Week 04 HW: Protein Design Part I
Part A. Conceptual Questions from Shuguang Zhang
1) How many molecules of amino acids are in 500 g of meat?
Assume meat is ~20–25% protein: 500 g meat → ~100–125 g protein.
Using ~100 Da per amino acid (given):
- 100 g / (100 g/mol) = 1.0 mol amino acids → ~6.0×10^23 molecules
- 125 g / (100 g/mol) = 1.25 mol amino acids → ~7.5×10^23 molecules
Answer: ~10^23
2) Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Food proteins are digested into amino acids/peptides, then your cells rebuild human proteins according to your genome and regulation. You recycle building blocks; you do not copy the animal’s body plan.
3) Why are there only 20 natural amino acids?
The translation system (genetic code + tRNAs + synthetases + ribosome) is optimized around a standard set that provides a broad, efficient chemical toolkit. Expanding it is costly because it would require coordinated changes across the whole decoding machinery (and most proteins). (Note: nature also uses rare genetically encoded additions like selenocysteine/pyrrolysine in some lineages.)
4) Can you make other non-natural amino acids? Design some new amino acids.
Yes—chemistry and engineered translation can incorporate noncanonical amino acids. Examples:
- Azido-alanine (Ala–N3): bioorthogonal “click” handle for labeling.
- p-benzoyl-phenylalanine: UV-activated crosslinker to trap interactions.
- Bipyridyl-alanine: metal-chelating side chain for catalysis/materials.
- Fluoroleucine: tunes hydrophobicity/stability and NMR/19F probes.
5) Where did amino acids come from before enzymes and before life started?
Abiotic synthesis from simple precursors (e.g., atmospheric/energy-driven reactions), mineral-catalyzed chemistry (e.g., hydrothermal settings), and extraterrestrial delivery (meteorites). Prebiotic chemistry can generate amino acids without enzymes.
6) If you make an α-helix using D-amino acids, what handedness would you expect?
A helix built from D-amino acids is the mirror of the L-form helix.
Answer: D-amino-acid α-helices are expected to be left-handed (L-amino-acid α-helices are typically right-handed).
7) Can you discover additional helices in proteins?
Yes. Beyond the canonical α-helix, proteins can contain 3₁₀ helices, π helices, and short helical turns. They can be identified by backbone hydrogen-bond patterns and secondary-structure assignment algorithms (e.g., DSSP/STRIDE) and validated by structural data (X-ray/cryo-EM/NMR).
8) Why are most molecular helices right-handed?
Because proteins use L-amino acids, and for L-residues the right-handed α-helix minimizes steric clashes and optimizes backbone H-bond geometry and side-chain packing. Left-handed helices are generally less favorable for L-residues.
9) Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-strands have backbone H-bond donors/acceptors; exposed “sheet edges” can form intermolecular H-bonds, effectively zipping molecules together.
Driving forces: backbone hydrogen bonding + hydrophobic packing (and release of ordered water), often producing very stable “stacked” β-structures.
Part B: Protein Analysis and Visualization
1) Briefly describe the protein and why I selected it
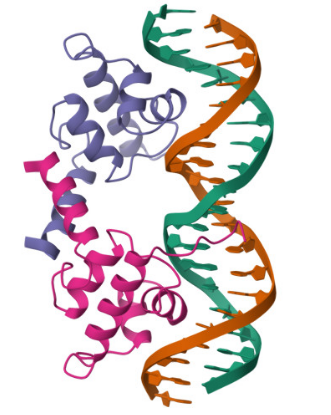
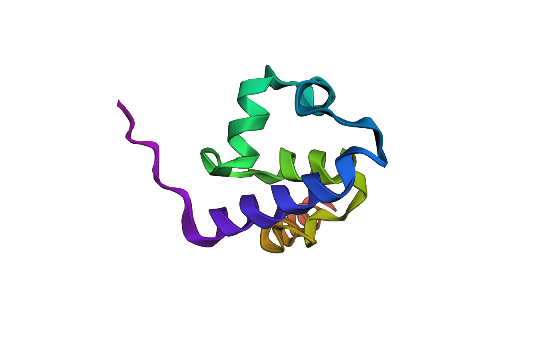
I selected the lambda repressor (cI) (PDB 1LMB) because it is a well-resolved DNA-binding transcriptional repressor and provides a direct structural model for the TlpA/TlpA39–P_tlpA thermal switch used in ultrasound-triggered bacterial circuits. In both systems, a repressor binds an operator/promoter to block transcription, and regulation occurs by changing the repressor’s ability to bind DNA; therefore, cI is an ideal, structurally validated example to analyze DNA binding, secondary structure, and regulatory interfaces using 3D visualization tools. (rcsb.org)
Bacteriophage lambda repressor (cI), N-terminal DNA-binding domain bound to operator DNA
RCSB PDB: 1LMB (X-ray, 1.80 Å)
2) Identify the amino acid sequence of your protein.
How long is it?
- Sequence length: 92 amino acids
What is the most frequent amino acid?
- Most frequent amino acid: A (Alanine) — 11 occurrences
How many protein sequence homologs are there for your protein?
Running UniProt BLAST with the 1LMB protein sequence returned 231 homologous sequences (hits) in UniProtKB. The top matches are annotated as phage repressors / HTH-type transcriptional regulators (lambda/lambdoid-like repressors).
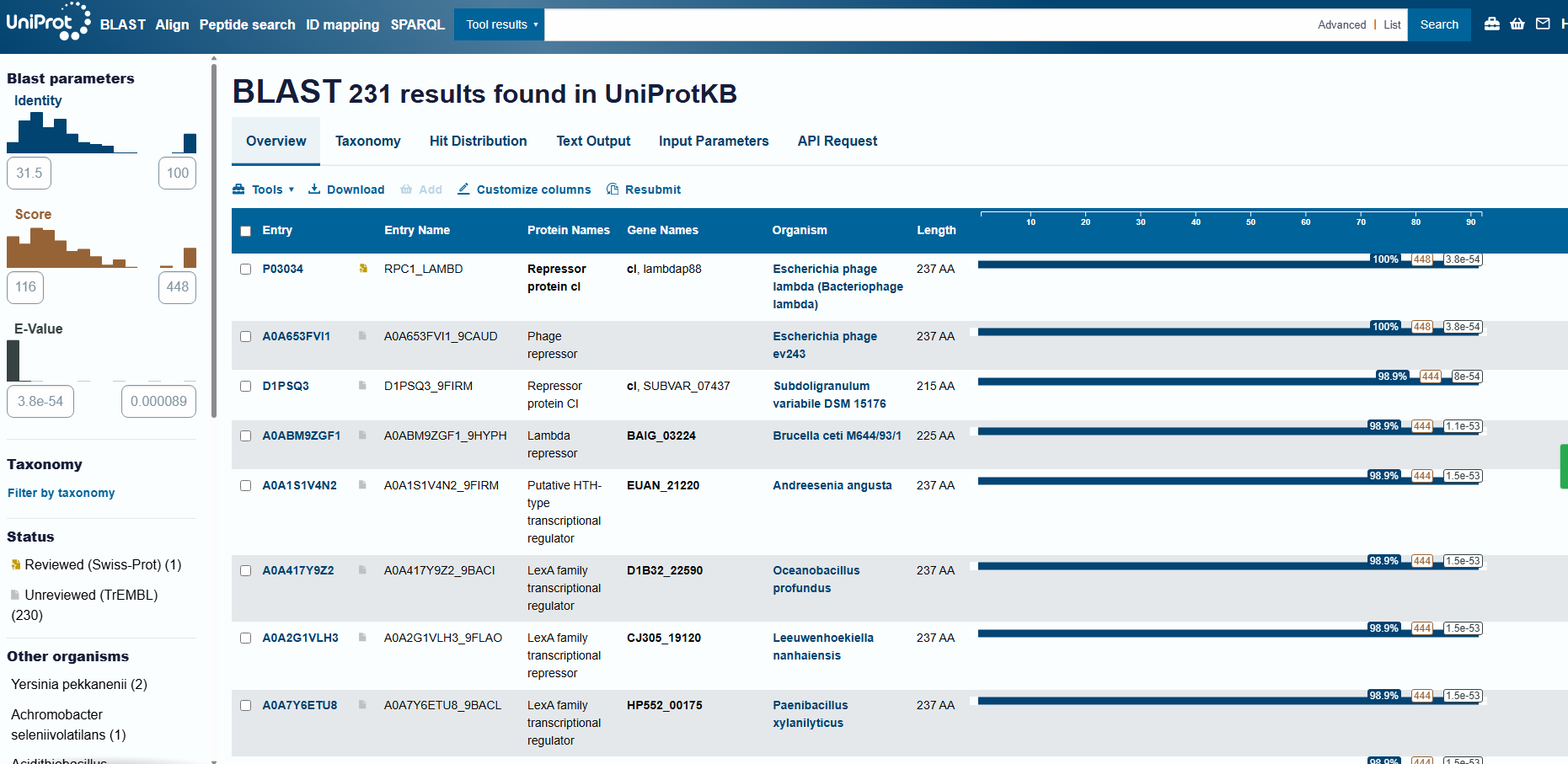
Protein family
cI is a helix-turn-helix (HTH) DNA-binding transcriptional repressor, part of the lambda/lambdoid phage repressor family that controls the lysis–lysogeny switch in temperate bacteriophages.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
- Deep Mutational Scans (ESM2)
I used ESM2 to generate an unsupervised deep mutational scan of my protein based on language-model likelihood scores (heatmap of all single substitutions).
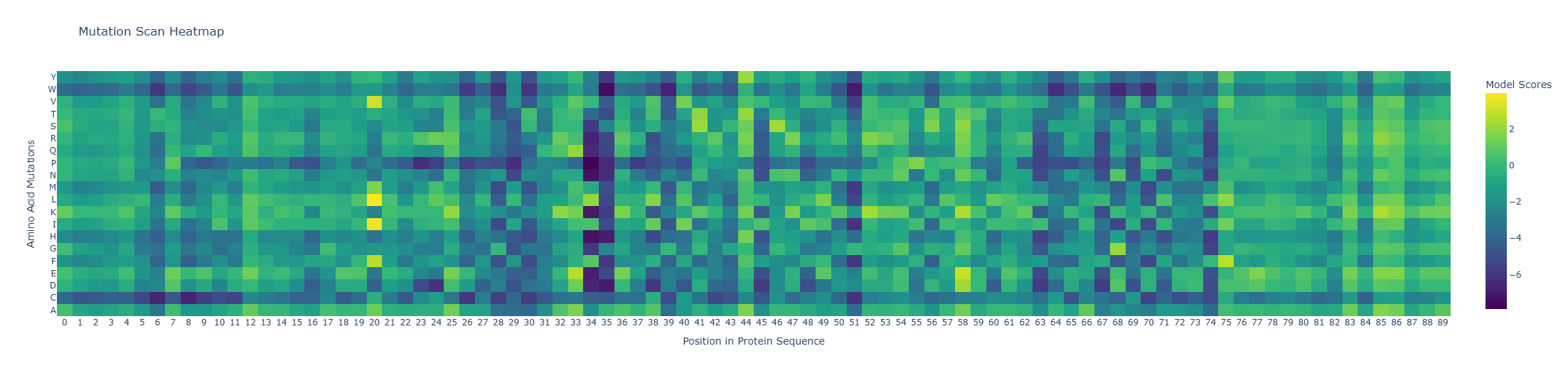
Pattern / standout example:
Because the heatmap is plotted as heatmap[:, 2:], the x-axis starts at residue 3 (1-based).
- Most deleterious mutation: D37→P (heatmap: x=34, y=P, z≈-7.89). Proline is strongly disfavored here, consistent with disruption of local secondary structure (proline is a common helix breaker).
- Most favorable mutation: K23→L (heatmap: x=20, y=L, z≈+3.95), suggesting this substitution is well tolerated in the local sequence context according to the model.
- Latent Space Analysis (protein embeddings + 3D t-SNE)
I embedded the provided dataset (n=15,177 proteins) using protein language model embeddings (320D) and reduced dimensionality with 3D t-SNE. The resulting map forms local neighborhoods where nearby points represent proteins with similar sequence features (t-SNE is most reliable for local similarity).
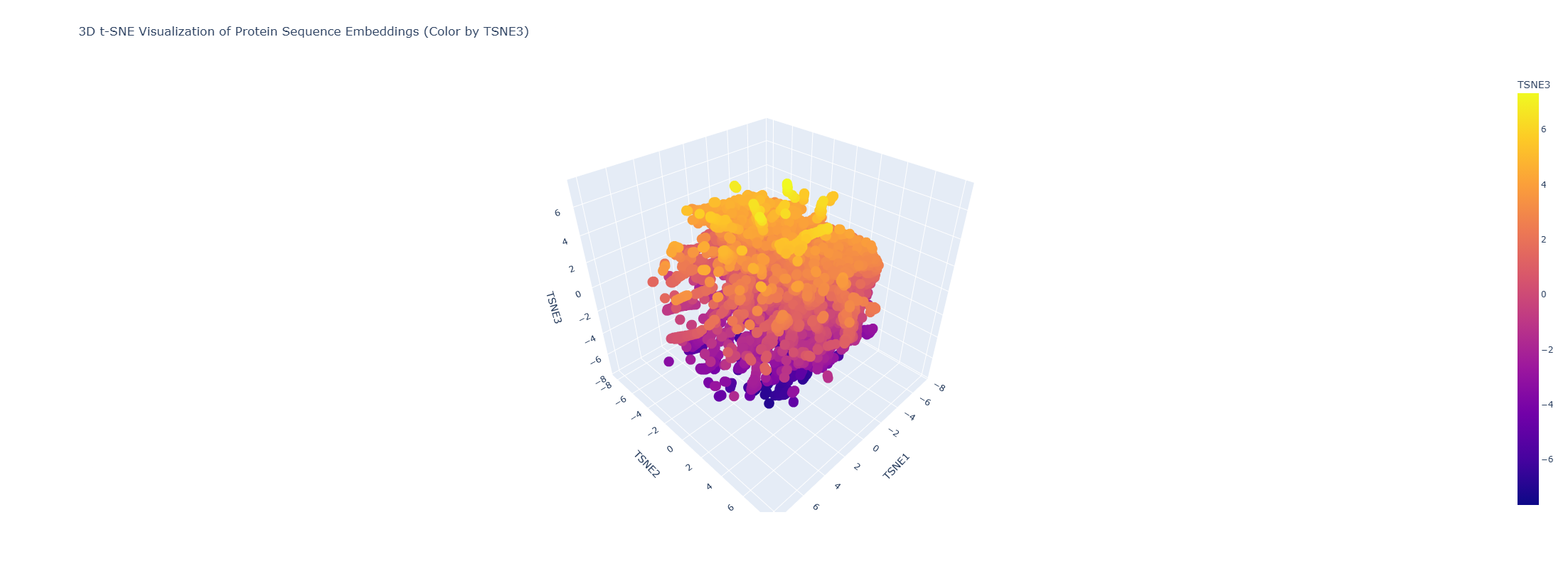
Do neighborhoods approximate similar proteins?
Yes. Proteins in the same neighborhood tend to share related sequence motifs and often similar functions/families.
Placing my protein in the map (via nearest neighbors):
My exact sequence is not present as a point in the provided dataset, so I embedded my sequence with the same model and located it by its closest neighbors in embedding space. The nearest neighbors include:
Nearest neighbors (top 5):
| index | distance | TSNE1 | TSNE2 | TSNE3 | annotation (short) |
|---|---|---|---|---|---|
| 1124 | 0.607 | 1.152 | 1.019 | -6.889 | lambda cI repressor, DNA-binding domain |
| 1152 | 1.080 | 1.113 | 1.027 | -6.865 | HTH-like match (Nostoc punctiforme) |
| 1149 | 1.168 | 1.099 | 1.009 | -6.878 | HTH-like match (E. coli) |
| 1153 | 1.224 | 1.065 | 0.997 | -6.873 | HTH-like match (P. aeruginosa) |
| 1128 | 1.257 | 1.105 | 1.005 | -6.878 | HigA antitoxin (HTH regulator) |
These neighbors are consistent with my protein being a helix-turn-helix (HTH) DNA-binding regulator, and indicate that my sequence lies in an HTH/transcription-factor neighborhood. Colab notebook (Latent space section)
C2. Protein Folding
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I folded the 92 aa lambda repressor DNA-binding domain using ESMFold and compared it to the experimental structure (PDB 1LMB). The prediction shows a mainly alpha-helical fold consistent with an HTH-like DNA-binding domain, so it matches the expected overall topology. Minor differences are expected because 1LMB is solved in a protein–DNA complex, while ESMFold predicts the protein without DNA.
 3. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
3. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I tested ESMFold predictions for (i) a point mutation predicted as favorable by the ESM2 scan (K23L), (ii) a strongly disfavored point mutation (D37P), and (iii) a large-segment replacement (10-aa alanine stretch: positions 31–40 → AAAAAAAAAA).
Across all three variants, the predicted structures remain predominantly alpha-helical and preserve the same overall fold/topology by qualitative visual comparison. Differences, if any, appear mainly local (subtle shifts in helix/loop geometry), rather than a global collapse or refolding.
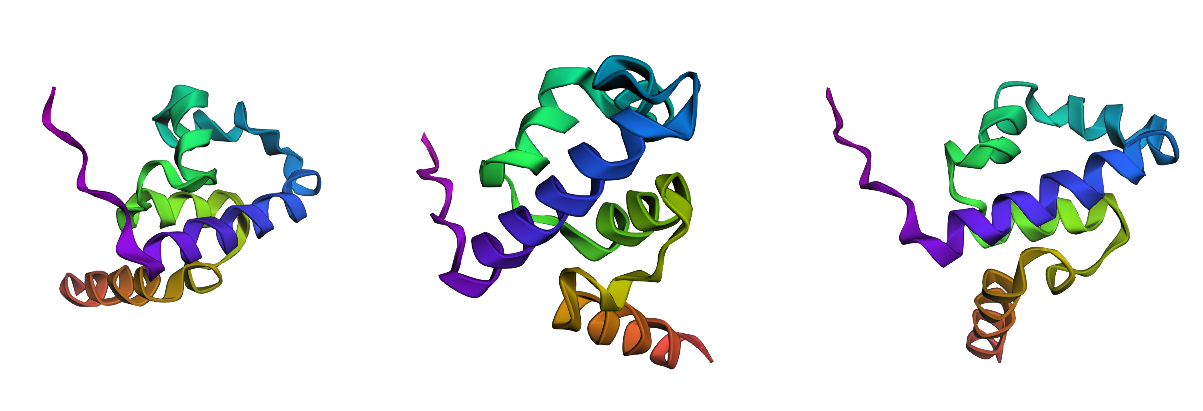 Conclusion: For this small HTH-like domain, the overall fold appears resilient to these mutations and to the tested segment-level replacement (at least at the level of ESMFold-predicted coordinates).
Conclusion: For this small HTH-like domain, the overall fold appears resilient to these mutations and to the tested segment-level replacement (at least at the level of ESMFold-predicted coordinates).
Note: ESM2 mutation scores reflect sequence plausibility, not a direct folding energy. In my tests, even a strongly disfavored mutation (e.g., D37→P) did not collapse the global fold in ESMFold, suggesting the overall topology is robust. The mutation scan is more informative for identifying specific constrained positions (likely functional/structural hotspots) than for predicting global unfolding from a single “worst-score” mutation.
C3. Protein Generation
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
I used the backbone coordinates from PDB 1LMB (protein chain 4) as input to ProteinMPNN to generate sequence candidates compatible with the same fold.
Original (WT):
STKKKPLTQEQLEDARRLKAIYEKKKNELGLSQESVADKMGMGQSGVGALFNGINALNAYNAALLAKILKVSVEEFSPSIAREIYEMYEAVS
ProteinMPNN (sample 0):
GPGRKPLTEEELEAAKKLKAIYEERKEELNLSQAKVAELLGVSQSTVSALFNGERAFNLEIAKKLAEILKIEVSEFSPELAKKIAEEEKKIE
Sequence recovery: 0.5109 (~51% of positions match the original).
Comparison (predicted vs original):
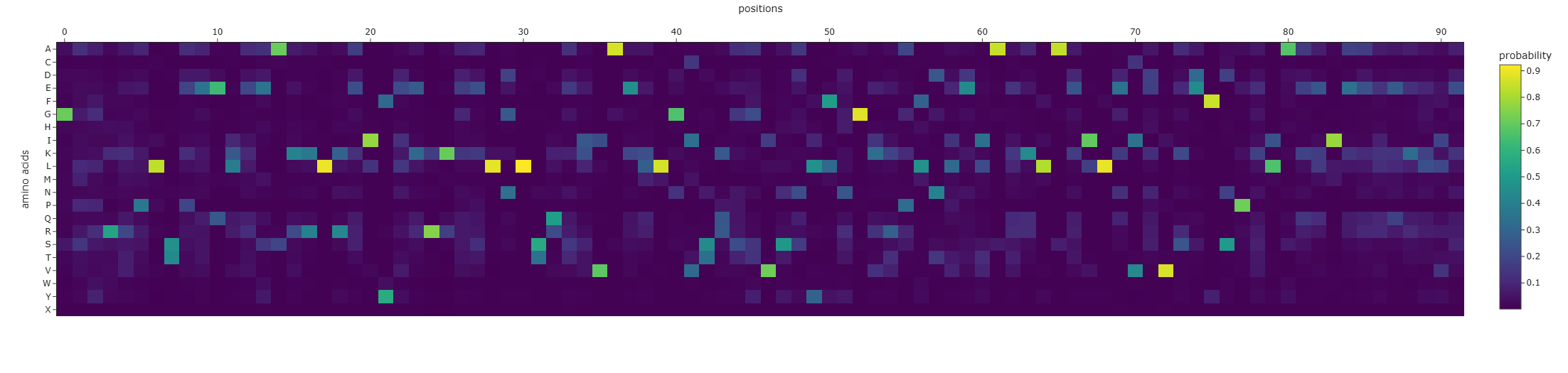 Predicted sequence probabilities:
Predicted sequence probabilities:
The per-position amino-acid probability heatmap shows a mix of:
- high-confidence positions (bright cells), where ProteinMPNN strongly prefers a specific residue given the backbone geometry, and
- low-confidence positions (diffuse/darker columns), where multiple residues are plausible (more sequence flexibility).
Overall, ProteinMPNN preserves the general biochemical character expected for this fold (many helix-compatible and charged residues) while allowing substantial substitutions at less constrained positions.
- Input this sequence into ESMFold and compare the predicted structure to your original.
I folded the ProteinMPNN-designed sequence with ESMFold and compared it to the original fold (left). The designed sequence (right) produces a very similar, predominantly alpha-helical topology, consistent with the same HTH-like backbone. Differences are mainly local (helix lengths/orientations and terminal regions), rather than a complete refolding.
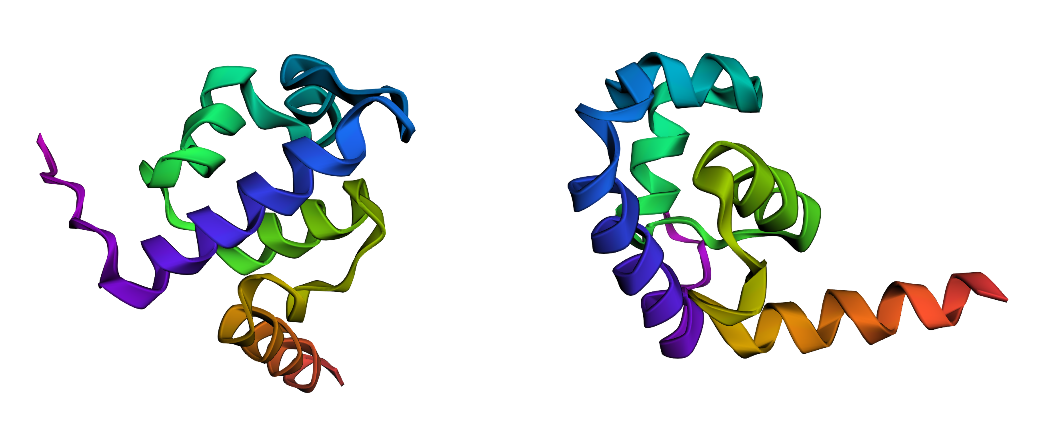 Conclusion: ProteinMPNN proposes a sequence that is compatible with the original backbone: despite ~50% sequence recovery, the predicted structure remains close to the original fold at the qualitative/topology level.
Conclusion: ProteinMPNN proposes a sequence that is compatible with the original backbone: despite ~50% sequence recovery, the predicted structure remains close to the original fold at the qualitative/topology level.
Part D. Group Brainstorm on Bacteriophage Engineering (Engineering MS2 Lysis Protein L via N-Terminal Modulation of DnaJ Dependence)
Selected Goals
Primary goal – Increased stability (functional robustness)
Identify sequence variants of MS2 lysis protein L that maintain structural plausibility and membrane-competent architecture.
Secondary goal – Higher titers (mechanism-linked)
Modulate the dependence of L on the host chaperone DnaJ by engineering the N-terminal regulatory segment that controls activation of the lysis protein.
Biological Motivation
Previous studies show that MS2 lysis protein L requires the host chaperone DnaJ for lytic activity, and that the N-terminal region plays a regulatory role in this dependence. However, no work has systematically explored how sequence variation in this region shapes the conformational constraints underlying host-assisted activation.
We hypothesize that DnaJ dependence emerges from sequence-encoded constraints within the N-terminal regulatory segment. By mapping mutational tolerance in this region, we aim to identify variants that alter host dependence while preserving the membrane-associated lytic function of L.
Computational Approach
Protein Language Models (ESM2 / ESM-3)
Perform an in silico mutational scan of the N-terminal region to identify sequence-plausible mutations. Language model likelihood scores provide a proxy for evolutionary constraints and help prioritize mutations that are unlikely to disrupt protein viability.
Structure Prediction (ESMFold or Boltz-1)
Predict structures for candidate variants and filter out mutations predicted to cause major structural disruption. These predictions act as a structural plausibility check rather than definitive structural validation.
Interaction Proxy (AlphaFold-Multimer)
Model complexes between MS2 L variants and the host chaperone DnaJ. While chaperone interactions are dynamic, these predictions provide a relative signal to compare potential effects of mutations on host interaction.
Sequence Conservation (BLAST + Clustal Omega)
Identify conserved residues to avoid mutating positions likely critical for function.
Potential Pitfalls
- Membrane proteins are challenging for structure predictors.
- Chaperone interactions may not be accurately captured by AlphaFold-Multimer.
- Variants that alter lysis timing could negatively affect phage burst size.
Pipeline
WT MS2 L sequence
↓
BLAST / Clustal → identify conserved N-terminal residues
↓
ESM2 mutational scan → generate candidate variants
↓
ESMFold / Boltz → remove structurally implausible variants
↓
AlphaFold-Multimer → compare predicted interaction with DnaJ
↓
Shortlist variants for experimental testing of lysis timing and phage titers