained surrogate, and “measured.” The surrogate was retrained on all three rounds.
- Export validation. At every round, the library was exported as both CSV (with per-construct metadata: ID, sequence, predicted AUC, uncertainty, category) and FASTA (in a format directly uploadable to Twist Bioscience’s gene-fragment ordering interface).
- Cross-round analysis. Best AUC, number of surviving candidates, and full AUC distributions were tracked across rounds to characterize loop behavior.
Synthetic Biology Techniques Used in Validation
This validation drew on several techniques from the course: DNA construct design was central — the project’s web application designs the variable regulatory region of a TX-TL-compatible linear-DNA construct, holding the T7 promoter, reporter CDS, and terminator fixed while varying the regulatory region as the optimization target. Designing a Twist order was practiced end-to-end: every library exports as a CSV/FASTA pair formatted to Twist Bioscience’s gene-fragment specification, including manufacturability pre-checks (GC content, homopolymers, repeats) so the output can be submitted without much effort. (Twist does similar checks in their webinterface, given that rejected variants arent included, this accelerates cycletime) Creating code for laboratory automation was applied throughout: the entire DBTL loop was implemented as a single web application that automates library generation, pre-screening, “measurement,” and surrogate retraining, removing manual handoffs between steps. Finally, databases and the Registry of Standard Biological Parts were used as the source of standard-part baselines (canonical RBS/promoter combinations) that anchor the comparison against random and Evo 2–generated designs, and bioethical considerations were applied throughout — including the recommendation that any library leaving the tool should pass biosecurity screening (e.g., via SecureDNA) before being submitted to a synthesis provider.
Data and Analysis
Three sequential rounds of the DBTL loop were executed in simulation, with the following summary results:
| Round | Generated | Surviving | Discarded | Best AUC | Surrogate Training N |
|---|---|---|---|---|---|
| 1 | 50 | 47 | 3 | ~101,425 | 47 |
| 2 | 50 | 49 | 1 | ~89,451 | 96 |
| 3 | 50 | 49 | 1 | ~95,790 | 144 |
Analysis. The validation produces three findings worth noting. First, the loop closes mechanically: each round successfully generated, filtered, “measured,” and trained on data, and the surrogate’s training set grew monotonically from 47 → 96 → 144 points, demonstrating that the data pipeline is intact end-to-end. Second, the best-AUC trajectory across rounds (101k → 89k → 96k) is non-monotonic, which is informative rather than a failure: it reflects the classic exploitation-vs-exploration tradeoff. Round 1 candidates are heuristic-ranked from a broad Evo 2 distribution and occasionally contain a strong outlier; Round 2 is re-ranked by a surrogate trained on only 47 points, which over-exploits a still-noisy estimate of the fitness landscape and slightly drops the top value; Round 3, with a more confident surrogate over 96 points, partially recovers. It is important to keep in mind that the AUC data is estimated, not experimentally validated, I did the analysis as is from the data I got, though I need to mention, that these estimations are error-prone, and therefore no experimental value can be derived from them Third, the surrogate-predicted AUC distributions (the green panels in slide 5) tighten across rounds while the simulated-fluorescence distributions (blue panels) shift in median, consistent with the ranker learning to identify the bulk of mid-to-high performers even when the single best construct is harder to recover. The headline takeaway is that the workflow is wet-lab-ready: the same loop, with the simulator swapped for a TX-TL reaction, can be run directly.
Unexpected Challenges and Mitigation Strategies
Several unexpected challenges arose, almost all on the software-engineering rather than the biological side. First, a significant fraction of the overall project time was consumed by getting the 40b Evo 2 model API endpoint to work reliably — the difficulty was not in the biology but in API authentication, rate limits, and response formatting, which is a useful warning about how much of “AI-guided synthetic biology” is in practice plumbing rather than science. Second, the originally planned approach of replacing the surrogate’s hand-crafted feature vector with embeddings drawn directly from the foundation model (so that the ranker would reason over learned sequence representations rather than tabulated descriptors) was attempted but produced mixed results, likely due to the small number of training points; the feature-vector RandomForest was retained as the more reliable choice for this scale of data, with embedding-based grading flagged as a development-aim follow-up. Third, the absence of wet-lab access (the project was executed from Germany while the node is in San Francisco, with the final month spent in China with limited resource access) forced the substitution of a simulator for the Test step; while this validates the loop’s mechanics, it does not validate the assumption that Evo 2–generated regulatory sequences actually translate to high cell-free expression — that assumption is the central scientific risk and can only be settled by the planned wet-lab campaign with the Biopunk Labs cohort. Fourth, the as-not-a-software-engineer reality that LLM-based coding agents are helpful but cannot yet produce enterprise-grade web applications via “vibe coding” alone meant a meaningful share of effort went into hardening the application — a limitation worth flagging because it generalizes: AI-guided biology workflows will only be as good as the (currently unglamorous) software infrastructure underneath them. Mitigation strategies for the remaining risks include running the planned wet-lab validation, swapping in real measurement data, retrying the embedding-based grader at larger N, and broadening the host-organism support to make the engine generalizable beyond E. coli TX-TL.
SECTION 6: ADDITIONAL INFORMATION
References
- Brixi, G., Durrant, M.G., Ku, J., et al. (2026). Genome modelling and design across all domains of life with Evo 2. Nature.
- Sun, Z.Z., Yeung, E., Hayes, C.A., Noireaux, V., and Murray, R.M. (2014). Linear DNA for rapid prototyping of synthetic biological circuits in an Escherichia coli based TX-TL cell-free system. ACS Synthetic Biology, 3(6), 387–397.
- Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, Noireaux V. Protocols for implementing an Escherichia coli based TX-TL cell-free expression system for synthetic biology. J Vis Exp. 2013 Sep 16;(79):e50762. doi: 10.3791/50762. PMID: 24084388; PMCID: PMC3960857.
- Yurchenko, A., Özkul, G., van Riel, N.A., van Hest, J.C., and de Greef, T.F. (2024). Mechanism-based and data-driven modeling in cell-free synthetic biology. Chemical Communications, 60(51), 6466–6475.
- iGEM Foundation. Registry of Standard Biological Parts. https://parts.igem.org
- National Center for Biotechnology Information (NCBI). GenBank. https://www.ncbi.nlm.nih.gov/genbank
- Twist Bioscience. Gene Fragments (eBlocks) specifications and ordering portal. https://www.twistbioscience.com
- SecureDNA. Biosecurity screening for synthetic DNA. https://www.securedna.org
- HTGAA 2025 Course Materials, Lecture and Recitation 1 (plasmid design); Wet-lab and cell-free systems recitations.
- Anthropic. Claude API (model: claude-opus-4-5) — used as the “Test” simulator and as the model-based grader in place of wet-lab measurement.
- NVIDIA / Arc Institute. Evo 2 model API endpoints (1b and 40b parameter variants) — used for generative sequence design in the variable regulatory region.
Supply List and Budget
The budget below covers the 12-week wet-lab continuation of the project — i.e., what would be needed to run three real DBTL rounds (3 × 50 constructs) and validate the workflow with the Biopunk Labs cohort. Software/compute costs already incurred during this course are listed separately at the bottom. All prices are approximate, in USD, list-price as of 2026 (community-lab pricing may differ). When not possible to find an appropriate quote, I used a GenAI Websearch to find an number.
DNA Synthesis (Build step) — the largest cost item
- Twist Bioscience Gene Fragments (eBlocks) — 3 rounds × 50 constructs × ~770 bp each = 150 fragments
- Per-fragment cost at this length: ~$30–50
- Subtotal: ~$5,000–7,500
- Optional: full sequence verification (Sanger) for top hits per round — $5/reaction × 30 reactions = **$150**
Cell-Free Expression (Test step)
- myTXTL Sigma 70 Master Mix (Arbor Biosciences / Daicel Arbor Biosciences) — 24-reaction kit, ~$400/kit
- 3 rounds × 50 constructs × 3 technical replicates = 450 reactions
- 19 kits needed → **$7,600**
- Alternative: NEB PURExpress In Vitro Protein Synthesis Kit (~$450 / 10 rxn) — more expensive per reaction but better defined for difficult proteins; flagged as backup
- 96-well black/clear-bottom plates for fluorescence reading — $10/plate × 15 plates = **$150**
- Plate seals (optical/breathable) — ~$100
- Sterile barrier pipette tips (10/20/200/1000 µL ranges) — ~$300
- Nuclease-free water, 1 L — ~$50
- Linear DNA stabilizer (GamS protein) — recommended for linear-template TX-TL per Sun et al. (2014), ~$200
Reporter Controls and Reference Parts
- Positive control plasmid (well-characterized strong RBS + sfGFP) from Addgene — $75/plasmid × 2 = **$150**
- Standard part references from the Registry of Standard Biological Parts — typically free with iGEM affiliation, otherwise $50/part × 3 = **$150**
- Recombinant sfGFP / deGFP purified protein (for fluorescence standard curve) — ~$300
Equipment (assumed available at Biopunk Labs / community lab; listed for completeness)
- Plate reader with fluorescence and kinetic readout capability (e.g., BioTek Synergy, Tecan Spark) — available
- –80 °C and –20 °C freezers for TX-TL kit storage — available
- Microcentrifuge, vortex, ice bucket, P2/P10/P20/P200/P1000 pipettes — available
- Thermal cycler (optional, for any PCR verification) — available
- Biosafety cabinet (BSL-1 work) — available
Software, Compute, and Cloud (per-project, 12 weeks)
- Evo 2 API access (NVIDIA NIM / Arc Institute endpoints, 40b and 1b) — usage-based, estimated for ~300 generation calls and 10 retraining iterations: **$300–500**
- Claude API access (for model-based grader and simulator substitution) — usage-based, estimated: ~$150–250
- Web application hosting (Streamlit/Vercel/Hugging Face Spaces tier) — ~$20/month × 3 = $60
- Storage / database (Supabase or equivalent for construct library and round-by-round data) — ~$25/month × 3 = $75
- Compute for surrogate model training (small RandomForest, runs locally on a laptop) — ~$0
Biosecurity and Compliance
- SecureDNA / IGSC-style sequence screening before each Twist order — typically bundled into vendor screening, but a dedicated screening service subscription if independently run: ~$200
- Institutional Biosafety Committee (IBC) review fees at a community lab — ~$100 (variable)
Personnel and Travel (optional, often unfunded for community-lab projects)
- Travel for in-person wet-lab work (Germany ↔ San Francisco for the Biopunk Labs validation campaign) — ~$1,500 (one trip, mid-project)
Budget Summary
| Category | Estimated Cost |
|---|---|
| DNA synthesis (Twist gene fragments, 150 fragments) | $5,000–7,500 |
| Cell-free reagents (TX-TL kits, GamS, consumables) | $8,400 |
| Controls and reference parts | $600 |
| Software, compute, and cloud APIs | $585–885 |
| Biosecurity screening and compliance | $300 |
| Verification sequencing (optional) | $150 |
| Subtotal (lab + compute) | ~$15,000–17,800 |
| Travel (optional) | $1,500 |
| Grand Total (full 12-week wet-lab project) | ~$15,000–19,300 |
Minimum Viable Budget (1-round proof-of-concept only)
If the goal is to validate only Round 1 with the top-tier candidates (e.g., the 10 Grade-A constructs plus controls):
- Twist gene fragments: 12 × $40 = $480
- TX-TL kit: 2 kits = $800
- Consumables, controls, plates: $400
- Software/API: $200
- Minimum viable total: ~$1,900
This minimum-viable path is the one currently being scoped with the Biopunk Labs cohort for the first real-world validation of construct #40 and the top Grade-A candidates from the Round-1 library.