Week 4 HW: Protein Design Part I
Part I: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
$1 Dalton = 1 g/mol$
The protein content of meat is about 20%.1 That means 500g of meat contain 100g protein.
$100g/100g/mol=1mol$
$1mol = Avogadro_constant = 6*10^{23}$
Under the given assumption your intake of molecules of amino acids equals the value of the avogadro constant.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
You don’t become what you eat because your body breaks food down into basic building blocks, then rebuilds them according to your own DNA instructions.
3. Why are there only 20 natural amino acids?
There aren’t only 20 natural amino acids. Two additional amino acids exist in nature:
- Selenocysteine (the 21st)
- Pyrrolysine (the 22nd)
For humans the 20 basic amino acids are essential for surviving. There are 20 canonical ones because evolution selected a set that is chemically sufficient, efficient, and stable.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes. Although natural proteins are built from only 20 amino acids, nature has expanded this repertoire by genetically encoding Selenocysteine and Pyrrolysine through stop codon reassignment. Inspired by this flexibility, scientists have engineered the genetic code to incorporate many non-natural amino acids, greatly expanding the chemical and functional diversity available in protein design. 2
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life and enzymes existed, amino acids were formed through abiotic chemical processes—that is, purely chemical reactions in the environment. There are a few major sources scientists think contributed:
Atmospheric and lightning synthesis: Simple gases like CH₄, NH₃, H₂, and H₂O could react under energy input (like lightning or UV light) to form amino acids. This was famously demonstrated in the Miller–Urey experiment.3
Hydrothermal vents: Hot, mineral-rich water at the ocean floor could drive chemical reactions that produce amino acids from simple carbon and nitrogen compounds.4
Extraterrestrial delivery: Amino acids have been found in meteorites, suggesting that some were formed in space and brought to Earth.4
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
It would be left‑handed.
If you make an α-helix using D-amino acids instead of the natural L-amino acids, the helix will have the opposite handedness. Natural α-helices made of L-amino acids are right-handed. α-helices made of D-amino acids will be left-handed. This happens because the chirality at the α-carbon determines the backbone geometry, so inverting the stereochemistry flips the helix’s screw direction. 5
7. Can you discover additional helices in proteins?
Yes, additional helices beyond the classical α-helix can exist and have been discovered or designed. Besides the common α-helix, proteins can form structures like the 3₁₀ helix and the π-helix, which differ in hydrogen bonding patterns and backbone geometry. Moreover, by incorporating non-natural or β-amino acids, researchers have created entirely new helical structures (“foldamers”) that do not occur in natural proteins. So both nature and protein engineers can access additional helical architectures by altering backbone chemistry or hydrogen-bonding patterns.
8. Why are most molecular helices right-handed
Because most helices consist of only L-amino acids. Most molecular helices, including α-helices in proteins and the DNA double helix, are right-handed because of the chirality of their building blocks. 5
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their extended backbone and repetitive hydrogen-bonding pattern allow multiple strands to stack together in a very regular way.
Why β-sheets aggregate: The exposed edges of a β-sheet present backbone C=O and N–H groups already arranged in the correct geometry to hydrogen bond with any compatible β-strand they encounter, so two sheets or strands can zip together without major rearrangement
What is the driving force: Backbone hydrogen bonding, Hydrophobic interactions, Van der Waals forces 6
Part II: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I selected the protein with UniProt accession A0A0F6TNJ6_DWV, which is part of the genome polyprotein of Deformed wing virus (DWV), a positive-strand RNA virus that infects honey bees and contributes to wing deformities and colony collapse. DWV expresses its proteins as one large polyprotein that is later cleaved into functional units including capsid and enzymatic proteins, and I chose it because viral polyproteins illustrate how a single chain can generate multiple sub-proteins with distinct 3D structures and functions that you can visualize in molecular viewers.
2. Identify the amino acid sequence of your protein.
amino acid sequence: LIVGYVPGLTASLQQQMDYMKLKSSSYVVFDLQESNSFTFEVPYVSYRPWWVRKYGGNYLPSSTDAPSTLFMYVQVPLIPMEAVSDTIDINVYVRGGSSFEVCVPVQPSLGLNWNTDFILRNDEEYRAKTGYAPYYAGVWHSFNNSNSLVFRWGSASDQIAQWPTISVPRGELAFLRIRDGKQAAVGTQPWRTMVVWPSGHGYNIGIPTYNAERARQLAQHLYGGGSLTDEKAKQLFVPANQQGPGKVSNGNPVWEVMRAPLATQRAHVQDFESRAQI
The most common amino acid is: V, which appears 26 times.
homologs: There are 24 protein sequence homologs for this protein
family: The protein A0A0F6TNJ6_DWV belongs to the Iflavirus polyprotein family, which is part of the larger Picornavirus-like superfamily of positive-sense single-stranded RNA viruses.
3. Identify the structure page of your protein in RCSB.
Structure of deformed wing virus, a honeybee pathogen: 5L7Q | pdb_00005l7q
- 2017
- Resolution 3.50 Å
- vp1, vp2, vp3
- Classification: VIRAL PROTEIN
4. Open the structure of your protein in any 3D molecule visualization software:
Part III: Using ML-Based Protein Design Tools
Deep Mutational Scans
1a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
The heatmap of my capsid protein:
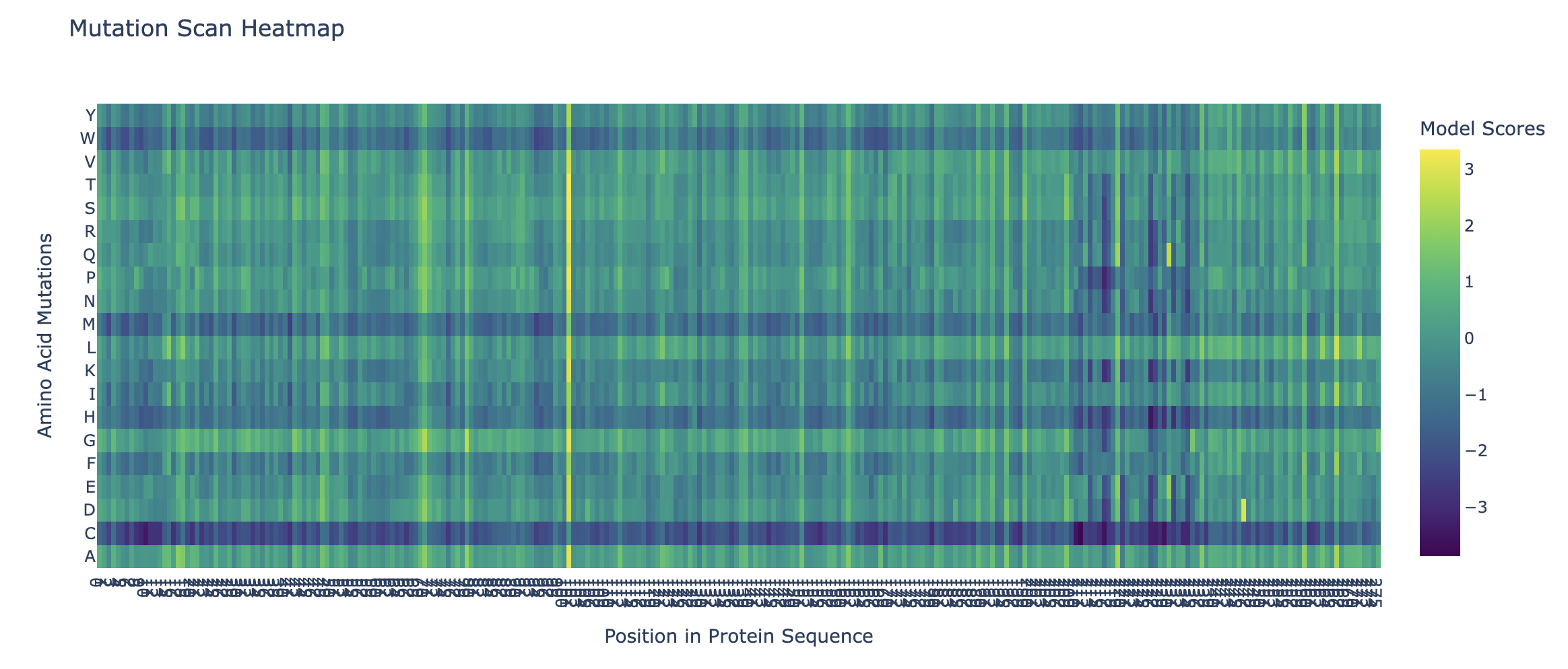
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
You can clearly observe:
Some vertical stripes (columns) are very purple
- These positions are mutation-intolerant.
- Likely structurally or functionally critical residues.
Some regions are mostly green
- These are mutation-tolerant regions.
- Likely surface-exposed loops or flexible regions.
The Cysteine row is strongly purple across many positions as well as Methionine and Tryptophan. This means that these three amino acids disfavor the occurence of mutations.
Looking at the colums there is one that is specifically interesting but i can’t see the x-axis right.
Latent Space Analysis
2a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
Shape of embeddings array after 3D t-SNE: (15177, 3)
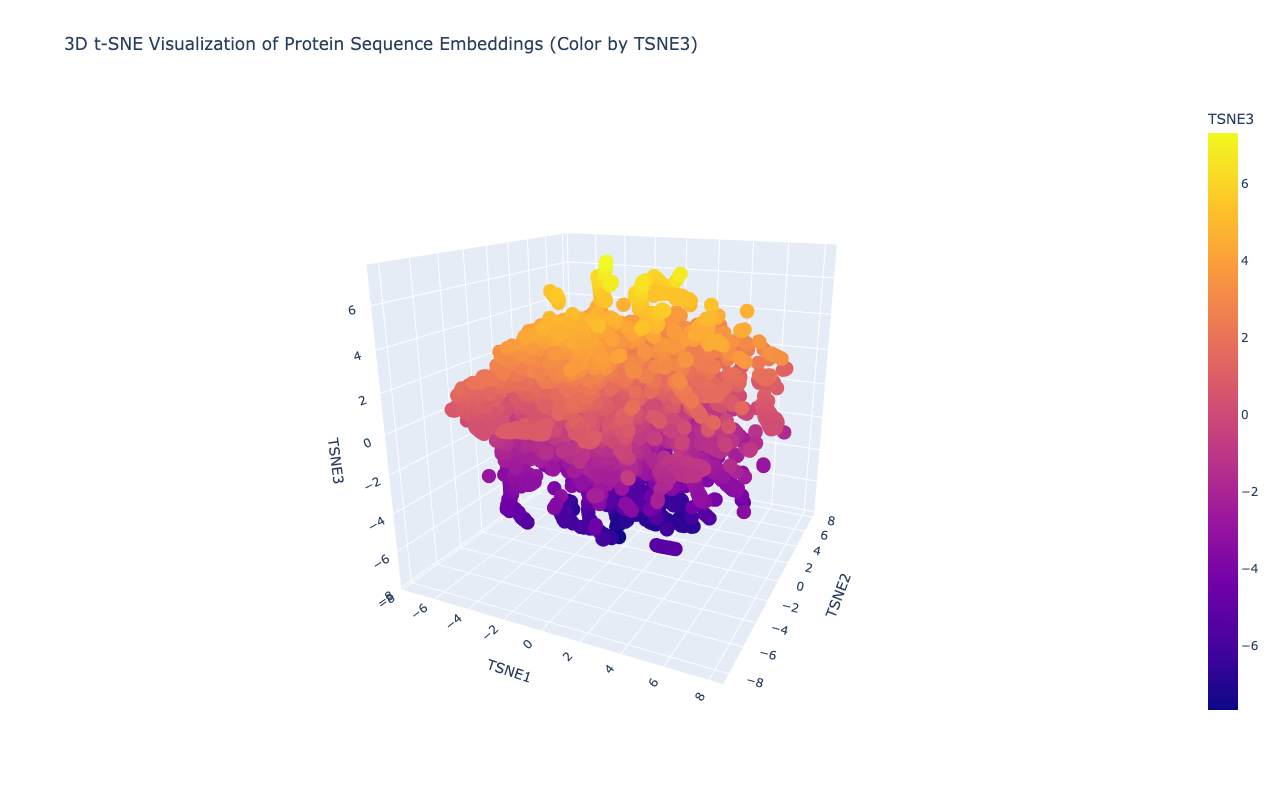
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes,typically they do. In protein language model embeddings:
- Proteins from the same family cluster together
- Proteins with similar functions cluster
- Proteins with similar folds cluster
- e.g. Viral polyproteins cluster with other viral polyproteins
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
After embedding the protein sequences into a reduced latent space, distinct neighborhoods emerged that correspond to proteins with similar evolutionary and functional characteristics. Proteins belonging to related viral families cluster closely, indicating that the language model captures conserved sequence motifs and domain architecture. My selected protein, the Deformed Wing Virus polyprotein (A0A0F6TNJ6_DWV), clusters with other positive-sense RNA viral polyproteins, reflecting shared conserved domains such as RNA-dependent RNA polymerase and viral protease regions.
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Total sequence length: 278 Running ESMFold inference for sequence with length 278… Prediction complete. ptm: 0.329 plddt: 46.496 Results saved to test_877d8/ CPU times: user 27.2 s, sys: 8.79 s, total: 36 s Wall time: 1min
Structure display:
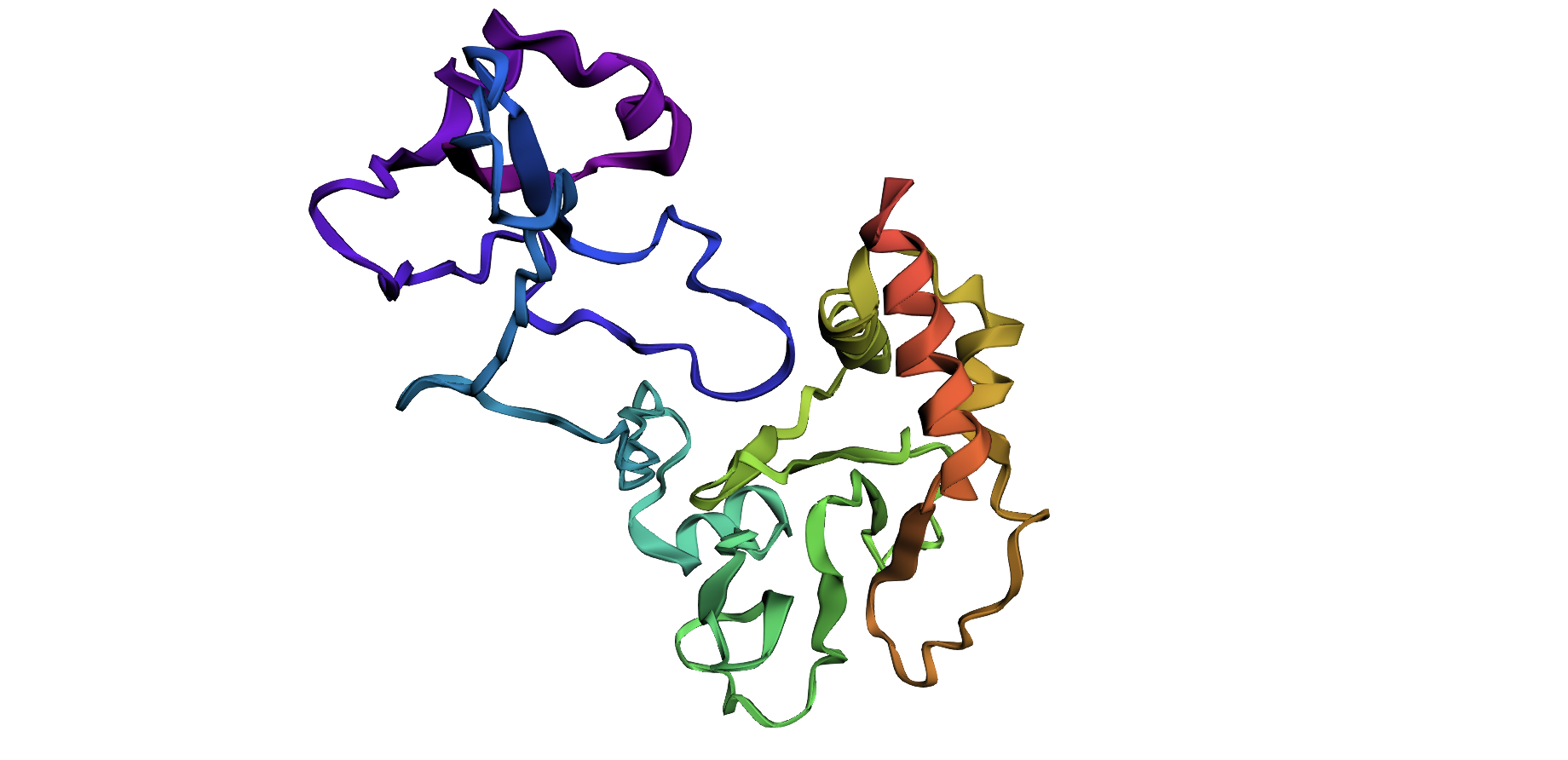
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I changed the sampling temperature to 0.5 and the number of sequences to 4. I
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
Zeng, Y., Chen, E., Zhang, X., Li, D., Wang, Q., & Sun, Y. (2022). Nutritional Value and Physicochemical Characteristics of Alternative Protein for Meat and Dairy—A Review. Foods, 11(21), 3326. https://doi.org/10.3390/foods11213326 ↩︎
Zitti, A., Jones, D. (2023). Expanding the genetic code: a non-natural amino acid story. The Biochemist, 45(1), 2–6. https://doi.org/10.1042/bio_2023_102 ↩︎
Ring D, Wolman Y, Friedmann N, Miller S.L. (1972) Prebiotic synthesis of hydrophobic and protein amino acids. PNAS, 69(3), 765-8. https://doi.org/10.1073/pnas.69.3.765 ↩︎
Higgs P.G., Pudritz R.E. (2009). A Thermodynamic Basis for Prebiotic Amino Acid Synthesis and the Nature of the First Genetic Code. Astrobiology, 9(5), 483-490. https://doi.org/10.1089/ast.2008.0280 ↩︎ ↩︎
Hoang, H. N., Abbenante, G., Hill, T. A., Ruiz-Gómez, G., Fairlie, D. P. (2012). Folding pentapeptides into left and right handed alpha helices. Tetrahedron, 68(23), 4513-4516, https://doi.org/10.1016/j.tet.2011.10.108 ↩︎ ↩︎
Richardson, J. S., Richardson, D.C. (2002). Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. PNAS, 99(5), 2754-9. https://doi.org/10.1073/pnas.052706099 ↩︎