FUS-Controlled Unidirectional Iron Sequestration in E. coli Nissle 1917 for Tumor Metabolic Collapse SECTION 1: ABSTRACT Solid tumors establish a metabolically distinct microenvironment (TME) characterized by hypoxia and necrosis—conditions that selectively favor the colonization of E. coli Nissle 1917 (EcN) (Stritzker et al., 2007). Once established, the bacteria encounter a niche defined by elevated lactate levels (Pérez-Tomás & Pérez-Guillén, 2020) and an intense demand for iron by cancer cells. While EcN typically upregulates enterobactin biosynthesis to compete for these resources, the host protein Lipocalin-2 neutralizes this siderophore, effectively limiting bacterial iron scavenging (Huang et al., 2024). This project engineers EcN to produce salmochelin—a Lipocalin-2-resistant siderophore (Fischbach et al., 2006) encoded by the iroBCDE/iroN locus, which is a key determinant of EcN’s competitive survival in iron-restricted environments (Massip et al., 2019). To implement this therapeutic logic, a FUS-controlled genetic circuit was designed in Asimov Kernel and digitally assembled in Benchling using the native pMUT2 backbone of EcN (CP023342.1). The circuit integrates a thermal activation cassette (TlpA39C/pTlpA) triggered by Focused Ultrasound, a lactate-gated lysis kill switch (BBa_K3848004) for tumor-specific biocontainment, and dual sRNA effectors targeting fur and iroN to drive unidirectional iron sequestration, that depletes the tumor’s labile iron pool, inducing cancer cell metabolic collapse (Pinnix et al., 2010; Saha et al., 2019). Boolean circuit logic was computationally simulated in Python and the complete 10,114 bp therapeutic vector was assembled in silico via Gibson Assembly at the s2 intergenic site of pMUT2, ensuring antibiotic-free stability through the endogenous RelB/RelE toxin-antitoxin system (Kan et al., 2021).
FUS-Controlled Unidirectional Iron Sequestration in E. coli Nissle 1917 for Tumor Metabolic Collapse
SECTION 1: ABSTRACT
Solid tumors establish a metabolically distinct microenvironment (TME) characterized by hypoxia and necrosis—conditions that selectively favor the colonization of E. coli Nissle 1917 (EcN) (Stritzker et al., 2007). Once established, the bacteria encounter a niche defined by elevated lactate levels (Pérez-Tomás & Pérez-Guillén, 2020) and an intense demand for iron by cancer cells. While EcN typically upregulates enterobactin biosynthesis to compete for these resources, the host protein Lipocalin-2 neutralizes this siderophore, effectively limiting bacterial iron scavenging (Huang et al., 2024). This project engineers EcN to produce salmochelin—a Lipocalin-2-resistant siderophore (Fischbach et al., 2006) encoded by the iroBCDE/iroN locus, which is a key determinant of EcN’s competitive survival in iron-restricted environments (Massip et al., 2019). To implement this therapeutic logic, a FUS-controlled genetic circuit was designed in Asimov Kernel and digitally assembled in Benchling using the native pMUT2 backbone of EcN (CP023342.1). The circuit integrates a thermal activation cassette (TlpA39C/pTlpA) triggered by Focused Ultrasound, a lactate-gated lysis kill switch (BBa_K3848004) for tumor-specific biocontainment, and dual sRNA effectors targeting fur and iroN to drive unidirectional iron sequestration, that depletes the tumor’s labile iron pool, inducing cancer cell metabolic collapse (Pinnix et al., 2010; Saha et al., 2019). Boolean circuit logic was computationally simulated in Python and the complete 10,114 bp therapeutic vector was assembled in silico via Gibson Assembly at the s2 intergenic site of pMUT2, ensuring antibiotic-free stability through the endogenous RelB/RelE toxin-antitoxin system (Kan et al., 2021).
SECTION 2: PROJECT AIMS
Aim 1 — Experimental Aim (This Project)
To design and computationally validate a genetic circuit in E. coli Nissle 1917, using Focused Ultrasound (FUS) as a remote thermal trigger to control dual sRNA-mediated iron sequestration, integrating a lactate-gated kill switch and antibiotic-free stability.
Aim 2 — Development Aim
To experimentally validate the therapeutic circuit by constructing and transforming the pMUT2-based plasmid into EcN, and quantifying iron sequestration efficiency through bacterial-cancer cell co-culture assays (e.g., HT-29 or CT26 lines). This will be performed under simulated TME conditions, using ΔFur and ΔiroN isogenic mutants as controls to define the optimal FUS duty cycle for cumulative tumor iron depletion.
Aim 3 — Visionary Aim
To deploy the validated therapeutic EcN as a Bacteria-Based Cancer Therapy (BBCT) for the treatment of solid tumors, leveraging tumor-selective colonization and physician-controlled FUS activation to achieve unidirectional iron sequestration across multiple tumor types and administration routes.
SECTION 3: BACKGROUND
Literature Context
Pita-Grisanti et al. (2023) / Torti & Torti (2013): Characterized the metabolic “iron addiction” of malignant cells, where tumors upregulate iron-binding import machinery (e.g., Transferrin receptor, Lipocalin-2) and downregulate the exporter ferroportin to drive proliferation. While exogenous iron chelation via bacterial siderophores triggers tumor metabolic collapse and p53/Bax-mediated apoptosis, traditional systemic treatments yield inconsistent clinical outcomes and severe, dose-limiting off-target toxicities due to a lack of spatial selectivity.
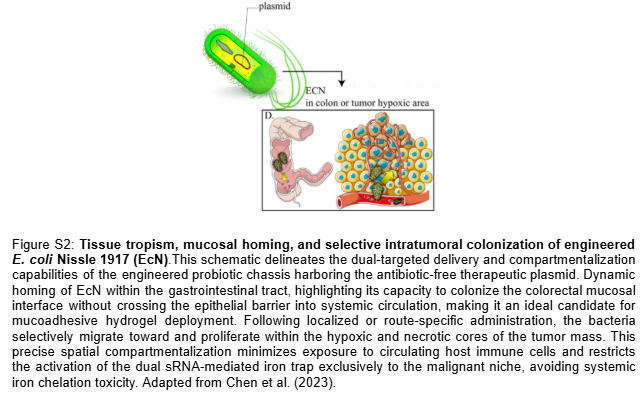
Stritzker et al. (2007): Evaluated tumor-specific colonization profiles, demonstrating that systemic administration of the probiotic Escherichia coli Nissle 1917 (EcN) results in exceptional intratumoral replication (>= 1 x 10^8 CFU/g tissue) specifically targeting the border of viable and necrotic tumor zones, without colonizing healthy organs like the liver or spleen. This innate, high-density tumor tropism solves the historical challenge of systemic chelation toxicity highlighted by Pita-Grisanti et al., validating EcN as an ideal localized cellular chassis to restrict aggressive iron starvation exclusively to the malignant microenvironment.
This research directly justifies the therapeutic rationale of iron sequestration as a selective anticancer strategy. By identifying this metabolic dependency, the authors provide the biological basis for intercepting iron trafficking to collapse tumor function.
Novelty and Innovation
While iron-chelating siderophores have been proposed as anticancer agents (Saha et al., 2019), and FUS has been used to activate thermal bioswitches in bacteria (Piraner et al., 2017), no prior design has combined FUS-controlled sRNA-mediated iron sequestration with simultaneous biocontainment in a single antibiotic-free plasmid system. This project introduces a multi-layered therapeutic paradigm for bacteria-based cancer therapy by integrating physical, metabolic, and genetic control systems into a single antibiotic-free platform. The core innovations comprise:
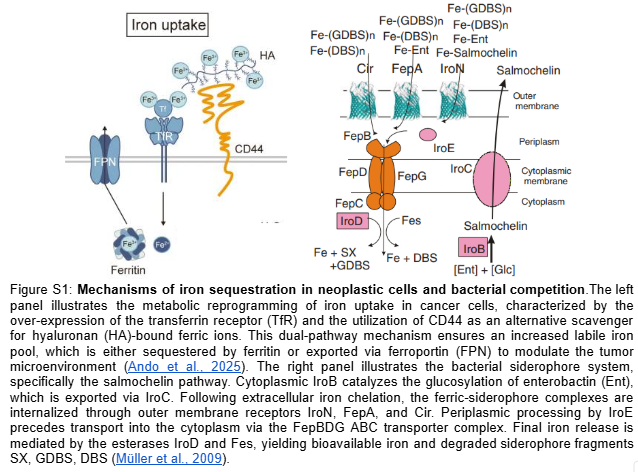
Unidirectional Siderophore Trap: A dual-targeting sRNA system simultaneously silences fur and iroN mRNAs. Knockdown of the Ferric Uptake Regulator (fur) constitutively desrepresses the iroA and ent operons to maximize salmochelin and enterobactin secretion, while the concurrent iroN blockade prevents iron re-import, rendering the chelated iron permanently inaccessible in the extracellular space, thereby starving the tumor microenvironment while preventing intracellular oxidative stress in EcN (Figure S1).
Boolean AND-Gate Activation: Precise spatial targeting pairs a Focused Ultrasound (FUS)-activated TlpA39C thermal gate—transitioning from its subcutaneous proof-of-concept [Piraner et al., 2017] to deep tissues—with an endogenous lactate-gated kill switch, restricting circuit expression strictly to the lactate-rich TME.
Clinical-Grade Biocontainment: The circuit is encoded on the native pMUT2 plasmid of E. coli Nissle 1917, stably maintained without antibiotic selection pressure via the endogenous RelB/RelE toxin-antitoxin system.
Compartmentalized Delivery: Translational scope extends beyond intravenous limits through route-specific biomaterials—degradable PLGA seeds for breast/prostate and mucoadhesive hydrogels for cervical/colorectal cancers—enabling localized administration without systemic bacterial dissemination.
Why This Project Matters
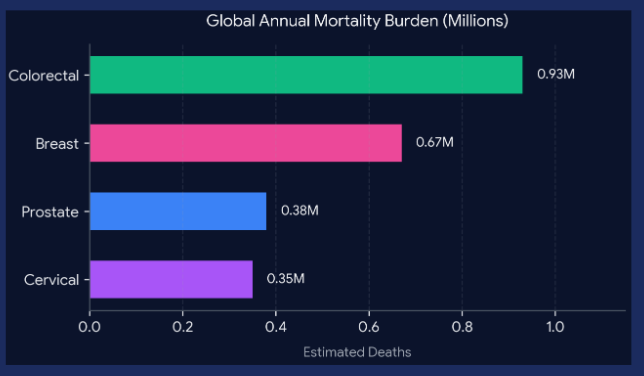
The Problem Addressed: Treatment-resistant solid tumors remain the leading cause of oncological mortality, with breast, colorectal, prostate, and cervical cancers accounting for over 2 million deaths annually (Bray et al., 2024). Conventional systemic therapies fail to eradicate these malignancies due to poor penetration into the dense, avascular tumor core and severe dose-limiting toxicities in healthy peripheral tissues.
Importance of the Problem and Critical Barriers: Neoplastic cells exhibit an aggressive “iron addiction,” selectively upregulating iron-binding import machinery to fuel rapid proliferation, DNA replication, and mitochondrial respiration (Liang & Ferrara, 2021). While exogenous iron chelation via traditional siderophores (e.g., enterobactin) has been validated to arrest cancer growth, its clinical translation represents a critical barrier due to low therapeutic efficacy and systemic off-target toxicities (Pita-Grisanti et al., 2023).
Advancement of Knowledge and Technical Capability: This project bypasses these historical delivery barriers by engineering E. coli Nissle 1917 (EcN) as a “Trojan Horse” that exploits innate tumor tropism to infiltrate the necrotic malignant core (Stritzker et al., 2007; Figure S2). We advance current genetic engineering capabilities by implementing a multi-layered “Passcode” logic architecture—inspired by Chan et al. (2016)—that pairs a physician-controlled Focused Ultrasound (FUS) thermal gate with an endogenous lactate-sensing kill switch. This Boolean AND-gate establishes an absolute biocontainment mechanism that prevents environmental escape and ensures that the engineered metabolic iron sink is activated strictly within the biochemical parameters of the tumor microenvironment (TME).
Field-Level Change and Paradigm Shift: If the aims are achieved, this research will shift the paradigm of bacteriotherapy from passive bacterial colonization to an active, precision-programmable living drug platform. Unlike existing optogenetic or chemical activation systems that cannot penetrate deep human tissues, the integration of clinically approved FUS provides millimeter-precision spatiotemporal control at depth (Piraner et al., 2017). Furthermore, the entire circuit operates on an antibiotic-free native plasmid system (Kan et al., 2021), proving that complex logic gates can be stably maintained in vivo without contributing to the global threat of antimicrobial resistance.
Broader Societal Contribution: On a societal scale, transitioning cancer treatment from systemic, high-toxicity chemotherapy to localized, autonomous microbial circuits could drastically reduce patient hospitalization rates, eliminate chemotherapy-related side effects, and provide an affordable, scalable option for refractory solid tumors worldwide.
Ethical Implications
The primary ethical principles governing this project are non-maleficence (preventing harm to the patient and the environment) and beneficence (providing a new treatment option for patients with limited alternatives). The most significant ethical concern is dual use: the same thermosensitive repressor (TlpA39C) and lactate sensor (LldR) that enable tumor-selective activation could theoretically be used to engineer pathogens that activate only within specific host thermal or metabolic environments. Responsible disclosure and selective publication of circuit performance data — particularly the exact thermal threshold and lactate concentrations at which each gate switches — are essential safeguards. Additionally, the use of a genetically modified organism with a kill switch does not eliminate all biocontainment risk: kill switches are known to fail through plasmid loss or loss-of-function mutations in toxin genes (Chan et al. 2016), and chromosomal integration is necessary for clinical-grade biocontainment.
Measures to ensure ethical conduct include: (1) all experiments with EcN in live animals must follow IACUC protocols with appropriate biosafety level (BSL-2) containment; (2) the kill switch must be validated for mutational escape frequency before any in vivo administration; (3) the project should engage regulatory review from the outset (ISP in Chile, FDA in the US) to establish safety criteria for engineered salmochelin-producing organisms; (4) equity considerations require that E. coli Nissle 1917 be used as the chassis — it is a probiotic with an established safety record and low-cost production profile, making future access possible for patients in developing regions. The ethical justification for using EcN as a delivery platform is further strengthened by recent clinical evidence (Gurbatri et al., 2024), which demonstrated that oral administration of EcN leads to selective colonization of tumor tissue in human colorectal cancer patients without significant adverse effects.
However, several unintended consequences and incorrect assumptions must be acknowledged. A critical unintended consequence is kill switch failure through loss-of-function mutations in the toxin genes or plasmid loss — if the lactate-gated lysis system fails, EcN could persist in healthy tissue post-therapy, raising significant safety concerns. A potentially incorrect assumption is that all targeted tumor subtypes maintain sufficient lactate concentrations to keep the bacteria viable within the TME, which could prematurely trigger the kill switch and compromise therapeutic efficacy. An alternative to the plasmid-based kill switch is chromosomal integration of the biocontainment system, which would eliminate plasmid loss as a failure mode and provide clinical-grade biocontainment, albeit at the cost of reduced modularity and more complex genetic engineering.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
This section describes the experimental design, tools, and technologies employed across the three aims of this project. Aim 1 constitutes the core experimental work of this project — encompassing computational circuit design, in silico plasmid assembly, and Boolean logic simulation. Aims 2 and 3 define the development and translational roadmap required to advance the therapeutic circuit from computational validation to experimental characterization and clinical deployment. Each aim is described in terms of its experimental rationale, methodology, and the specific tools and technologies employed.
4.1 Aim 1: Genetic Circuit Design and Validation
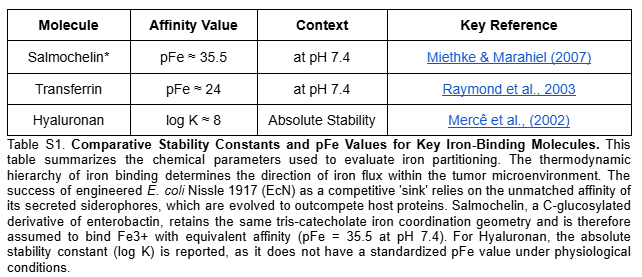
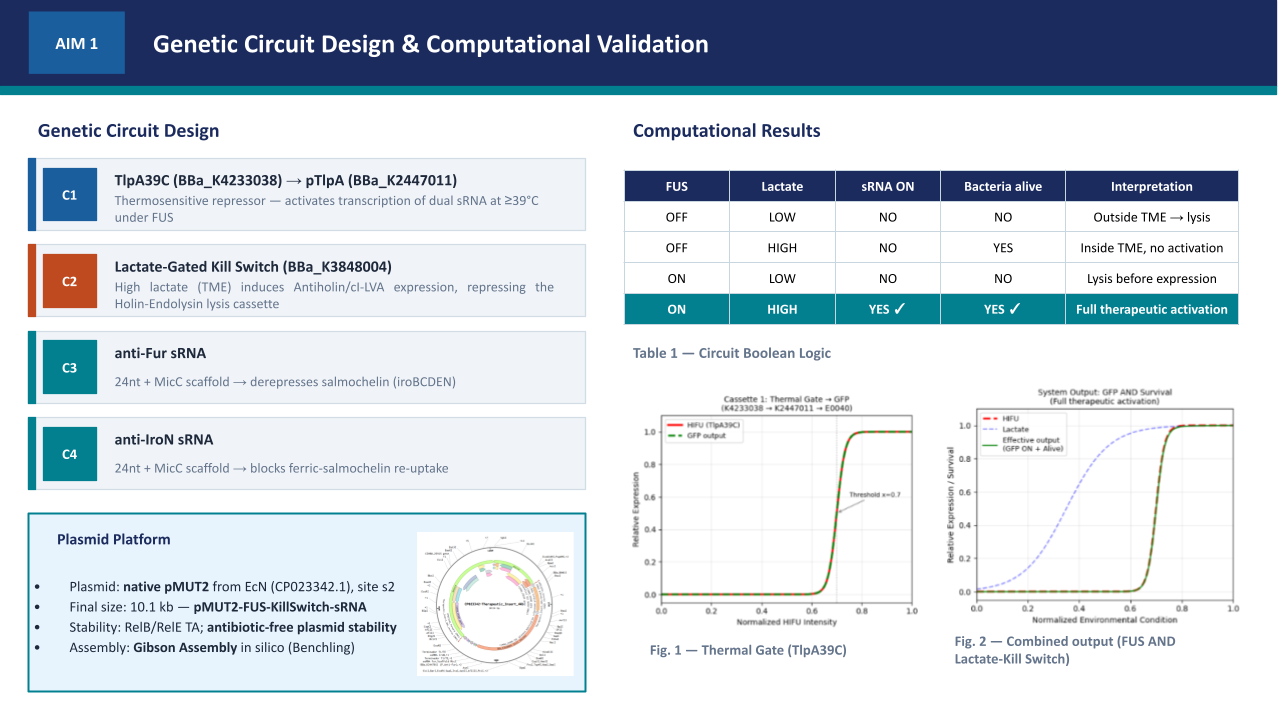
Aim 1.1 — Circuit Design and Plasmid Assembly: The therapeutic circuit was designed in Asimov Kernel and digitally assembled in Benchling via simulated Gibson Assembly at the s2 intergenic site of the native pMUT2 backbone (CP023342.1), generating a 10,114 bp therapeutic vector. The design integrates two independent functional modules: a FUS-controlled thermal activation cassette (BBa_K4233038 -> BBa_K2447011) driving dual sRNA effectors targeting fur and iroN to promote unidirectional iron sequestration, and a lactate-gated kill switch (BBa_K3848004) for biocontainment. The 24nt antisense sequences were derived from the EcN genome (NZ_CP007799.1) — fur (position 757641–758087, minus strand) and iroN (position 1171492–1173669, plus strand) — and fused to a MicC scaffold (Na et al., 2013) for Hfq-mediated silencing. The pMUT2 backbone sequence was extracted from NCBI (CP023342.1), providing a ColE2-like origin of replication and endogenous RelB/RelE toxin-antitoxin system that ensures antibiotic-free plasmid stability (Kan et al., 2021). Specifically, the anti-Fur sRNA derepresses the endogenous ent and iro operons (entABCDEFH and iroBCDEN), promoting enterobactin biosynthesis and its IroB-mediated glucosylation to produce salmochelin — a Lipocalin-2-resistant siderophore that sequesters extracellular Fe3+ with higher affinity (Table S1). Simultaneously, the anti-IroN sRNA silences the outer membrane reimporter, preventing recovery of the ferric-salmochelin complex by the bacteria and creating a unidirectional iron trap that depletes the tumor extracellular labile iron pool.
Aim 1.2 — Computational Logic Validation: Circuit logic was validated computationally in Python using Hill function modeling of the thermal gate transfer function (k=60, x0=0.70, ~39°C) and kill switch sigmoid response (k=12, x0=0.35, ~10–12 mM lactate), confirming Boolean AND gate behavior — full therapeutic activation occurs exclusively when FUS is applied within the TME where lactate exceeds the kill switch threshold. Results are reported as normalized transfer functions across all four Boolean input states.
4.2 Aim 2: Experimental Validation and Therapeutic Characterization
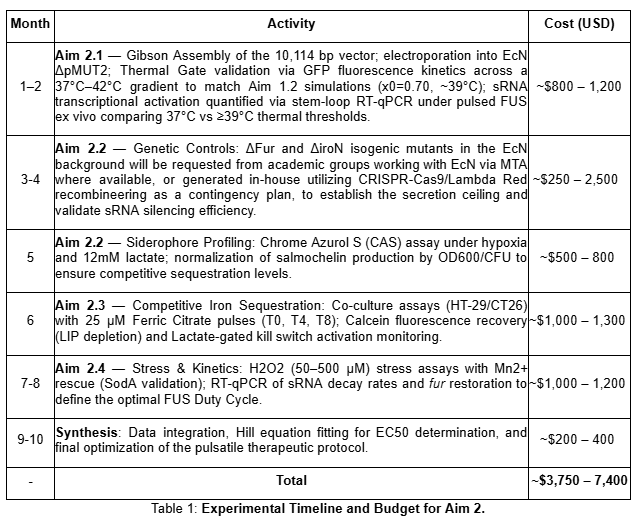
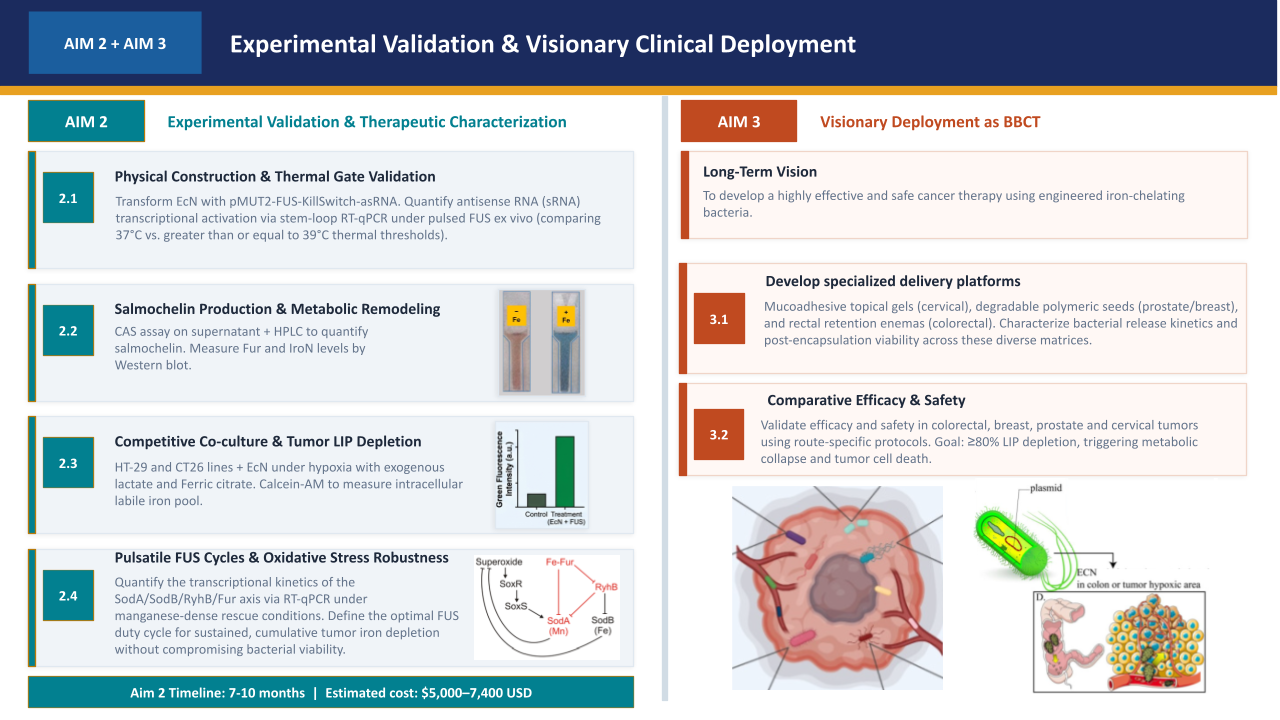
The experimental validation of the therapeutic circuit requires four sequential phases, each building on the results of the previous one. The detailed activities, estimated costs, and execution timeline for these phases are summarized in Table 1.
Aim 2.1 — Physical Assembly and Thermal Gate Validation: The 10,114 bp therapeutic vector (pMUT2-FUS-KillSwitch-asRNA) will be physically assembled via Gibson Assembly, integrating the 4.6 kb synthetic insert into the s2 intergenic site of the pMUT2 backbone as specified in the Benchling reference. Following electroporation into EcN DpMUT2 and sequence verification, the TlpA39C/pTlpA thermal gate will be characterized via GFP fluorescence kinetics. We will determine the thermal activation threshold under a graduated stress (37°C to 42°C) to verify if the physical chassis replicates the Hill function model (x0=0.70, ~39°C) computationally simulated in Aim 1.2. Additionally, sRNA transcriptional activation will be directly quantified via stem-loop RT-qPCR under pulsed FUS ex vivo, comparing 37°C versus >=39°C thermal thresholds, to confirm that TlpA39C derepression leads to effective anti-Fur and anti-IroN sRNA expression above the thermal gate.
Aim 2.2 — Characterization of Salmochelin Production and Metabolic Remodeling Validation: Salmochelin biosynthesis will be quantified under simulated TME conditions—hypoxia (1% O2) and exogenous lactate (10–12 mM)—using the Chrome Azurol S (CAS) assay for quantitative siderophore detection. This phase validates the metabolic remodeling described by Salvail et al. (2010), where sRNA-mediated regulation optimizes siderophore production by redirecting metabolic flux—specifically redirecting serine building blocks from cysteine biosynthesis toward the nonribosomal peptide synthesis (NRPS) pathway for enterobactin/salmochelin assembly. To isolate the specific contribution of the dual sRNA effectors (anti-fur and anti-iroN) designed in Aim 1.1, we will compare the FUS-induced EcN against Dfur and DiroN isogenic mutants, constructed via CRISPR-Cas9 or Lambda Red recombineering. This phase establishes the ‘secretion ceiling’ by normalizing production rates by OD600/CFU, ensuring that the sRNA-induced metabolic optimization reaches competitive levels of iron sequestration comparable to constitutive genetic deletions.
Aim 2.3 — Competitive Co-culture, LIP Depletion, and Biocontainment Validation: Iron depletion efficacy is assessed in bacterial-cancer cell co-culture assays using HT-29 or CT26 lines. To simulate the continuous iron-recycling role of M2 macrophages (Liang & Ferrara, 2021), the TME will be challenged with calibrated pulses of 25 uM Ferric Citrate every 4 hours (at T0, T4, and T8) over a 12-hour observation window (Sohn et al., 2010). Ferric Citrate is selected due to its physiological relevance and low stability constant (log K approx 11.85), as characterized by Szlasa et al. (2022), ensuring a steady supply of bioavailable iron for competitive uptake. This experimental setup creates a dynamic competitive environment where the rate of bacterial sequestration must exceed the exogenous iron influx to prevent cancer cell uptake. Intracellular Labile Iron Pool (LIP) depletion will be measured via calcein fluorescence recovery, with data fitted to a Hill equation model to calculate the fold induction and the EC50 required to achieve cancer cell metabolic collapse under competitive pressure. Crucially, as cancer cell death leads to a reduction in exogenous lactate levels, the corresponding activation of the lactate-gated lysis kill switch will be monitored. Bacterial viability (CFU/mL) will be quantified post-therapeutic effect to validate that the depletion of the tumor-derived lactate signal successfully induces chassis biocontainment once the metabolic mission is complete.
Aim 2.4 — Characterization of Pulsatile Duty Cycles and Stress Robustness: This sub-aim defines the operational limits of the genetic circuit. We will quantify the ‘Time-to-Collapse’ of the bacterial chassis under therapeutic activation by exposing FUS-induced EcN to TME-simulated oxidative stress (H2O2 50–500 uM; Seaver & Imlay, 2001). Based on the regulatory feed-forward loop described by Semsey (2014), the circuit-induced silencing of fur derepresses the endogenous RyhB sRNA, which in turn represses the iron-dependent superoxide dismutase SodB (Massé & Gottesman, 2002), increasing ROS sensitivity under therapeutic activation. We will validate if 100 uM Mn2+ supplementation rescues viability by supporting SodA (Mn-SOD) activity (Privalle & Fridovich, 1988), establishing the optimal ON/OFF duty cycle required to maintain a negative iron balance in the TME without reaching the bacterial lethal threshold.
Expected Results Aim 2
The execution of Aim 2 is expected to demonstrate a high-fidelity correlation between computational design and experimental performance. In Aim 2.1, a sigmoidal GFP induction matching the Hill model x0=0.70 will confirm thermal gate precision. Aim 2.2 will show that FUS-induced EcN achieves salmochelin secretion rates per cell (normalized by OD600) comparable to the Dfur ceiling; crucially, the unidirectional trap will be validated by the extracellular accumulation of the ferric-salmochelin complex, mimicking the DiroN phenotype. In Aim 2.3, co-culture assays will show calcein fluorescence recovery in HT-29/CT26 cells, proving the sink outcompetes cancer iron uptake even under 25 uM ferric citrate pulses, followed by bacterial lysis upon lactate depletion. Finally, Aim 2.4 will establish the bacterial ‘Time-to-Collapse’ via Live/Dead kinetic assays using SYTO 9 and propidium iodide fluorescence in a plate reader, with parallel CFU/mL counts at defined time intervals, under H2O2 stress (50–500 uM). This collapse threshold defines the maximum FUS pulse duration — the window within which EcN must sequester sufficient iron to drive cancer cell metabolic collapse before ROS-induced bacterial death terminates the therapeutic effect. Mn2+ rescue experiments will confirm whether SodA activity can extend this operational window, enabling optimization of the ON/OFF duty cycle for cumulative iron depletion across multiple therapeutic sessions.
4.3 Aim 3: Visionary Deployment as Siderophore-Driven Bacteria-Based Cancer Therapy
To develop a highly effective and safe cancer therapy in humans using engineered iron-chelating bacteria deployed across four tumor types through route-specific local administration.
Aim 3.1 — Develop Specialized Delivery Platforms: Three route-specific delivery matrices will be developed and characterized. For breast and prostate cancer, degradable polymeric PLGA seeds will be fabricated and implanted via ultrasound-guided needle. For cervical cancer, a mucoadhesive vaginal gel will be formulated for topical application and for colorectal cancer, a mucoadhesive gel will be deposited directly onto the tumor site via colonoscopy.
PLGA Seeds (breast and prostate): PLGA seeds will be fabricated as cylindrical implants of approximately 0.8mm diameter by extrusion or micro-molding, compatible with standard implantation needle gauge used in clinical protocols. Multiple seeds will be implanted per session to ensure adequate spatial coverage of the tumor volume. EcN will be incorporated into the PLGA matrix during fabrication, and seed morphology and integrity will be confirmed by SEM prior to use. To characterize matrix degradation and validate tumor colonization, a dual-phase protocol will be executed. In vitro, PLGA seeds will be incubated in PBS at 37°C; polymer erosion and pore morphology will be evaluated via gravimetric mass loss and scanning electron microscopy (SEM) at defined timepoints (T0, T6, T12, T24, T48h), while viable bacterial release kinetics will be quantified by CFU counting on LB agar. In vivo, seeds will be intratumorally implanted into murine solid tumor models. Tumor colonization efficiency and spatial distribution within necrotic cores will be determined via histological Gram staining and quantitative CFU counts per gram of tumor tissue, while systemic biosafety will be confirmed by monitoring bacterial clearance in healthy off-target organs.
Mucoadhesive Gels (cervical and colorectal): Mucoadhesive gels will be formulated using biocompatible polymers such as chitosan or carbopol, optimized for viscosity and adhesion to mucosal surfaces. For cervical cancer, the gel will be designed for topical vaginal application. For colorectal cancer, the gel will be deposited directly onto the tumor site via colonoscopy using an open-channel spray catheter or endoscopic delivery catheter compatible with high-viscosity formulations. To characterize matrix performance and validate tumor colonization, a dual-phase protocol will be executed. In vitro, bacterial release kinetics will be characterized by depositing the gel in a two-compartment diffusion chamber with a semipermeable membrane simulating mucosal tissue. CFU will be quantified in the receptor compartment at defined timepoints (T0, T6, T12, T24, T48h). Mucosal penetration depth will be additionally assessed by confocal microscopy of fluorescently labeled EcN in ex vivo tissue sections. In vivo, gels will be administered to murine tumor models via their respective routes. Tumor colonization efficiency and spatial distribution will be confirmed by histological Gram staining and quantitative CFU counts per gram of tumor tissue, while systemic biosafety will be assessed by monitoring bacterial clearance in healthy off-target organs.
Post-encapsulation Viability: To confirm that the encapsulation process does not compromise bacterial fitness prior to in vivo deployment, post-encapsulation viability will be assessed for all three delivery matrices. CFU counts will be compared before and after encapsulation to quantify viable bacterial recovery. Growth curves of encapsulated versus free EcN will be measured under identical culture conditions (LB medium, 37°C, 200 rpm) to assess whether encapsulation affects growth kinetics. Additionally, plasmid retention will be confirmed by differential plating to verify the endogenous addiction system, ensuring that pMUT2 stability is maintained throughout the encapsulation process without the use of antibiotic selection pressure.
Aim 3.2 — Comparative Efficacy & Safety: Therapeutic efficacy and safety will be validated across four tumor types in human clinical settings using route-specific delivery protocols. For each indication, EcN will be administered via its corresponding delivery matrix — PLGA seeds for breast and prostate, mucoadhesive vaginal gel for cervical, and colonoscopic mucoadhesive gel for colorectal — followed by a colonization window and physician-controlled pulsed FUS activation. The primary clinical endpoint is >= 80% labile iron pool depletion within the tumor, triggering metabolic collapse and arrest of tumor cell proliferation without systemic iron depletion or off-target toxicity. Secondary endpoints include tumor volume reduction, patient tolerability, and absence of systemic bacterial dissemination. The long-term vision is to establish this platform as a physician-controlled, antibiotic-free, route-specific bacteria-based cancer therapy applicable across multiple solid tumor types, offering a non-toxic alternative to systemic iron chelation with minimal invasiveness and maximum tumor selectivity.
4.4 Techniques Checklist
4.5 Expanded Technique Description
Gibson Assembly: This is the core cloning strategy employed in this project to physically construct the 10,114 bp therapeutic vector pMUT2-FUS-KillSwitch-asRNA. The 4.6 kb synthetic insert — comprising four insulated transcriptional cassettes (TlpA39C thermal gate, lactate-gated kill switch, anti-Fur sRNA, and anti-IroN sRNA) — will be assembled into the s2 intergenic site of the native pMUT2 backbone, using primers designed with 30–40 bp homology overhangs at each junction. The assembled vector will be transformed into EcN DpMUT2 by electroporation, and successful assembly confirmed by colony PCR and Sanger sequencing across all four cassette junctions.
Benchling: This platform was used as the primary platform for in silico plasmid design and annotation of the therapeutic vector. The native pMUT2 backbone sequence (CP023342.1) was imported from NCBI and the s2 intergenic insertion site identified based on Kan et al. (2021). All four transcriptional cassettes were sequentially annotated, and Gibson Assembly primers were designed directly within Benchling, verifying homology overhang lengths, melting temperatures, and correct cassette orientation before any physical cloning was attempted. The final annotated plasmid map reported in the Results section of this project was generated and exported from Benchling.
4.6 Industry Council Companies
Asimov (Kernel): Used directly for the in silico design, simulation, and cellular logic modeling of the genetic circuit in Aim 1.1.
Twist Bioscience: Used for the high-fidelity de novo DNA synthesis of the 4.6 kb insert comprising the four insulated transcriptional cassettes.
Addgene: Used as the non-profit academic repository to acquire the physical pMUT2-based backbone vector from Kan et al. (2021) for use as the cloning backbone in Aim 2.
New England Biolabs: Provider of the NEBuilder HiFi DNA Assembly Master Mix, restriction enzymes, and Q5 High-Fidelity DNA Polymerase required for physical vector construction.
Thermo Fisher Scientific: Provider of electroporation cuvettes, cell culture media for human cancer lines, DNA purification kits, and Calcein-AM fluorescent probes for the intracellular LIP assay.
Millipore Sigma: Provider of basic microbiological reagents, LB medium, and chemical components required for the CAS assay and hydrogen peroxide H2O2 oxidative stress trials.
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
5.1 Genetic Circuit Design and Computational Logic Validation
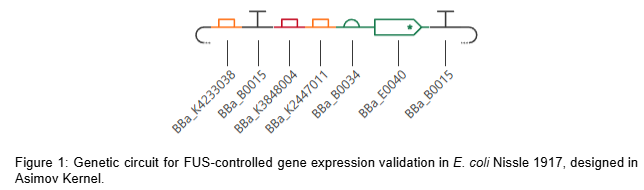
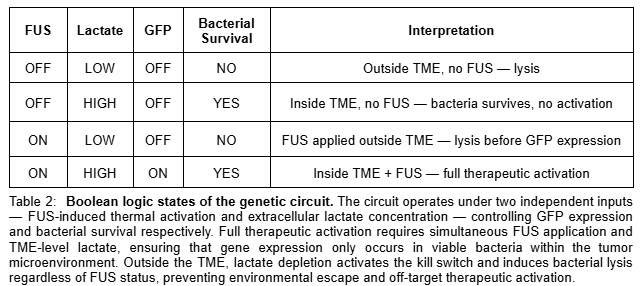
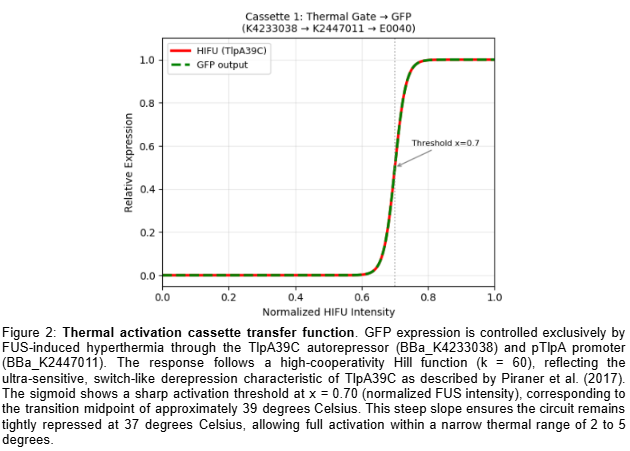
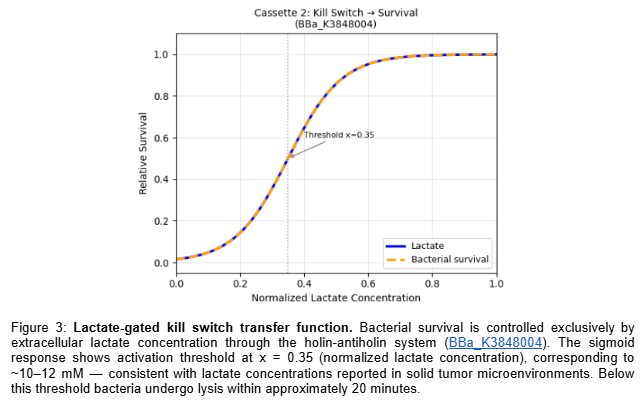
The circuit comprises two independent cassettes (Figure 1). The first is a thermal activation cassette in which TlpA39C autorepressor (BBa_K4233038 - pTlpA2-RBS-TlpA39C) controls transcription through pTlpA (BBa_K2447011 - Temperature sensitive promoter pTlpA), driving GFP expression (BBa_E0040) as fluorescent output for the computational simulation of the thermal gate transfer function. The second is a lactate-gated lysis kill switch (BBa_K3848004) for biocontainment, which operates via an asymmetric holin-antiholin toxin-antitoxin system. In the presence of lactate — as found in the tumor microenvironment — a lactate-sensitive promoter drives expression of antiholin and cI repressor, the latter blocking transcription of the lytic cassette encoding holin and endolysin. Upon exit from the TME and consequent lactate depletion, cI repressor is rapidly degraded via its LVA degradation tag, derepressing holin and endolysin expression. Holin perforates the inner membrane, allowing endolysin to degrade the cell wall, inducing complete bacterial lysis within approximately 20 minutes. The asymmetric RBS design — medium strength for antiholin/cI and strong for holin/endolysin — ensures rapid and irreversible lysis upon TME exit, preventing environmental escape. Circuit logic was designed in Asimov Kernel and the Boolean transfer function was computationally simulated in Python, confirming the expected multi-input logic gate outputs across all physiological conditions (Table 2).
5.2 Plasmid Design for FUS-Controlled Fur and IroN Silencing and Iron Sequestration in EcN
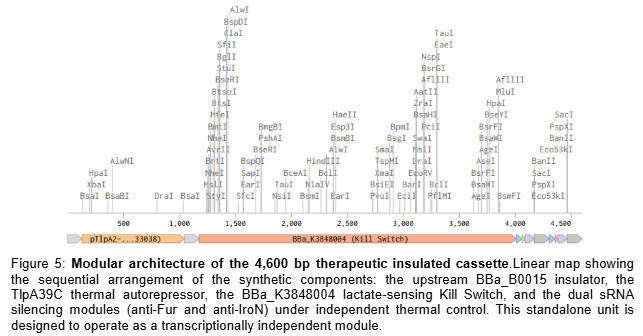
The computational validation of the thermal gate transfer function in Section 5.1 establishes the logic framework for therapeutic activation. The therapeutic plasmid replaces the GFP reporter with two independent synthetic antisense RNA cassettes targeting the fur gene (genomic position 757641-758087, NZ_CP007799.1, negative strand) and the iroN gene (genomic position 1171492-1173669, NZ_CP007799.1, positive strand) of EcN Nissle 1917. Silencing fur derepresses the endogenous iroBCDEN operon under FUS-controlled conditions, driving salmochelin biosynthesis. Salmochelin, a C-glucosylated enterobactin produced by IroB, evades host Lipocalin-2 sequestration and competes for extracellular iron within the TME (Huang et al., 2024). Simultaneous silencing of iroN — the outer membrane receptor responsible for reimporting the ferric-salmochelin complex — prevents iron recovery by the bacteria, creating a unidirectional iron sequestration trap that depletes the extracellular labile iron pool of the tumor. Each sRNA cassette consists of a 24nt antisense targeting sequence fused to a MicC scaffold (BBa_K5176030), which recruits the endogenous Hfq chaperone to stabilize the sRNA and facilitate target mRNA degradation (Dam et al., 2017). To ensure optimal folding and independent function of each sRNA, both cassettes are placed under separate individual thermal promoters in tandem: BBa_K4233038 -> BBa_K2447011 -> [24nt anti-Fur + MicC] -> T1/TE -> BBa_K2447011 -> [24nt anti-IroN + MicC] -> T1/TE. The specific DNA sequences for these elements are detailed in Table 3.
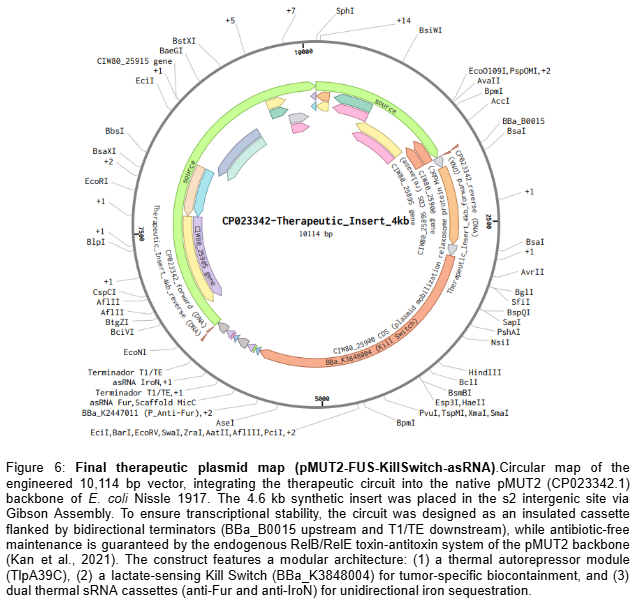
To implement this logic, the complete therapeutic module—Therapeutic_Insert_4kb (4,600 bp)—was digitally assembled in Benchling as an insulated genetic unit. The internal architecture was engineered in a modular fashion, starting with an upstream BBa_B0015 bidirectional terminator to shield the circuit from backbone-derived noise. This is followed by the TlpA39C thermal autorepressor (BBa_K4233038) and the lactate-sensing Kill Switch (BBa_K3848004), which provides tumor-specific biocontainment. The downstream section of the insert contains the dual effector modules: the anti-Fur and anti-IroN asRNA cassettes, each independently driven by the BBa_K2447011 thermal promoter and fused to the MicC scaffold. To ensure transcriptional independence, the entire 4.6 kb module is flanked by a final T1/TE terminator (Figure 5). This 4.6 kb module was then integrated into the native pMUT2 (CP023342.1) plasmid of EcN via a simulated Gibson Assembly at the s2 intergenic site. By utilizing the native pMUT2 backbone, the final 10,114 bp therapeutic vector (Figure 6) ensures 100% antibiotic-free stability in vivo through its endogenous RelB/RelE toxin-antitoxin system, providing a robust clinical-grade platform as characterized by Kan et al. (2021).
To ensure maximum genetic stability and rapid prototyping, the therapeutic circuit was housed in the native pMUT2 plasmid of EcN instead of pursuing genomic integration. This decision is supported by the findings of Kan et al. (2021), who demonstrated that pMUT-based vectors maintain 100% stability in vivo without antibiotic selection. Specifically, our 4.6 kb synthetic insert falls well within the validated capacity of the pMUT2 backbone, which has been shown to accommodate up to 7 kb of recombinant DNA (total plasmid size ~13.1 Kbp) without compromising bacterial fitness (Kan et al., 2021). By utilizing the s2 intergenic site, the construct avoids disrupting essential mobility or replication functions, providing a robust, clinical-grade platform for the FUS-controlled iron sequestration system.
5.3 Validation Aspect
The computational logic of the therapeutic genetic circuit was validated by developing a Python-based Boolean simulation of the TlpA39C thermal gate and lactate-gated kill switch transfer functions using Hill function modeling. Additionally, the complete 10,114 bp therapeutic plasmid was digitally assembled and validated in Benchling via simulated Gibson Assembly.
5.4 Validation Protocol
Retrieved the TlpA39C thermal gate parameters from Piraner et al. (2017): Hill coefficient k=60, activation midpoint x0=0.70, corresponding to ~39°C.
Retrieved the lactate-gated kill switch parameters from BBa_K3848004 characterization data: k=12, x0=0.35, corresponding to ~10-12 mM lactate.
Implemented both transfer functions in Python using NumPy, defining normalized input arrays for FUS intensity and lactate concentration respectively.
Plotted each transfer function independently using Matplotlib to generate Figures 3 and 4.
Computed the combined Boolean AND gate output by multiplying the thermal gate and kill switch outputs across all four input state combinations.
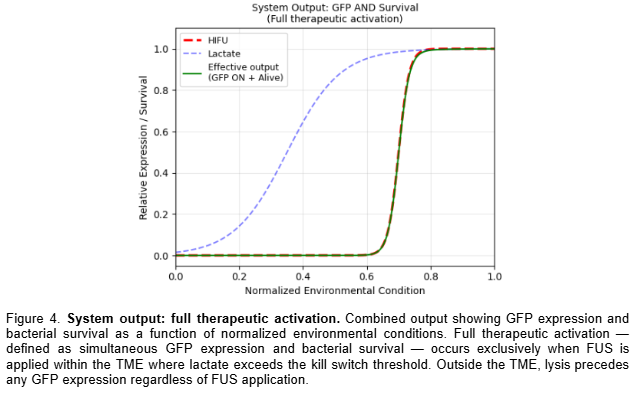
Plotted the combined system output as Figure 5, confirming full therapeutic activation exclusively under simultaneous FUS and TME-level lactate conditions.
All figures were exported and incorporated into the Results section of this report.
The EcN genome (NZ_CP007799.1) was retrieved from NCBI and the 24nt antisense targeting sequences for fur and iroN were extracted and designed.
Both antisense sequences were fused in silico to the MicC scaffold to generate the complete sRNA cassettes.
The four transcriptional cassettes were sequentially assembled in Benchling via simulated Gibson Assembly into the pMUT2 backbone at the s2 intergenic site, generating the 10,114 bp therapeutic vector.
Junction integrity, homology overhang lengths (40 bp), cassette orientation, and transcriptional insulation were verified within Benchling.
The final annotated plasmid map (Figure 6) and linear insert map (Figure 5) were generated and exported from Benchling.
5.5 Synthetic Biology Techniques Used
The validation of this project utilized two core synthetic biology techniques. First, computational modeling in Python using Hill function mathematics to simulate the thermal gate and kill switch transfer functions, confirming Boolean AND gate behavior across all four input states. Second, DNA construct design, where the complete 10,114 bp therapeutic vector was digitally constructed via simulated Gibson Assembly at the s2 intergenic site of pMUT2, verifying junction integrity and cassette orientation.
5.6 Data and Analysis
Simulated data was generated and presented in Section 5 of this report, including the thermal gate transfer function (Figure 2), the lactate-gated kill switch sigmoid response (Figure 3), and the combined Boolean system output (Figure 4). These transfer functions were computationally derived using Hill function modeling in Python and confirm that full therapeutic activation occurs exclusively under simultaneous FUS and TME-level lactate conditions.
5.7 Challenges and Limitations
Two principal limitations have been identified in this project and are addressed through specific experimental and design strategies.
The first and most critical limitation concerns the metabolic burden imposed on EcN by therapeutic circuit activation. Upon FUS-induced derepression of fur, the endogenous RyhBsRNA is upregulated, which in turn represses SodB — the iron-dependent superoxide dismutase — rendering the bacteria significantly more susceptible to reactive oxygen species (ROS) at the precise moment of maximum therapeutic activity. This creates a fundamental tension between therapeutic efficacy and bacterial viability: the circuit must sequester sufficient iron to drive tumor metabolic collapse before oxidative stress reaches the bacterial lethal threshold. To address this, Aim 2.4 is specifically designed to define the optimal FUS duty cycle — alternating activation windows with metabolic recovery periods — that maintains a negative iron balance in the tumor microenvironment without compromising bacterial survival. The potential role of Mn2+ in sustaining SodA-mediated oxidative stress resistance during therapeutic activation will also be evaluated, leveraging the elevated endogenous Mn2+ concentrations characteristic of solid tumor microenvironments and EcN’s active Mn2+ uptake via MntH transporters. Whether endogenous TME Mn2+ is sufficient or whether supplementation is required remains an open question to be resolved experimentally in Aim 2.4.
The second limitation concerns tumor localization for FUS application. Following PLGA seed degradation and bacterial colonization — which occurs over 24-48 hours — no physical reference marker remains to guide millimetric FUS targeting at the time of therapy. This is partially addressed through prior imaging coordinates established by ultrasound-guided implantation for breast and prostate, and by direct endoscopic or colposcopic tumor mapping for colorectal and cervical cancers respectively, combined with real-time FUS guidance. An alternative strategy under evaluation is the incorporation of a second plasmid (pMUT1) encoding acoustic reporter genes (ARG1) — gas vesicles detectable by diagnostic ultrasound — that would enable real-time bacterial localization within the tumor prior to FUS activation (Bourdeau et al., 2018). However, this approach introduces additional metabolic burden on EcN and its feasibility must be evaluated against the therapeutic efficiency of the primary circuit in Aim 2 before adoption.
SECTION 6: ADDITIONAL INFORMATION
6.1 References
Seaver, L.C., & Imlay, J.A. (2001). Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. Journal of Bacteriology, 183(24), 7173-7181. DOI: 10.1128/JB.183.24.7173-7181.2001
Salvail, H., Lanthier-Bourbonnais, P., Sobota, J.M., Caza, M., Benjamin, J.A., Mendieta, M.E., Lépine, F., Périard, G., Imlay, J.A., & Massé, E. (2010). A small RNA promotes siderophore production through transcriptional and metabolic remodeling. PNAS, 107(34), 15223-15228. DOI: 10.1073/pnas.1007805107
Massé, E., & Gottesman, S. (2002). A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. PNAS, 99(7), 4620-4625. DOI: 10.1073/pnas.032066599
Semsey, S. (2014). A mixed incoherent feed-forward loop allows conditional regulation of response dynamics. PLOS ONE, 9(3), e91243. DOI: 10.1371/journal.pone.0091243
Sohn, Y.S., Mitterstiller, A.M., Breuer, W., Weiss, G., & Cabantchik, Z.I. (2011). Rescuing iron-overloaded macrophages by conservative relocation of the accumulated metal. British Journal of Pharmacology, 164(2), 406-418. DOI: 10.1111/j.1476-5381.2010.01120.x
Liang, W., & Ferrara, N. (2021). Iron metabolism in the tumor microenvironment: contributions of innate immune cells. Frontiers in Immunology, 11, 626812. DOI: 10.3389/fimmu.2020.626812
Szlasa, W., Gachowska, M., Kiszka, K., Rakoczy, K., Kiełbik, A., Wala, K., Puchała, J., Chorążykiewicz, K., Saczko, J., & Kulbacka, J. (2021). Iron chelates in the anticancer therapy. Chemical Papers. DOI: 10.1007/s11696-021-02001-2
Privalle, C.T., & Fridovich, I. (1988). Inductions of superoxide dismutases in Escherichia coli under anaerobic conditions. Journal of Biological Chemistry, 263(9), 4274-4279. PMID: 3279033
Bourdeau, R., Lee-Gosselin, A., Lakshmanan, A. et al. (2018) Acoustic reporter genes for noninvasive imaging of microorganisms in mammalian hosts. Nature 553, 86-90. https://doi.org/10.1038/nature25021
Torti, S. V., & Torti, F. M. (2013). Iron and cancer: more ore to be mined. Nature Reviews Cancer, 13(5), 342-355. https://doi.org/10.1038/nrc3495
Pita-Grisanti V. et al. (2022). Understanding the Potential and Risk of Bacterial Siderophores in Cancer. Frontiers in Oncology. https://doi.org/10.3389/fonc.2022.867271
Stritzker et al., (2007) Tumor-specific colonization, tissue distribution, and gene induction by probiotic Escherichia coli Nissle 1917 in live mice. International Journal of Medical Microbiology. Volume 297, Issue 3. DOI: 10.1016/j.ijmm.2007.01.008
Saha P. et al. (2019). Enterobactin, an iron chelating bacterial siderophore, arrests cancer cell proliferation. Biochemical Pharmacology, Volume 168, 71-81. https://doi.org/10.1016/j.bcp.2019.06.017
Chen et al. (2023). Advances in Escherichia coli Nissle 1917 as a customizable drug delivery system for disease treatment and diagnosis strategies. Materials Today Bio Volume 18. https://doi.org/10.1016/j.mtbio.2023.100543
Piraner D.I., Abedi M.H., Moser B.A., Lee-Gosselin A. & Shapiro M.G. (2017). Tunable thermal bioswitches for in vivo control of microbial therapeutics. Nature Chemical Biology 13:75-80. DOI: 10.1038/nchembio.2233
Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229-263. doi:10.3322/caac.21834
Kan, A., Gelfat, I., Emani, S., Beliveau, A., Way, J. C., Silver, P. A., & Joshi, N. S. (2021). Plasmid Vectors for in Vivo Selection-Free Use with the Probiotic E. coli Nissle 1917. ACS Synthetic Biology, doi: 10.1021/acssynbio.0c00466
Pérez-Tomás & Pérez-Guillén, (2020). Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers (Basel). 3;12(11):3244. doi: 10.3390/cancers12113244
Huang et al., (2024). Overcoming the nutritional immunity by engineering iron-scavenging bacteria for cancer therapy. eLife 12:RP90798. https://doi.org/10.7554/eLife.90798.3
Fischbach et al., (2006). The pathogen-associated iroA gene cluster mediates bacterial evasion of lipocalin 2. Microbiology 103 (44) 16502-16507. https://doi.org/10.1073/pnas.0604636103
Massip C, Branchu P, Bossuet-Greif N, Chagneau CV, Gaillard D, Martin P, et al. (2019) Deciphering the interplay between the genotoxic and probiotic activities of Escherichia coli Nissle 1917. PLoS Pathog 15(9): e1008029. https://doi.org/10.1371/journal.ppat.1008029
Pinnix et al., (2010). Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci Transl Med. 4;2(43):43ra56. doi: 10.1126/scisignal.3001127
Dam et al., (2017). Dual Regulation of the Small RNA MicC and the Quiescent Porin OmpN in Response to Antibiotic Stress in Escherichia coli. Antibiotics, 6(4), 33. https://doi.org/10.3390/antibiotics6040033
Na et al., (2013). Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat Biotechnol. 31(2):170-4. DOI: 10.1038/nbt.2461
Gurbatri et al., (2024). Engineering tumor-colonizing E. coli Nissle 1917 for detection and treatment of colorectal neoplasia. Nat Commun 15, 646. https://doi.org/10.1038/s41467-024-44776-4
Ando et al., (2025). CD44: a key regulator of iron metabolism, redox balance, and therapeutic resistance in cancer stem cells. Stem Cells. 27;43(6):sxaf024. doi: 10.1093/stmcls/sxaf024.
Müller et al., (2009). Salmochelin, the long-overlooked catecholate siderophore of Salmonella. Biometals. 22:691-695 DOI 10.1007/s10534-009-9217-4
Miethke & Marahiel. (2007) Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev. 71(3):413-51. doi: 10.1128/MMBR.00012-07.
Raymond et al., (2003) Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. U.S.A. 100 (7) 3584-3588.https://doi.org/10.1073/pnas.063001810
Mercê, A. L. R., Carrera, L. C. M., Romanholi, L. K. S., & Recio, M. A. L. (2002). Aqueous and solid complexes of iron(III) with hyaluronic acid: Potentiometric titrations and infrared spectroscopy studies. Journal of Inorganic Biochemistry, 89(3-4), 212-218. https://doi.org/10.1016/S0162-0134(01)00422-6
Chan C.T., Lee J.W., Cameron D.E., Bashor C.J. & Collins J.J. (2016). Deadman and Passcode microbial kill switches for bacterial containment. Nature Chemical Biology 12:82-86. https://doi.org/10.1038/nchembio.1979
6.2 Supply List and Budget Summary
Phase 1 - Academic R&D and Preclinical Validation (Aims 2 and 3.1): The benchwork and murine in vivo evaluation operate under university laboratory scales, requiring localized funding for specialized materials and facility fees:
Phase 2 - Visionary Clinical Translation and Human Deployment (Aim 3.2)
Clinical-Grade Scaling & Regulatory Assays: Transitioning the platform to human clinical trials (Aim 3.2) expands the financial scope beyond university grant limits. This phase requires scaling to GMP (Good Manufacturing Practices) bacterial cultivation, international multicenter clinical trials across four target cancers, and structural regulatory filings (FDA/ISP Chile).
Projected Phase 2 Capital: $15,000,000 – $50,000,000 USD (To be secured via institutional venture capital, biopharma partnerships, or translational milestone-based government grants, moving from bench-top prototypes to clinical-grade living therapeutics). If human clinical trials (Aim 3.2) are executed through sovereign biotechnology consortia or established Contract Research Organizations (CROs) in emerging regions such as India, China, or Latin American medical hubs, the clinical capital requirements can be reduced by 60% to 90%.
Parallel Veterinary Translation: To further accelerate the translational timeline and mitigate clinical risk, the platform can be strategically deployed within veterinary oncology. Pet dogs and cats develop spontaneous, immunologically complex solid tumors that mimic human pathophysiology far better than engineered murine models. Because the regulatory framework managed by the FDA-CVM operates under a more flexible regulatory framework relative to the multi-year, sequential human phase checkpoints, validating this bacteria-based therapy in veterinary clinical trials can be achieved within 18 to 36 months with a minimal capital requirement of $300,000 – $500,000 USD. This comparative oncology approach offers an immediate, high-volume market to save millions of companion animals worldwide, while generating robust, real-world efficacy and safety data to fundamentally de-risk and fast-track subsequent human clinical approvals.
6.3 Final presentation HTGAA-2026
6.4 Using Programmable Bacteria to Detect and Treat Cancer
The integration of engineered cellular platforms represents a milestone in oncological synthetic biology. To contextualize the real-world execution of autonomous diagnostic and therapeutic loops, this section benchmarks our genetic circuit strategies against the pioneering work of synthetic biologist Tal Danino regarding programmable probiotics: