Week 05 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Original sequencesp|P00441|SODC_HUMAN Superoxide dismutase
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Variant: A4V
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Using the mutant SOD1 sequence as input, PepMLM Colab generated four 12-residue candidate binders
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | WRYPVAGARHWE | 18.89836973999799 |
| 1 | KLYYPVVVAWWK | 17.203301905376957 |
| 2 | HRYPVVVAALKE | 11.315016775827807 |
| 3 | WLYGAAVLRHGE | 15.526728984710877 |
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
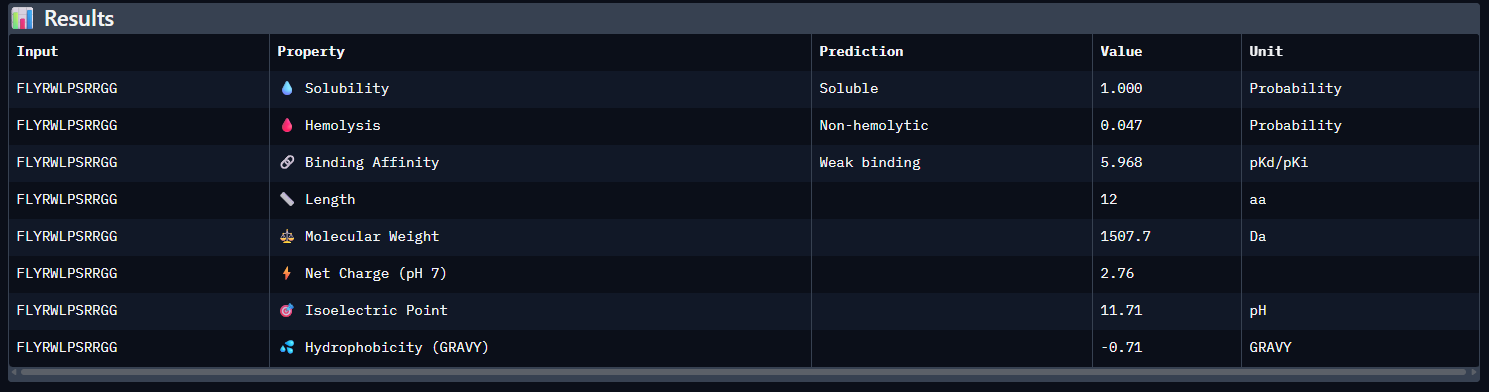
PepMLM’s pseudo-perplexity scores indicate the model’s confidence in the generated binders, with lower values corresponding to higher confidence. Among the four generated peptides, HRYPVVVAALKE showed the highest confidence (lowest pseudo-perplexity, 11.315), whereas WRYPVAGARHWE showed the lowest confidence (highest pseudo-perplexity, 18.898). The reference peptide FLYRWLPSRRGG was included for comparison, but no pseudo-perplexity score for it was provided in the displayed output.
Part 2: Evaluate Binders with AlphaFold3
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
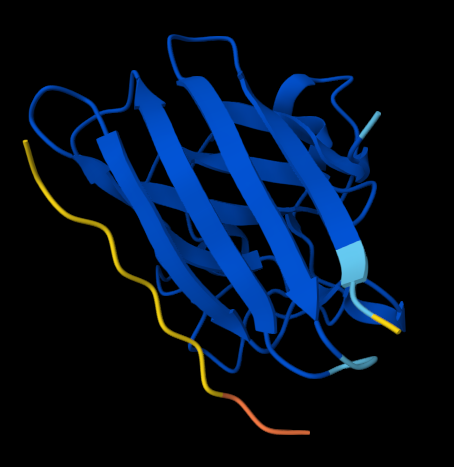
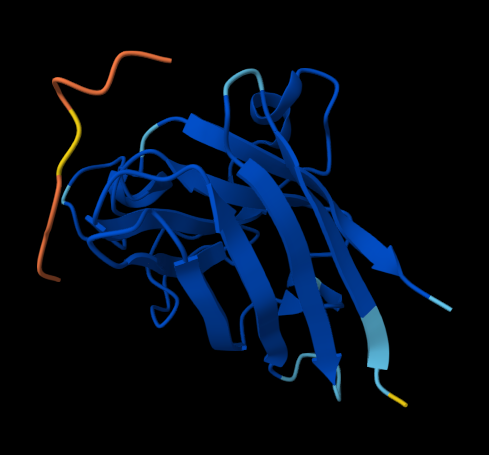
KLVYPVVVAWWK (ipTM = 0.59) FLYRWLPSRRGG (ipTM = 0.33)
Figure 1. AlphaFold-predicted SOD1 A4V complexes shown side by side for comparison. Left: complex with the PepMLM-generated peptide KLVYPVVVAWWK. Right: complex with the known SOD1-binding peptide FLYRWLPSRRGG.
- Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
WRYPVAGARHWE — ipTM = 0.35. The peptide appears loosely surface-bound on the SOD1 surface, with no clear evidence of a well-defined buried binding mode. It does not convincingly localize near the N-terminus/A4V region and instead appears to contact the β-barrel surface in a weakly resolved manner.
KLVYPVVVAWWK — ipTM = 0.59. This peptide showed the strongest predicted interface among the tested candidates. It appears mainly surface-bound and extended along the β-barrel region, rather than deeply buried in a pocket. It does not clearly localize near the N-terminus where A4V sits, and no strong interaction with the dimer-interface region is evident.
HRYPVVVAALKE — ipTM = 0.48. The peptide appears surface-associated with low-to-moderate interface confidence. It does not seem to bind near the N-terminal A4V region and instead contacts an exposed outer region of SOD1, consistent with a surface-bound interaction rather than a partially buried one.
WLYGAAVLRHGE — ipTM = 0.31. This peptide shows a very weak predicted interface and appears largely extended and surface-associated, without a defined binding pocket. It does not localize near the A4V-containing N-terminus, nor does it show a clear approach to the dimer interface. The interaction appears predominantly surface-bound.
FLYRWLPSRRGG — ipTM = 0.33. The known binder also showed a low-confidence interface in this AlphaFold prediction. The peptide appears loosely surface-bound rather than buried, with no strong evidence of localization near the N-terminus/A4V site or a clearly resolved interaction at the dimer-interface region.
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Overall, the predicted SOD1 A4V–peptide complexes showed low-to-moderate ipTM values, indicating that none of the modeled interfaces was predicted with high confidence. Among the PepMLM-generated candidates, KLVYPVVVAWWK produced the highest ipTM (0.59), followed by HRYPVVVAALKE (0.48), whereas WRYPVAGARHWE (0.35) and WLYGAAVLRHGE (0.31) showed weaker predicted interfaces. The known SOD1-binding peptide FLYRWLPSRRGG gave an ipTM of 0.33. Therefore, the best PepMLM-generated peptide, KLVYPVVVAWWK, exceeded the known binder in this AlphaFold-based comparison, and HRYPVVVAALKE and WRYPVAGARHWE also matched or surpassed it, whereas WLYGAAVLRHGE did not.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Results
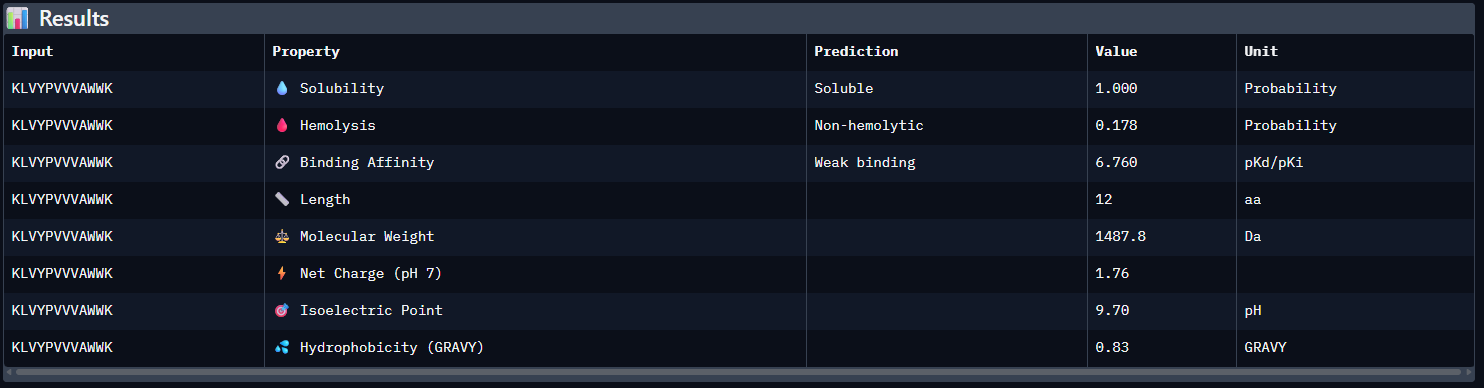
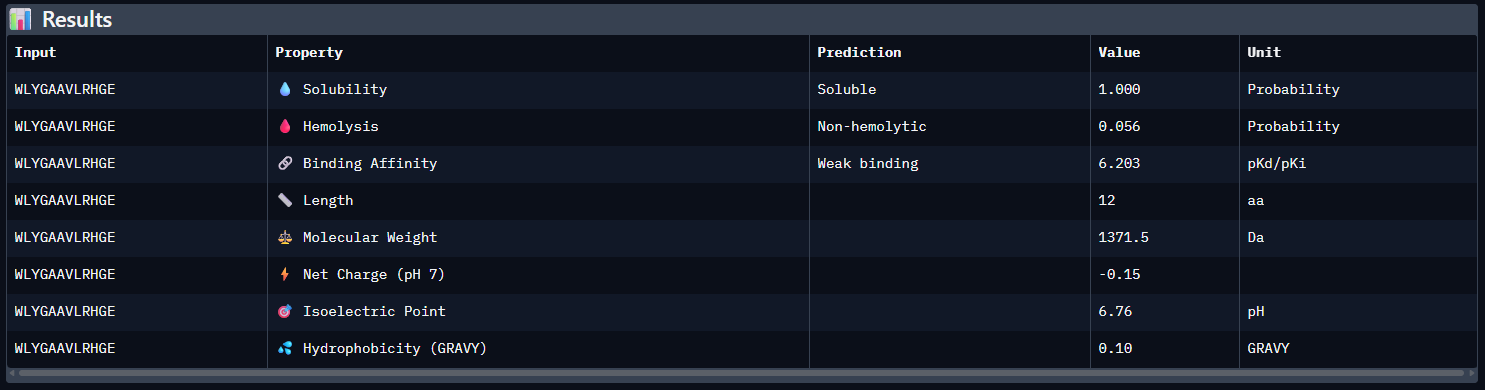
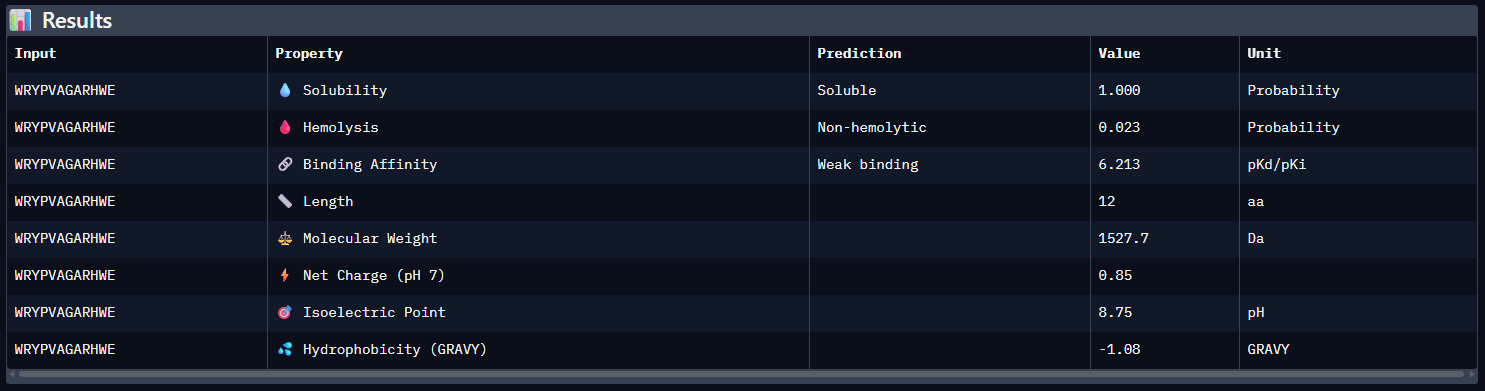
- Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
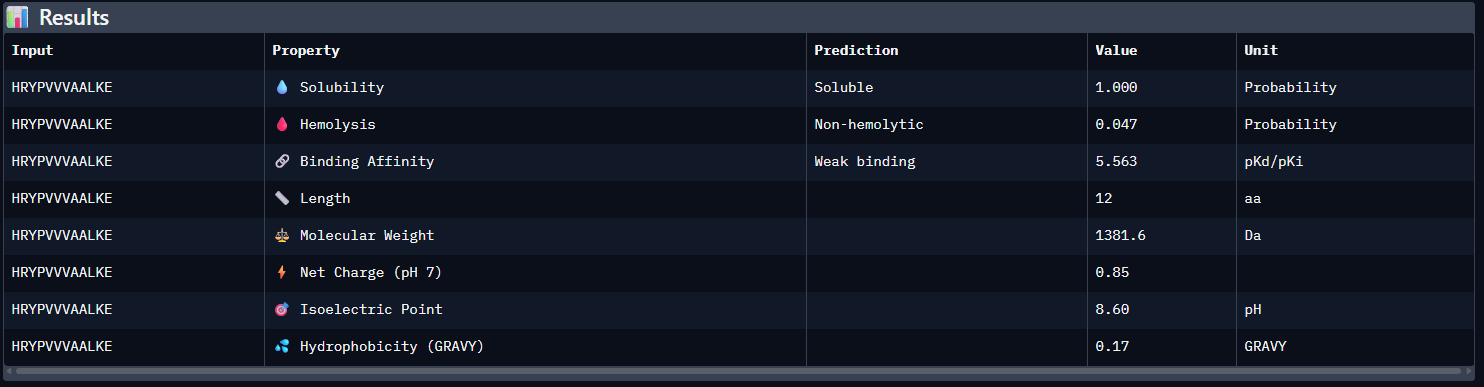
Compared with the AlphaFold3 models, the sequence-based peptide property predictions show only partial agreement with the structural results. The peptide with the highest structural confidence, KLVYPVVVAWWK (ipTM = 0.59), also has the strongest predicted binding affinity among the candidates (pKd/pKi = 6.760), so in that case the two methods are consistent. However, this trend is not perfect across all peptides: for example, WRYPVAGARHWE has a low AlphaFold3 interface score (ipTM = 0.35) but still a moderately favorable predicted affinity (6.213), while the known binder FLYRWLPSRRGG showed both low structural confidence (ipTM = 0.33) and the weakest predicted affinity (5.968). Importantly, all peptides were predicted to be soluble and non-hemolytic, so none of the better binders appears disqualified by poor solubility or overt hemolysis risk. Among them, KLVYPVVVAWWK appears to best balance predicted binding and therapeutic properties, since it combines the highest ipTM, the strongest predicted affinity, full solubility, and a non-hemolytic prediction, although its hemolysis probability (0.178) is somewhat higher than that of the other candidates and would still merit attention in follow-up validation.
- Choose one peptide you would advance and justify your decision briefly.
I would advance KLVYPVVVAWWK. Among the tested peptides, it showed the highest AlphaFold3 interface confidence (ipTM = 0.59) and the strongest predicted binding affinity (pKd/pKi = 6.760), making it the most consistent top candidate across both structural and sequence-based evaluations. It was also predicted to be soluble and non-hemolytic, which supports its therapeutic potential. Although its hemolysis probability was somewhat higher than that of the other candidates, it remained below the threshold for a hemolytic prediction, so overall it provided the best balance between predicted binding performance and developability.
Part 4: Generate Optimized Peptides with moPPIt
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
| Peptide | Method | Target motif | Affinity score | ipTM | Interpretation |
|---|---|---|---|---|---|
| KLVYPVVVAWWK | PepMLM | None | 6.760 | 0.59 | Best overall PepMLM candidate |
| HRYPVVVAALKE | PepMLM | None | 5.563 | 0.48 | Intermediate |
| WRYPVAGARHWE | PepMLM | None | 6.213 | 0.35 | Weak interface |
| WLYGAAVLRHGE | PepMLM | None | 6.203 | 0.31 | Weak interface |
| FLYRWLPSRRGG | Known binder | None | 5.968 | 0.33 | Reference; weaker than best PepMLM candidate |
| RYTDIQQYCGKW | moPPIt | 29–35 | 6.423 | 0.42 | Moderate but not strong |
| GQSDYCTRQGKI | moPPIt | 29–35 | 5.933 | 0.52 | Best moPPIt structural result so far |
| KRGKTCLECYQY | moPPIt | 29–35 | 7.286 | 0.28 | Strong property scores but poor structural support |
| GCGYSRSYTKYE | moPPIt | 107–115 | 7.286 | 0.44 | Good score profile, but only moderate structural support |
| GDRSEYCSQKKQ | moPPIt | 107–115 | 6.418 | 0.53 | Best moPPIt structural result; moderate interface confidence |
| EQSRYGHKQDER | moPPIt | 107–115 | 5.221 | 0.36 | High motif score but weak structural support |
Note: Residues 29–35; 107-115 were selected as a hypothesis-driven target motif based on their apparent mutational sensitivity in the ESM2 deep mutational scan. This choice does not demonstrate that the region is surface-exposed or experimentally validated as a peptide-binding site, but it highlights a segment that may be structurally or functionally important and therefore worth testing in a controlled design setting.
Before any clinical development, these peptides would require stepwise preclinical evaluation. First, their intended mechanism of action would need to be clarified: whether they are meant to bind mutant SOD1 merely as recognition molecules, to block a pathogenic interaction surface, to interfere with dimerization, or to reduce misfolding or aggregation. They would then need to be tested experimentally in biochemical and cellular assays to confirm real binding to mutant SOD1, measure affinity and selectivity relative to wild-type SOD1, and determine whether binding produces a meaningful functional effect. This should be followed by preclinical studies addressing stability, protease susceptibility, uptake or delivery, toxicity, hemolysis, immunogenicity risk, pharmacokinetics, and efficacy in relevant animal models of SOD1-associated disease before considering any first-in-human study. In other words, these computational results could justify preclinical follow-up, but they are far from sufficient to support direct clinical advancement.
Part C: Final Project: L-Protein Mutants
1. Project Objective and Biological Rationale
The primary objective of this assignment is to introduce rational single-point mutations into the MS2 bacteriophage lysis protein (L-protein) to overcome host resistance mechanisms. Resistant E. coli strains modify their DnaJ chaperone to prevent interaction with the N-terminal soluble domain of the L-protein, halting its folding cascade and evading viral lysis. To bypass this evolutionary barrier, we executed a comparative analysis benchmarking the theoretical predictions of the ESM-2 language model against an empirical wet-lab screening dataset (experimental_df). This dual-layer validation serves to discover functional variants that restore lysis or optimize membrane perforation kinetics.
2. Computational vs. Experimental Correlation Analysis
A critical outcome of this project is the identification of a severe dislocation between computational fitness predictions and empirical biological reality. General protein language models highly overrate thermodynamic stability in hydrophobic environments, leading to high-scoring false positives:
- C29R Discrepancy: ESM-2 ranked the C29R substitution as the second-highest stabilization peak (Score: 2.395), yet empirical screening confirms a total loss of function (Lysis = 0, Protein Levels = 0).
- Transmembrane Region Overestimation: Substitutions at position 50 (such as K50L, Score: 2.561) and position 53 (N53L, Score: 1.864) were predicted as highly advantageous. However, the experimental ledger reveals that any mutation at position 50 (K50E, K50N, K50I, K50Q) or position 53 (N53S, N53D, N53H, N53I, N53Q) results in complete functional inactivation (Lysis = 0).
Correlation Conclusion: While the machine learning model accurately identifies deleterious mutations in the early N-terminus (positions 1-10), it fails to predict functional phenotypes within the transmembrane helix. Consequently, uncalibrated computational scores were discarded, and variant selection was driven strictly by empirical functional validation.
3. Quantitative Selection Matrix and Biofrequent Justification
To isolate the optimal single-point mutations under strict structural constraints, the empirical screening dataset was filtered exclusively for active phenotypic outcomes. The matrix below aggregates every targeted mutation within the library that demonstrated successful bacterolytic execution:
| Posición | Mutación | Región | Lysis | Proteína |
|---|---|---|---|---|
| 13 | P -> L | Soluble | 1 | 1 |
| 15 | S -> A | Soluble | 1 | 1 |
| 18 | R -> G | Soluble | 1 | 1 |
| 18 | R -> I | Soluble | 1 | 1 |
| 19 | R -> S | Soluble | 1 | 0 |
| 19 | R -> H | Soluble | 1 | 0 |
| 20 | R -> W | Soluble | 1 | 0 |
| 20 | R -> L | Soluble | 1 | 0 |
| 23 | K -> E | Soluble | 1 | 0 |
| 25 | E -> V/G/D | Soluble | 1 | 0 |
| 26 | D -> G | Soluble | 1 | 0 |
| 30 | R -> Q/L | Soluble | 1 | 1 |
| 31 | R -> I | Soluble | 1 | 1 |
| 44 | L -> P | TM | 1 | 1 |
| 45 | A -> P | TM | 1 | 1 |
| 46 | I -> F | TM | 1 | 1 |
From this validated functional baseline, five distinct single-point substitutions were selected to satisfy the experimental criteria across the topography of the L-protein:
Transmembrane Region (Residues 41 to 75)
- Mutation 1 (L44P): Lysis=1, Protein=1 experimentally. Proline introduces a structural kink in the TM helix that probably facilitates membrane perforation. The computational score does not appear in the top rankings, but the experimental data is clear.
- Mutation 2 (A45P): Lysis=1, Protein=1 experimentally. Same rationale — proline in TM generates helical curvature that favors insertion and pore formation. Two consecutive prolines in TM is a known mechanism of holin activation.
Soluble Region (Residues 1 to 40)
- Mutation 3 (R18G): Lysis=1, Protein=1 experimentally. Eliminating the positive charge of arginine at position 18 of the soluble domain keeps the protein functional and expressed. Glycine is the most flexible amino acid — it can improve the conformational dynamics of the soluble domain for interaction with DnaJ.
- Mutation 4 (P13L): Lysis=1, Protein=1 experimentally. Proline at position 13 can create counterproductive structural rigidity in the soluble domain. Replacing it with leucine could increase local flexibility and improve interaction with DnaJ.
Free Variant (Domain Interface Boundary)
- Mutation 5 (R30Q): Lysis=1, Protein=1 experimentally. Arginine at position 30, soluble/TM boundary. Glutamine maintains the ability to form hydrogen bonds but eliminates the positive charge, potentially optimizing the transition between domains.
4. Homo-Oligomeric Assembly Modeling via AlphaFold2-Multimer
To evaluate the running structural hypothesis — where the L-protein functions by assembling into a multimeric pore complex to perforate the host lipid bilayer — the engineered A45P sequence was modeled as an 8-chain homomultimer in ColabFold. The predictive execution reached convergence after 3 recycles, with the top-ranked structural configuration (Rank 1, Model 2: pLDDT = 33.6, ipTM = 0.141) yielding an insightful spatial architecture:
- C-Terminal Pore Core: The optimized transmembrane regions successfully aligned in a parallel arrangement, compacting into a defined cylindrical helical bundle that establishes the structural foundation of the lytic channel.
- N-Terminal Soluble Funnels: The flexible loop domains extended outward from the membrane channel boundaries in a symmetrical funnel formation. While confidence metrics remain low (red structural domains) due to the computational absence of the coordinating host lipid matrix or native chaperones, the successful clustering of the core helices validates the structural viability of the library to execute cell-wall disruption.
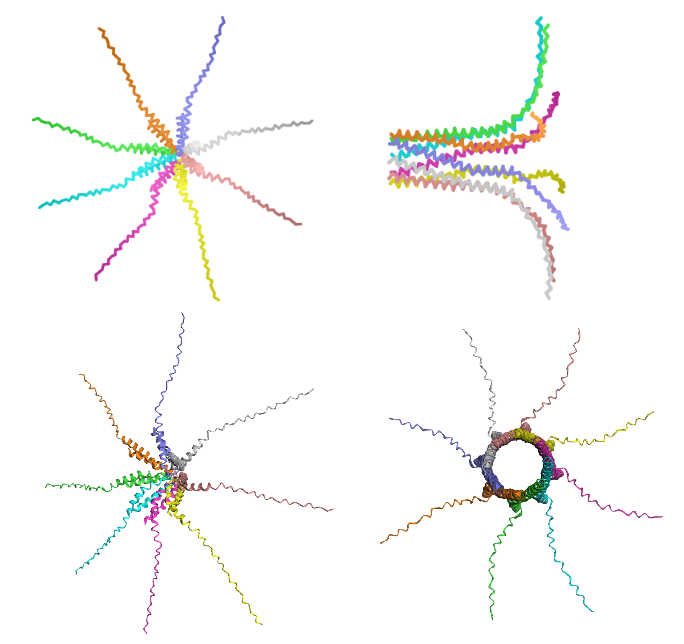
Figure 1: Comparative multi-chain structural re-ranking of the engineered A45P L-protein octamer.
Left (Top & Bottom): Rank 5 structural configuration (Model 5), representing the lowest confidence prediction (pLDDT = 26.4). The architecture shows a symmetric star-like or spider morphology where the flexible soluble domains project outward horizontally. Right (Top & Bottom): Rank 1 structural configuration (Model 2), validating the top-performing prediction (pLDDT = 33.6, ipTM = 0.141). The side view (top-right) shows a parallel alignment of the transmembrane alpha-helices into a cylindrical bundle, while the top-down axial view (bottom-right) shows a central, hollow pore channel.
4.1 Negative Control Structural Profiling (K50L)
The non-functional K50L variant was executed under identical multi-chain parameters to analyze its structural topography. The execution completed successfully (Rank 1, Model 2), yielding an alternative configuration across its statistical models (Rank 1: pLDDT = 32.2, ipTM = 0.153).
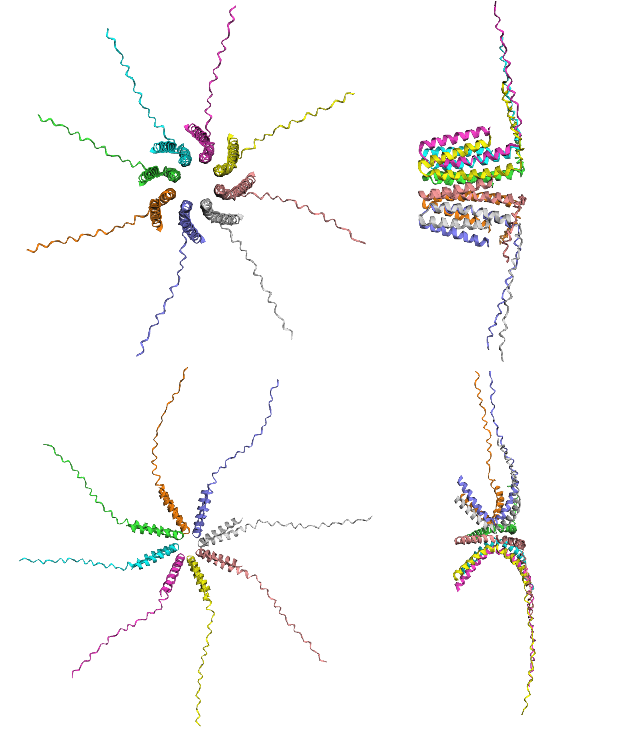
Figure 2: Homo-oligomeric channel length and shearing analysis of the K50L negative control.
Top (Left & Right): Rank 1 structural configuration (Model 2), representing the top-performing prediction by ipTM metrics. The top-down axial view (top-left) shows the geometric formation of a hollow central channel, while the side view (top-right) shows longitudinal shearing and straight alignment in the transmembrane helices. Bottom (Left & Right): Rank 5 structural configuration (Model 5), representing the lowest confidence prediction by ipTM metrics. The axial prediction (bottom-left) captures a centralized pore layout, while the side view (bottom-right) shows a tilted cross-like geometry where the vertical length of the core is compressed.
4.2 Discussion: Conformational Sampling Limits and Integrated Biophysical Hypotheses
The descriptive data from these multimeric assemblies reveals that relying on a limited batch of five predictive iterations is statistically insufficient to declare a single, definitive structural state. Crucially, the low-throughput structural channel profiles must be interpreted alongside the genetic boundaries outlined by Chamakura et al. (2017). The literature establishes that the N-terminal soluble domain of the native L-protein functions as an autologous inhibitor that requires host DnaJ coordination to prevent a destructive steric clash. In a resistant host (dnaJ P330Q), this chaperone rescue is abolished, leaving the baseline lytic cascade completely blocked (Lysis = 0).
When analyzing our computational results, the localized variations in cylinder length and vertical shearing observed in the inactive K50L control function as a compelling exploratory hypothesis regarding geometric mismatch. However, this structural interpretation is not mutually exclusive with the chemical constraints highlighted in the literature. Lysine 50 represents a highly atypical, positively charged residue buried directly within the hydrophobic transmembrane helix. While AlphaFold2-Multimer heavily prioritizes the straight, aliphatic packing of Leucine to generate an idealized circular barrel, substituting this Lysine removes a critical electrostatic hotspot. This charge deletion likely abrogates the mandatory chemical interaction with the unknown membrane target of the L-protein, rendering the rigid cylinder functionally inert regardless of its simulated geometric dimensions.
Conversely, our engineered variant A45P introduces a targeted Proline substitution. The Proline residue acts as a dynamic molecular hinge that breaks ideal helical symmetry, introducing structural flexibility that is scored with low local pLDDT metrics. This proline-mediated bending may provide the necessary conformational elasticity to bypass the autologous steric blockade of the unchaperoned N-terminus or dynamically realign the remaining interaction vectors with the membrane target, successfully rescuing lytic execution (Lysis = 1). Ultimately, this parallel simulation demonstrates that structural pore formation in predictive software is a geometric baseline rather than a guarantee of biological execution, establishing that future high-throughput sampling must balance macro-geometric length parameters with discrete electrostatic interface constraints.