Week 2 HW: DNA-read-write-and-edit
1. Benchling & In-silico Gel Art
1.1 Extraction of Restriction Site Data
Restriction site positions for 10 enzymes were extracted from Benchling for the λ phage genome.
Based on these cut positions, fragment sizes were calculated for each individual enzyme digestion.
 (Image 1: Benchling screenshot showing restriction sites)
(Image 1: Benchling screenshot showing restriction sites)
1.2. Construction of the Combinatorial Space
All possible enzyme combinations were generated from the 10 enzymes
(2¹⁰ − 1 = 1023 combinations).
For each combination:
- Fragment sizes were computed.
- A discrete size axis was built from all unique fragment lengths.
- A binary matrix (combination × fragment sizes) was constructed, indicating presence/absence of each fragment.
This forms the complete “puzzle space” of available molecular weight distributions.
1.3. Definition of the Target Pattern
A desired visual pattern was manually designed using an interactive executable that allows activation/deactivation of bands on the real fragment size axis.
The output is a binary target matrix (lanes × fragment sizes).
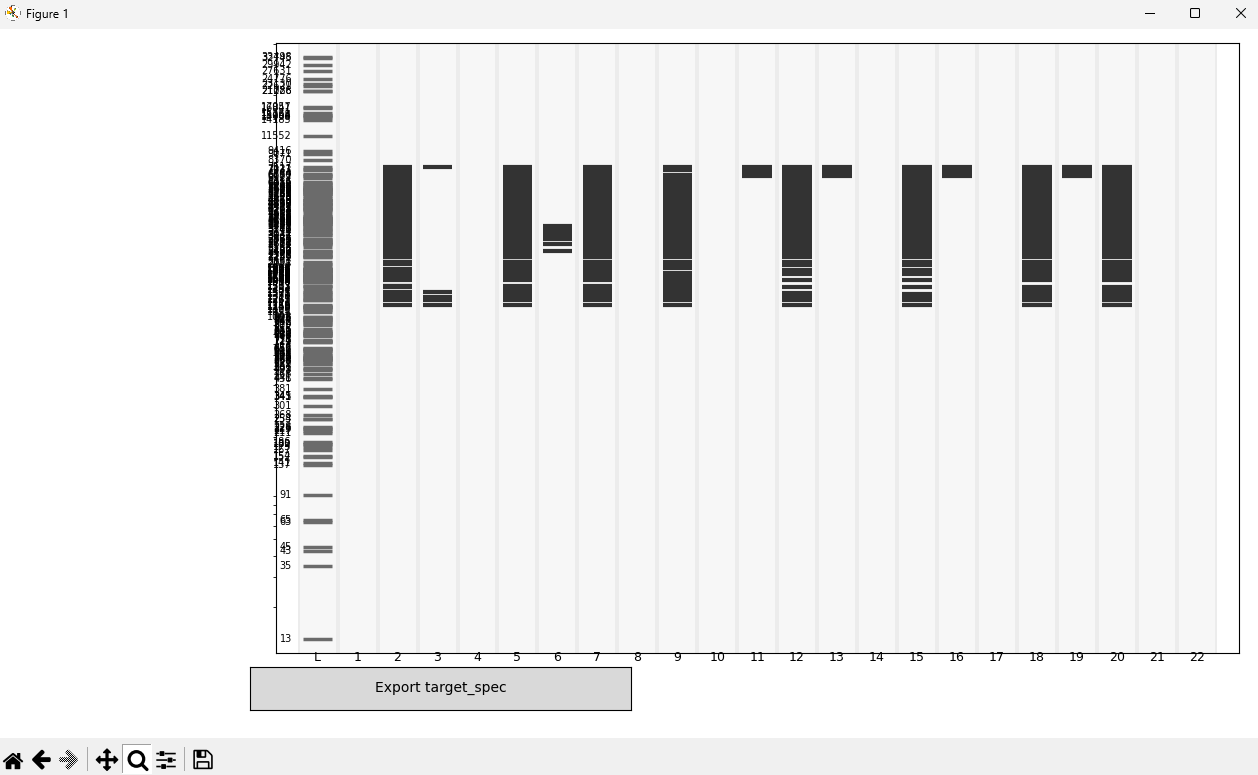 (Image 2: manually designed target pattern)
(Image 2: manually designed target pattern)
The notebook used to manually design the target band pattern can be accessed here:
➡️ Manual Pattern Generator Notebook
1.4. Similarity Comparison and Optimization
For each lane of the target pattern:
- It was compared against all 1023 possible enzyme combinations.
- A contrast-based similarity metric was computed (rewarding matches and penalizing undesired bands).
- A score was assigned.
- The highest-scoring combination was selected.
The resulting set of enzyme combinations per lane represents the optimal reconstruction of the desired pattern.
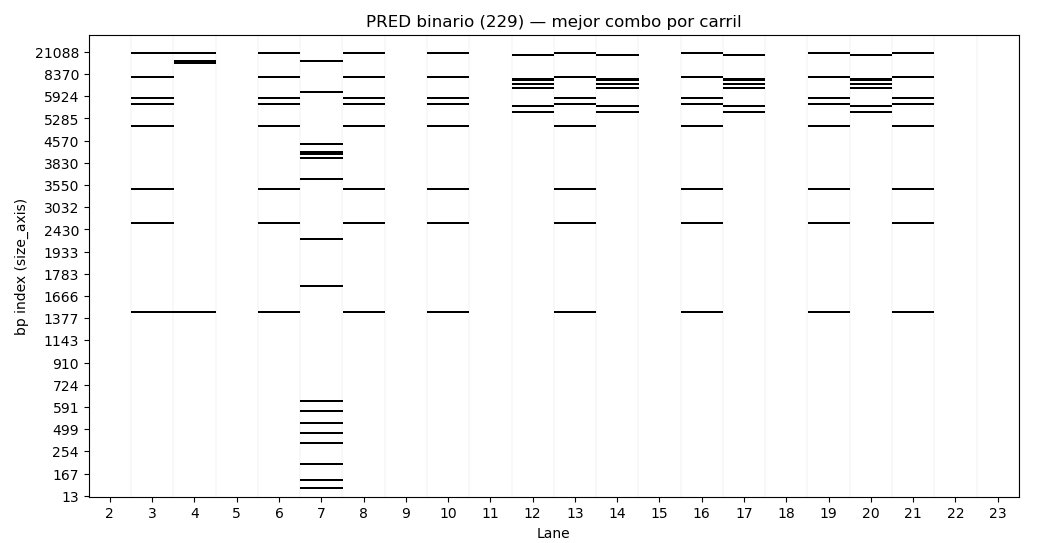 (Image 3: algorithm output + selected enzyme combinations)
(Image 3: algorithm output + selected enzyme combinations)
The notebook implementing the combinatorial search and similarity scoring can be accessed here:
➡️ Optimization Algorithm Notebook
1.5 Result
After optimization and manual refinement, the following enzyme combinations were selected per lane:
Manually Forced Lanes
- Lane 4 → (‘XhoI’, ‘KpnI’)
- Lane 7 → (‘PvuII’, ‘XhoI’)
Best-Scoring Lanes (Algorithm Output)
- Lane 03 → (‘EcoRI’, ‘KpnI’)
- Lane 6 → (‘EcoRI’, ‘KpnI’)
- Lane 8 → (‘EcoRI’, ‘KpnI’)
- Lane 10 → (‘EcoRI’, ‘KpnI’)
- Lane 12 → (‘BamHI’,)
- Lane 13 → (‘EcoRI’, ‘KpnI’)
- Lane 14 → (‘BamHI’,)
- Lane 16 → (‘EcoRI’, ‘KpnI’)
- Lane 17 → (‘BamHI’,)
- Lane 19 → (‘EcoRI’, ‘KpnI’)
- Lane 20 → (‘BamHI’,)
- Lane 21 → (‘EcoRI’, ‘KpnI’)
The final configuration, combining algorithm-selected and manually adjusted lanes, produces a readable macroscopic pattern that spells:
This demonstrates that complex visual structures can be reconstructed using only physically valid restriction enzyme digestion combinations drawn from the complete 1023-piece combinatorial space.
3. DNA Design Challenge
3.1. Gene Selection: E. coli IroB
For my final project, I am designing a therapeutic bacterium for solid tumor treatment using E. coli Nissle 1917 as the chassis. My goal is to synthesize the iroBCDEN operon, which is responsible for Salmochelin production, allowing the bacteria to scavenge iron more efficiently in the tumor microenvironment.
For this sequence design exercise, I am focusing on the key enzyme: IroB (C-glycosyltransferase). To ensure optimal gene expression, proper protein folding, and to minimize metabolic burden, I selected the native IroB sequence from Escherichia coli (Accession: WP_016242764.1). Utilizing a sequence native to the chassis species is a biologically superior approach compared to importing foreign variants.
3.2. Protein Sequence Input
By starting directly with the pure amino acid sequence, I utilized a Reverse Translation approach to build a pristine DNA sequence tailored for my chassis.
Protein Sequence (WP_016242764.1):
MRILFVGPPLYGLLYPVLSLAQAFRVNGHEVLIASGGQFAQKAAEAGLVVFDAAPGLDSEAGYRHHEAQR KKSNIGTQMGNFSFFSEEMADHLVEFAGHWRPDLIIYPPLGVIGPLIAAKYDIPVVMQTVGFGHTPWHIK GVTRSLTDAYRRHNVGTTPRDMAWIDVTPPSMSILENDGEPIIPMQYVPYNGGAVWEPWWERRPERKRLL VSLGTVKPMVDGLDLIAWVMDSASEVDAEIILHISANARSDLRSLPSNVRLVDWIPMGVFLNGADGFIHH GGAGNTLTALHAGIPQIVFGQGADRPVNARVVAERGCGIIPGDVGLSSNMINAFLNNRSLRKASEEVAAE MAAQPCPGEVAKSLITMVQKG
3.3. Codon Optimization
Using the Twist Bioscience Expression Optimization tool, I reverse-translated the E. coli protein sequence into an optimized DNA sequence. (https://www.idtdna.com/CodonOpt)
Optimization Parameters & Constraints:
- Chassis: Escherichia coli.
- Genetic Logic Compatibility: I explicitly removed internal restriction sites (
GGTCTCfor BsaI andGAAGACfor BbsI). This is a critical engineering step to ensure the synthesized gene is fully compatible with the Golden Gate Assembly method, which is required for assembling my NAND logic gate circuit.
Optimized IroB DNA Sequence:
ATG AGA ATT TTA TTT GTT GGA CCG CCG CTC TAC GGC CTG CTG TAT CCG GTG CTG AGC CTG GCG CAG GCG TTC CGC GTC AAC GGC CAC GAG GTG CTG ATT GCC TCC GGC GGG CAG TTT > GCG CAG AAA GCG GCG GAA GCC GGT CTG GTG GTG TTT GAT GCC GCG CCG GGC CTG GAC TCT GAA GCG GGT TAC CGC CAT CAC GAA GCG CAG CGC AAA AAA AGC AAC ATT GGC ACC CAG > ATG GGT AAC TTC AGC TTC TTC TCT GAA GAA ATG GCC GAT CAC CTG GTT GAG TTT GCC GGT CAC TGG CGT CCG GAC CTG ATT ATC TAT CCG CCG CTG GGT GTG ATT GGT CCG CTG ATT > GCG GCA AAA TAT GAC ATC CCG GTG GTT ATG CAG ACC GTC GGC TTT GGT CAC ACG CCG TGG CAC ATC AAA GGC GTG ACC CGC AGC CTG ACC GAT GCC TAT CGC CGT CAC AAC GTT GGC > ACC ACA CCG CGT GAT ATG GCG TGG ATC GAC GTC ACA CCG CCA AGC ATG AGC ATC CTG GAA AAC GAC GGT GAG CCG ATC ATT CCG ATG CAG TAT GTG CCG TAC AAC GGT GGT GCG GTG > TGG GAG CCG TGG TGG GAG CGT CGT CCG GAG CGC AAG CGC CTG CTG GTG AGC CTG GGT ACG GTG AAA CCG ATG GTG GAC GGT CTG GAT CTG ATT GCC TGG GTG ATG GAC AGC GCC AGC > GAA GTT GAT GCG GAG ATC ATC CTG CAC ATC TCT GCC AAC GCG CGC AGC GAC CTG CGC TCG CTG CCG AGC AAC GTG CGC CTG GTT GAT TGG ATT CCG ATG GGT GTG TTC CTG AAC GGT > GCG GAC GGC TTT ATC CAC CAC GGT GGT GCG GGT AAC ACC CTG ACT GCG CTG CAT GCC GGT ATT CCG CAG ATT GTC TTT GGT CAG GGT GCT GAC CGC CCG GTT AAT GCG CGT GTG GTG > GCG GAG CGT GGC TGT GGG ATC ATC CCG GGT GAT GTC GGC CTG TCC AGC AAC ATG ATC AAC GCC TTC CTG AAC AAC CGC TCG CTG CGT AAA GCC TCT GAA GAG GTT GCG GCA GAA ATG > GCG GCG CAG CCG TGC CCG GGT GAG GTG GCC AAA TCG CTG ATC ACC ATG GTT CAG AAA GGG
3.4. Protein Production Technologies
To produce the IroB protein from the newly designed and optimized DNA sequence, two main technological approaches can be employed: cell-dependent (in vivo) and cell-free (in vitro) systems.
1. Cell-Dependent Method (In vivo expression)
This is the traditional recombinant protein production method. The optimized iroB DNA sequence would be cloned into an expression plasmid containing a strong promoter and a Ribosome Binding Site (RBS). This plasmid is then transformed into a bacterial host, such as Escherichia coli BL21(DE3) for massive lab-scale production, or directly into our therapeutic chassis, E. coli Nissle 1917. The living bacteria will act as bio-factories, using their native cellular machinery to express the protein during their growth phase.
2. Cell-Free Protein Synthesis (CFPS) (In vitro expression)
Alternatively, a cell-free system (such as the PURE system or an E. coli cell extract) can be used. This technology strips away the living cell and uses only the essential biological machinery (RNA polymerases, ribosomes, tRNAs, amino acids, and energy molecules) mixed in a tube. By adding our linear or plasmid DNA directly into this mixture, the IroB protein can be synthesized in a few hours. This method is highly advantageous for rapid prototyping and testing of genetic circuits, as it bypasses the need for cell transformation and culturing.
DNA sequence to Protein
- Transcription: Under the control of a hypoxia-sensitive promoter (part of my NAND gate logic), the bacterial RNA Polymerase enzyme recognizes and binds to the specific promoter sequence located just upstream of our iroB gene. The enzyme unwinds the double-stranded DNA and uses the template strand to synthesize a single-stranded messenger RNA (mRNA). It reads through our optimized sequence, creating an exact RNA copy, and stops when it reaches a terminator sequence.
- Translation: Once the mRNA is transcribed, the bacterial ribosome recognizes and binds to the Ribosome Binding Site (RBS) on the mRNA. The ribosome scans the mRNA until it finds the start codon (
ATG). Because we performed codon optimization via reverse translation, the sequence is perfectly calibrated for E. coli. Transfer RNAs (tRNAs) carrying specific amino acids will efficiently recognize the optimized mRNA codons (three-letter nucleotide sequences) without stalling. The ribosome links these amino acids together through peptide bonds, moving along the mRNA until it reaches the stop codon (TAA,TAG, orTGA). At this point, the newly synthesized IroB C-glycosyltransferase protein folds into its 3D structure and is released to perform its catalytic function. - Function: Once folded, IroB will begin glycosylating enterobactin within the cytoplasm to produce the therapeutic Salmochelin.
3.5 How does it work in nature/biological systems?
Historically, the rule in biology was “one gene, one protein.” However, we now know that a single gene can produce multiple different protein variants (isoforms) through mechanisms that alter the mRNA transcript before it is translated. At the transcriptional (and early post-transcriptional) level, there are two primary mechanisms for this:
1. Alternative Splicing
In eukaryotic cells, genes are composed of coding regions (exons) and non-coding regions (introns). When RNA polymerase transcribes the gene, it creates a precursor mRNA (pre-mRNA) that contains both. During a process called alternative splicing, a cellular complex called the spliceosome removes the introns and joins the exons together. However, the spliceosome can choose to include or skip certain exons. Depending on which combination of exons is spliced together to form the mature mRNA, the ribosome will translate completely different protein isoforms, each potentially having different structural domains or functions, all originating from the exact same DNA sequence.
2. Alternative Promoters (Alternative Transcription Start Sites)
A single gene can possess multiple promoters (the DNA sequence where RNA polymerase binds to initiate transcription). Depending on which promoter the cell activates—often influenced by environmental signals or tissue type—transcription will start at different points along the gene. If transcription starts at a downstream promoter, the resulting mRNA will be shorter and will lack the initial genetic instructions. When translated, this produces a truncated version of the protein, often missing specific signaling sequences or regulatory domains present in the full-length version.
4. Plasmid Construction and In Silico Validation (Phase I: iroB)
This section documents the construction of the initial expression vector in Benchling, starting from the optimized iroB gene and culminating in a verified plasmid assembly ready for future expansion.
4.1. Genetic Cassette Design and Optimization
The expression cassette was built systematically in Benchling, starting with the codon-optimized sequence for the iroB gene. To ensure functionality, modularity, and future purifiability, specific genetic parts were integrated:
- Components: The cassette includes a Promoter (Pro), Ribosome Binding Site (RBS), the iroB CDS, and a strong transcriptional Terminator (Ter).
- C-Terminal tag: A 7xHis-tag was added immediately upstream of the STOP codon to allow for future protein purification via affinity chromatography.
- Modular Restriction Sites: To create a standardized “BioBrick-like” part, the entire cassette was flanked with unique restriction enzyme sites: NotI (
GCGGCCGC) at the 5’ end and XbaI (TCTAGA) at the 3’ end.
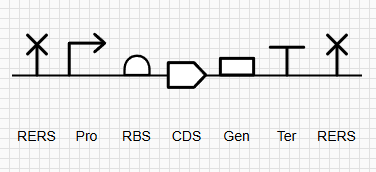 Figure 1: SBOL Diagram of the iroB Expression Cassette. The symbols denote (from left to right): NotI RERS, Promoter, RBS, iroB CDS, 7xHis-Tag, Terminator, and XbaI RERS.
Figure 1: SBOL Diagram of the iroB Expression Cassette. The symbols denote (from left to right): NotI RERS, Promoter, RBS, iroB CDS, 7xHis-Tag, Terminator, and XbaI RERS.
Technical Note: The XbaI site was placed immediately following the terminator to encompass the entire modular cassette. It is important to note that since the selected terminator sequence ends in ‘TA’ (and not ‘GA’), the formation of a Dam methylation site (
GATCTAGA) is avoided. This serendipitous sequence alignment ensures that the enzyme will not be blocked by methylation, allowing for efficient cleavage during laboratory procedures.
4.2. Vector Selection and In Silico Assembly
The commercial vector pTwist Amp High Copy (2221 bp) was selected as the backbone for this phase. (https://www.twistbioscience.com/products/genes/vectors?tab=catalog-vectors)
Benchling Assembly Process:
- Backbone Preparation: The pTwist map was imported, identifying the Multiple Cloning Site (MCS:region between coordinates ~73 and ~245) as the optimal insertion point. Specifically, coordinate 200 was selected to ensure that critical elements like the origin of replication (ori) and the ampicillin resistance gene (AmpR) remained undisturbed.
- Insert Preparation: The 1326 bp iroB modular cassette was defined using the flanking NotI (5’ end) and XbaI (3’ end) recognition sites. This allows any researcher to infer the intended Forward direction of the gene by identifying the positions of these specific landmarks on the plasmid map. Modularity is also ensured, allowing the entire expression cassette to be excised and transferred to different vectors in future iterations of the project.
- Assembly Simulation: Using Benchling’s molecular biology tools, a Gibson Assembly was simulated to insert the designed iroB modular cassette into the pTwist MCS, resulting in a final circular plasmid of exactly 3561 bp.
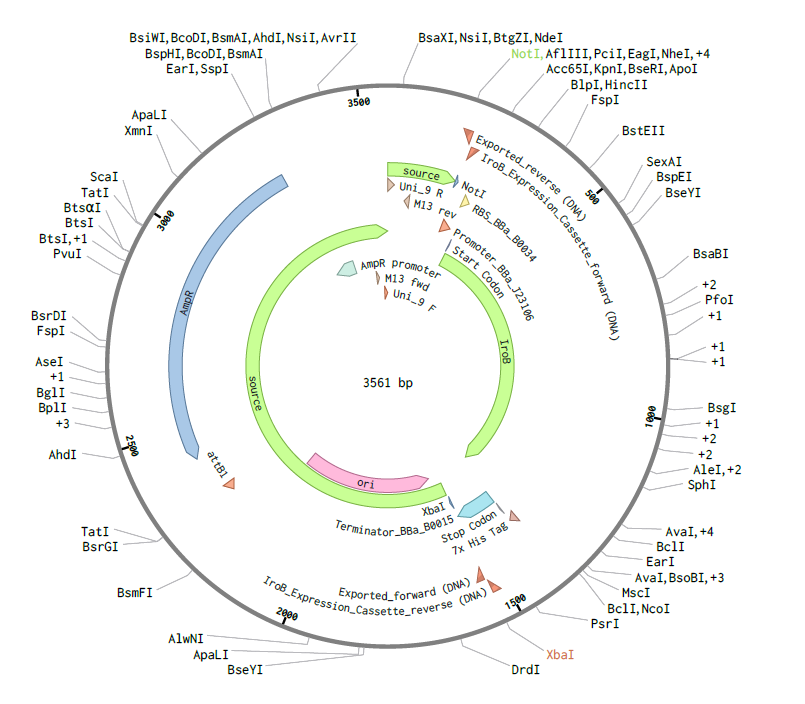 Figure 2: Circular map of the assembled pTwist-iroB-cassette plasmid (3561 bp).
Figure 2: Circular map of the assembled pTwist-iroB-cassette plasmid (3561 bp).
4.3. Results and Validation: Virtual Digest
To validate the structural integrity of the design, a Virtual Enzymatic Digest was performed using NotI and XbaI. The simulation results account for the redistribution of nucleotides at the restriction sites following the cleavage:
- Fragment 1 (Vector Backbone - pTwist): 2228 bp. This represents the original 2221 bp pTwist sequence plus 7 bp derived from the flanking restriction site architecture.
- Fragment 2 (iroB Expression Cassette): 1333 bp. This comprises the 1326 bp optimized cassette plus 7 bp from the remaining restriction site sequences.
The sum of these fragments confirms a total plasmid length of 3561 bp.
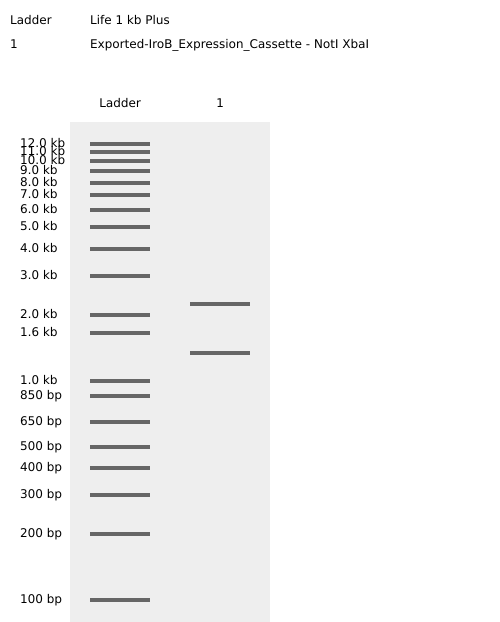 Figure 3: Virtual agarose gel electrophoresis (1% agarose). Lane 1: DNA Ladder. Lane 2: pTwist-iroB digested with NotI/XbaI, yielding two distinct and sharp bands at 2228 bp and 1333 bp. This result confirms successful in silico assembly and validates that the iroB optimized sequence is free of internal restriction sites for the selected enzymes.
Figure 3: Virtual agarose gel electrophoresis (1% agarose). Lane 1: DNA Ladder. Lane 2: pTwist-iroB digested with NotI/XbaI, yielding two distinct and sharp bands at 2228 bp and 1333 bp. This result confirms successful in silico assembly and validates that the iroB optimized sequence is free of internal restriction sites for the selected enzymes.
4.4. Future Work: Iterative Design
This validated plasmid serves as the foundational “chassis” for the project. The next engineering phases involve:
Promoter Re-engineering: Replacing the current constitutive promoter with a Boolean Logic (e.g., NAND gate) promoter designed to respond to hypoxia and ultrasound-linked stimuli.
Operon Completion: Sequentially assembling the remaining genes (iroC, iroD, iroE, and iroN) into the cassette to generate a single polycistronic iroBCDEN operon.
Clinical-Grade Vector Redesign: The current backbone includes an antibiotic-resistance marker, which is not ideal for therapeutic applications due to biosafety and regulatory concerns. Future versions of the construct should transition to a non-antibiotic plasmid maintenance system appropriate for clinical use.
Biocontainment Strategy (Kill Switch Evaluation): A toxin–antitoxin-based containment module is a candidate approach. In such systems, continuous expression of an antitoxin neutralizes a stable toxin; loss or inhibition of the antitoxin can result in growth arrest or cell death. The stability, leakiness, and escape frequency of this strategy must be experimentally evaluated.
Expression Burden Mitigation: Full expression of the iroBCDEN complex may impose significant metabolic and translational burden. Strategies such as orthogonal translation systems (e.g., orthogonal ribosomes) or alternative burden-mitigation approaches should be assessed to improve stability and performance.
5. Theoretical Questions: DNA Read, Write, & Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
- Target: I would sequence the genome of the Panther Chameleon (Furcifer pardalis) and the Common Octopus (Octopus vulgaris), focusing on the gene families of reflectins and the regulatory proteins of iridophores/chromatophores.
- Why: Current biological reporters, such as Fluorescent Proteins (FPs), have a fundamental limitation: high stability leads to poor temporal resolution. Once an FP is expressed or activated, it remains fluorescent for hours, “smearing” the signal and masking real-time dynamics. This makes it impossible to observe rapid “on/off” pulses in neural circuits (like serotonergic vs. octopaminergic crosstalk) or the precise timing of a synthetic logic gate.
By sequencing these organisms, I aim to discover the genetic basis of reversible structural color. Unlike fluorescence, which requires high-energy lasers that cause phototoxicity and photobleaching, reflectins change their optical properties through rapid conformational shifts. In the context of my cancer-targeting project, these proteins could serve as “dynamic reporters” for my NAND logic gate. They would allow me to observe, in vitro, the exact moment the bacteria detect ultrasound or hypoxia and—crucially—see the signal vanish the instant the stimulus stops. This would provide a level of kinetic resolution and biophysical feedback that is currently unattainable with standard fluorescence, enabling the study of fast enzymatic transitions and synaptic-like communications without “staining” the entire experimental field.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
- Selected Technology: PacBio HiFi Sequencing combined with Iso-Seq.
- Rationale: Since the goal is to find functional proteins with specific structural kinetics, I need to resolve not just the genome, but the full-length isoforms of the proteins being expressed in the skin cells. PacBio HiFi provides the extreme accuracy (99.9%) and long reads necessary to assemble these complex, repetitive protein domains without the errors of short-read platforms.
Detailed Technical Questions:
- Is your method first-, second- or third-generation or other? How so?
- It is a third-generation technology (Single Molecule, Real-Time). It sequences individual DNA molecules as they are synthesized by a polymerase in a Zero-Mode Waveguide (ZMW), allowing for real-time observation of base incorporation.
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- Input: High Molecular Weight (HMW) genomic DNA and full-length mRNA (for Iso-Seq) from dermal tissue.
- Preparation steps: 1. Extraction: Specialized lysis to maintain long-strand integrity. 2. SMRTbell Library Prep: Ligation of hairpin adapters to create circular DNA templates. 3. Size Selection: Ensuring only long fragments (>10kb) are loaded to maximize information per read.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- Process: A polymerase at the bottom of a ZMW incorporates fluorescently labeled nucleotides. As each base is added, it emits a light pulse.
- Base Calling: The system records the color and duration of these pulses. Because the template is circular, the polymerase reads it multiple times (Circular Consensus Sequencing), which allows the software to “correct” any random errors and produce a HiFi read of extremely high quality.
- What is your output of your chosen sequencing technology?
- The output is a BAM file containing highly accurate, long-read sequences, ready for de novo assembly of the structural color gene clusters.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
- Target: I want to synthesize a modular genetic circuit containing a NAND logic gate that integrates two environmental sensores (ultrasound-responsive promoters and hypoxia-inducible factors) to drive the expression of the iroB cluster for salmochelin production, including a safety kill-switch.
- Why: This construct is the core of my final project: a targeted cancer therapy. The goal is to engineer bacteria that only produce potent iron-sequestering siderophores (salmochelins) within the tumor microenvironment (hypoxia) and under external activation (ultrasound). This ensures the therapy is localized, minimizing systemic toxicity. Synthesizing this specific construct via Twist would allow me to perform the first in vitro validations of the logic gate’s precision.
- Sequence: I don’t have the complete cassette sequence yet; I need to redesign the promoter, add the Kill-Switch and add the iroBCDEN complex. Preliminary sequence: TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGCCATTAAAGAGGAGAAAGGTACCATGATGAGAATTTTATTTGTTGGACCGCCGCTCTACGGCCTGCTGTATCCGGTGCTGAGCCTGGCGCAGGCGTTCCGCGTCAACGGCCACGAGGTGCTGATTGCCTCCGGCGGGCAGTTTGCGCAGAAAGCGGCGGAAGCCGGTCTGGTGGTGTTTGATGCCGCGCCGGGCCTGGACTCTGAAGCGGGTTACCGCCATCACGAAGCGCAGCGCAAAAAAAGCAACATTGGCACCCAGATGGGTAACTTCAGCTTCTTCTCTGAAGAAATGGCCGATCACCTGGTTGAGTTTGCCGGTCACTGGCGTCCGGACCTGATTATCTATCCGCCGCTGGGTGTGATTGGTCCGCTGATTGCGGCAAAATATGACATCCCGGTGGTTATGCAGACCGTCGGCTTTGGTCACACGCCGTGGCACATCAAAGGCGTGACCCGCAGCCTGACCGATGCCTATCGCCGTCACAACGTTGGCACCACACCGCGTGATATGGCGTGGATCGACGTCACACCGCCAAGCATGAGCATCCTGGAAAACGACGGTGAGCCGATCATTCCGATGCAGTATGTGCCGTACAACGGTGGTGCGGTGTGGGAGCCGTGGTGGGAGCGTCGTCCGGAGCGCAAGCGCCTGCTGGTGAGCCTGGGTACGGTGAAACCGATGGTGGACGGTCTGGATCTGATTGCCTGGGTGATGGACAGCGCCAGCGAAGTTGATGCGGAGATCATCCTGCACATCTCTGCCAACGCGCGCAGCGACCTGCGCTCGCTGCCGAGCAACGTGCGCCTGGTTGATTGGATTCCGATGGGTGTGTTCCTGAACGGTGCGGACGGCTTTATCCACCACGGTGGTGCGGGTAACACCCTGACTGCGCTGCATGCCGGTATTCCGCAGATTGTCTTTGGTCAGGGTGCTGACCGCCCGGTTAATGCGCGTGTGGTGGCGGAGCGTGGCTGTGGGATCATCCCGGGTGATGTCGGCCTGTCCAGCAACATGATCAACGCCTTCCTGAACAACCGCTCGCTGCGTAAAGCCTCTGAAGAGGTTGCGGCAGAAATGGCGGCGCAGCCGTGCCCGGGTGAGGTGGCCAAATCGCTGATCACCATGGTTCAGAAAGGGCATCACCATCACCATCATCACTAACCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
- Selected Technology: Silicon-based high-throughput chemical synthesis (Twist Bioscience platform).
- Rationale: For a complex circuit involving multiple promoters, a CDS (iroB), and a kill-switch, I need extreme precision and the ability to synthesize large quantities of different variations of the circuit. Twist’s technology uses a silicon platform that miniaturizes the traditional phosphoramidite chemistry, allowing for the synthesis of thousands of genes simultaneously with high fidelity and low cost, which is ideal for prototyping complex logic gates like mine.
Detailed Technical Questions:
- What are the essential steps of your chosen synthesis method?
- Phosphoramidite Cycle: The DNA is built base-by-base (A, T, C, G) on a silicon chip. Each addition follows a cycle of 4 steps: De-protection (preparing the strand), Coupling (adding the base), Capping (preventing errors), and Oxidation (stabilizing the bond).
- Assembly: Since the chemical process can only print short pieces (oligos), these pieces are harvested from the chip and assembled into the full 2.5 kb circuit.
- Error Correction: The final DNA is “polished” using enzymes to ensure there are no mutations, delivering a 100% accurate sequence.
- What are the limitations of your synthesis method (if any) in terms of speed, accuracy, scalability?
- Speed: The chemical synthesis is fast, but the complete process (assembly, quality control, and shipping) takes about 2 to 3 weeks.
- Accuracy: As the DNA strand gets longer, the chance of errors increases, which is why we must assemble smaller, verified fragments to build a large circuit.
- Scalability: It is highly scalable (thousands of genes at once), but it still depends on traditional chemicals, unlike newer enzymatic methods.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
- Target DNA: The genome of a therapeutic strain of Escherichia coli (like E. coli Nissle 1917).
- Why: I need to integrate my synthetic NAND-iroB circuit into the bacterial chromosome. If the circuit stays on a plasmid, it could be lost or vary in copy number. By editing the bacterial genome to insert the circuit into a “safe harbor” locus, I ensure the therapy is stable and the kill-switch works perfectly every time.
(ii) What technology or technologies would you use to perform these DNA edits and why?
- Selected Technology: CRISPR-Cas9 mediated Recombineering.
- Rationale: CRISPR-Cas9 is extremely precise at “cutting” the DNA at a specific site in the bacterial genome, and Recombineering allows us to “paste” my large 2.5 kb synthetic circuit into that gap with high efficiency.
Detailed Technical Questions:
- How does your technology of choice edit DNA? What are the essential steps?
- Targeting: A Guide RNA (gRNA) leads the Cas9 protein to the exact spot in the bacterial genome.
- Cutting: Cas9 creates a double-strand break (DSB) in that spot.
- Insertion: Using the cell’s repair machinery (and a template I provide), the synthetic NAND-iroB circuit is “pasted” into the cut, becoming a permanent part of the bacteria’s DNA.
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- Design steps: I need to design the gRNA (to target the genome) and the Homology Arms (flanking sequences that match the insertion site).
- Input: The Cas9 enzyme, the gRNA, and my synthetic DNA construct (the circuit synthesized by Twist).
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- Efficiency: In bacteria, the rate of successful “pasting” (HDR) can be low, often requiring selection markers (like antibiotic resistance) to find the edited cells.
- Precision: There is a small risk of off-target effects, where Cas9 might cut in the wrong place, though this is rare in simple bacterial genomes.