Week 5 HW: protein design part ii
Part A: SOD1 Binder Peptide Design
Part I: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V Mutation -> means one needs to change the A at position 4 to an V. This protein sequence only had one at the fifth position so I changed this Alanine.
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
- 1 KRVYVVVVEHKE 43.699289
- 2 WRYPVVAAALGX 7.252220
- 3 WLYYPAAVELGX 10.791046
- 4 HRYYPTAVRHWK 13.771030
- 5 FLYRWLPSRRGG
The aim of PepMLM is to finetune the ESM-2 protein language model to fully reconstruct the binder region, achieving low perplexities matching or improving upon validated peptide–protein sequence pairs. The lower the perplexity, the better the model. Perplexity in protein models measures how well a language model predicts the next amino acid in a sequence, acting as a proxy for how “natural” or physically plausible a protein sequence is. Lower perplexity indicates the model understands the protein structure constraints better, frequently used to estimate sequence fitness and stability.1
1.: Kantroo, P., Wagner, G. P., Machta, B. B. (2025) Pseudo-perplexity in one fell swoop for protein fitness estimation, PRX Life 3, https://doi.org/10.1103/zhx7-hcmm
Part II: Evaluate Binders with AlphaFold3
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
1: ipTM = 0.34, pTM = 0.88
PAE Graph
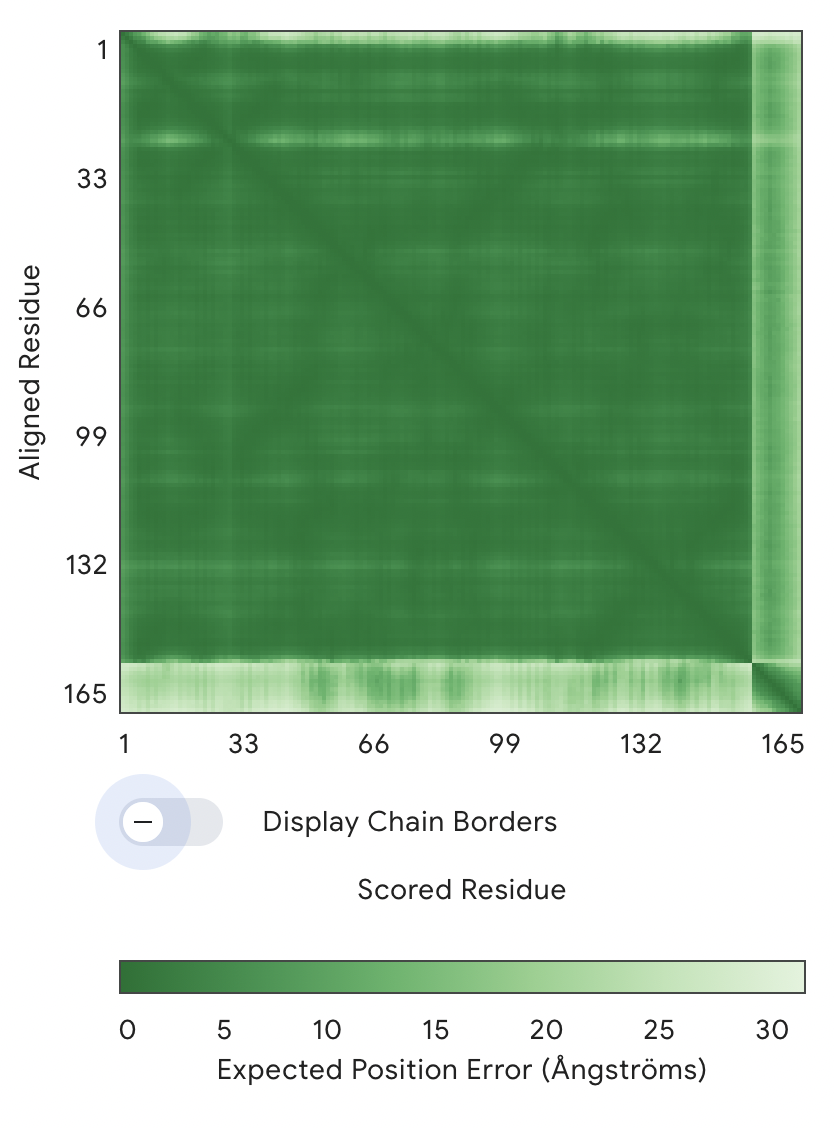
- The main large green square (1–154 residues) = SOD1 protein
- The thin strip on the right/bottom (~154–165) = peptide
Key observation:
The SOD1–SOD1 region is dark green → very low error → high confidence structure The peptide vs SOD1 region is much lighter (higher error) → low confidence in relative positioning → AlphaFold is uncertain where the peptide sits → The interaction is likely weak, flexible, or non-specific
As a result, no specific binding site can be confidently assigned. The peptide does not appear to localize near the N-terminal region where the A4V mutation is located, nor is there clear evidence that it engages the β-barrel core or the dimer interface. Instead, the peptide appears to be loosely associated with the protein surface, likely in a flexible and non-specific manner rather than forming a stable, partially buried interaction.
For the next two peptides, I replaced the Xs with As for 2 and 3. A X means there is not amino acid defined.
2: ipTM = 0.37, pTM = 0.83
3: ipTM = 0.36, pTM = 0.83
4: ipTM = 0.26, pTM = 0.84
5: ipTM = 0.39, pTM = 0.83
Overall, all the ipTM scores I received weren’t convincing. ipTM (interface predicted TM-score) measures how confidently AlphaFold predicts the interaction between chains -> the lower the worse the prediction.
Part III: Evaluate Properties of Generated Peptides in the PeptiVerse
PeptiVerse is a universal therapeutic peptide property prediction platform.
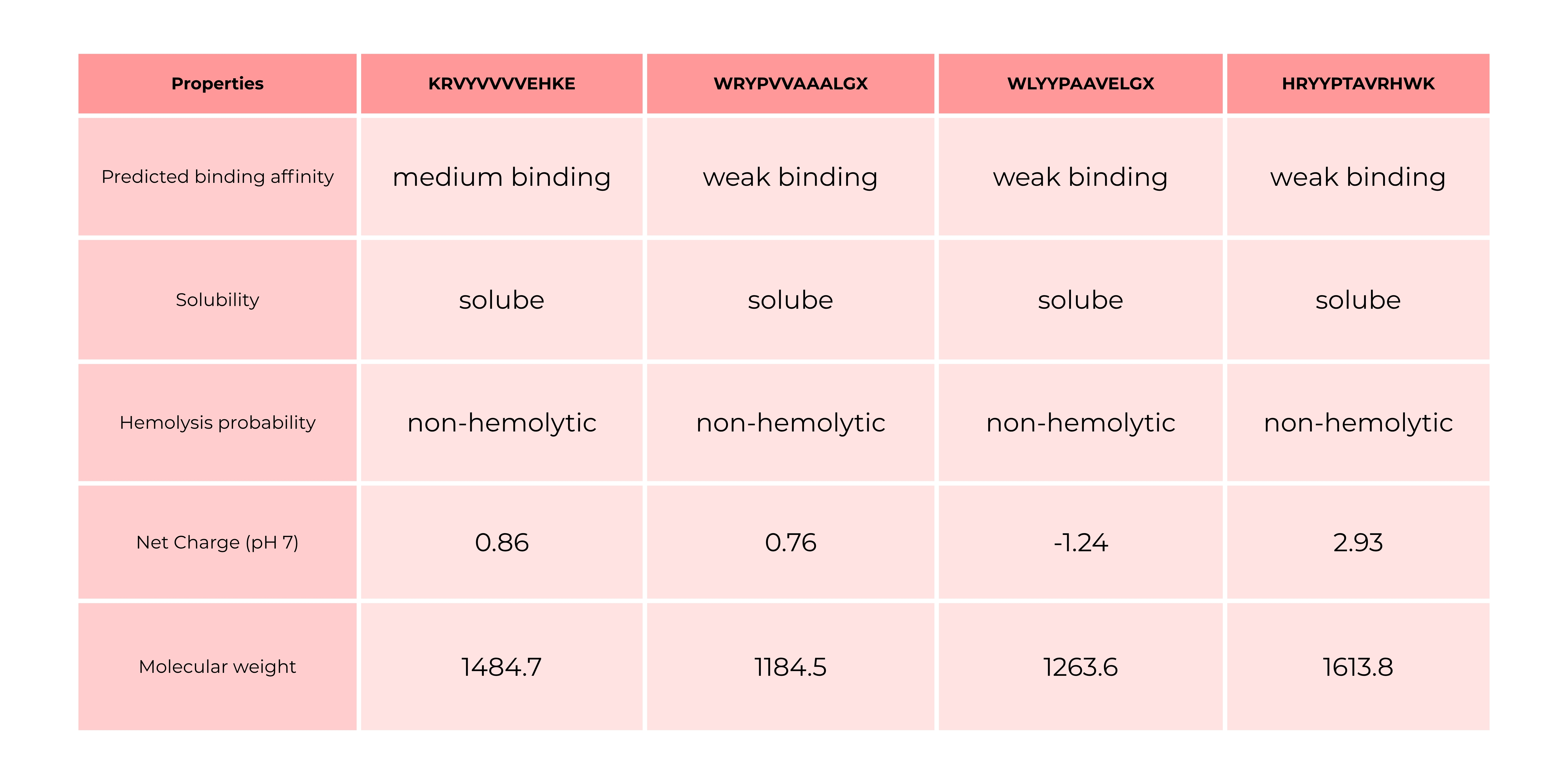
The comparison between AlphaFold3 predictions and PeptiVerse results shows that structural confidence (ipTM) does not necessarily correlate with predicted binding affinity. Peptides with higher ipTM scores did not consistently exhibit stronger predicted affinity, highlighting differences between structure-based and sequence-based approaches. Interestingly, the only peptide predicted to have medium binding affinity was the one with an ipTM of 0.34 and a perplexity of 43, which were the highest values among the tested candidates. All other peptides were predicted to bind only weakly, even in cases where they showed comparable or slightly better ipTM and perplexity scores. This suggests that neither ipTM nor perplexity alone is sufficient to reliably predict binding strength, and that combining multiple evaluation methods is necessary for a more comprehensive assessment.
In that case, I would go with peptide 1 because it binds the best.
Part IV: Generate Optimized Peptides with moPPIt
- 1: KKTKTYKETRGD
- 2: RTGSETGTEEKY
- 3: TKTKRERGYNKQ
- 4: QATKKKKETNKE
The moPPit peptides differ significantly from the PepMLM-generated peptides in both composition and physicochemical properties. While the PepMLM peptides contain a mix of hydrophobic and aromatic residues (e.g., W, Y, V, L), suggesting potential for structured binding and interaction with hydrophobic regions of SOD1, the moPPit peptides are dominated by charged and polar residues, particularly lysine (K), arginine (R), and glutamic acid (E). This makes the moPPit peptides more hydrophilic and likely more soluble, but also suggests that they may interact more nonspecifically through electrostatic interactions rather than forming well-defined binding interfaces. In contrast, the PepMLM peptides appear more “protein-like” and may be better suited for specific binding, although some sequences include undefined residues (X), introducing uncertainty. The moPPit peptides, with their high positive charge, may resemble cell-penetrating or antimicrobial peptides, which can increase membrane interaction but also raise concerns about cytotoxicity or hemolysis. Before advancing any of these peptides toward clinical studies, a comprehensive evaluation would be required. This would include computational validation such as structure prediction (e.g., via AlphaFold) and binding assessment, as well as sequence-based predictions of solubility, toxicity, and hemolysis (e.g., using PeptiVerse). Promising candidates should then be tested experimentally through in vitro assays to measure binding affinity, stability, and aggregation effects on SOD1. Additionally, toxicity assays, including hemolysis and cell viability tests, would be essential to assess safety.
Part C: Final Project: L-Protein Mutants
Option 1: Random Mutagenesis
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
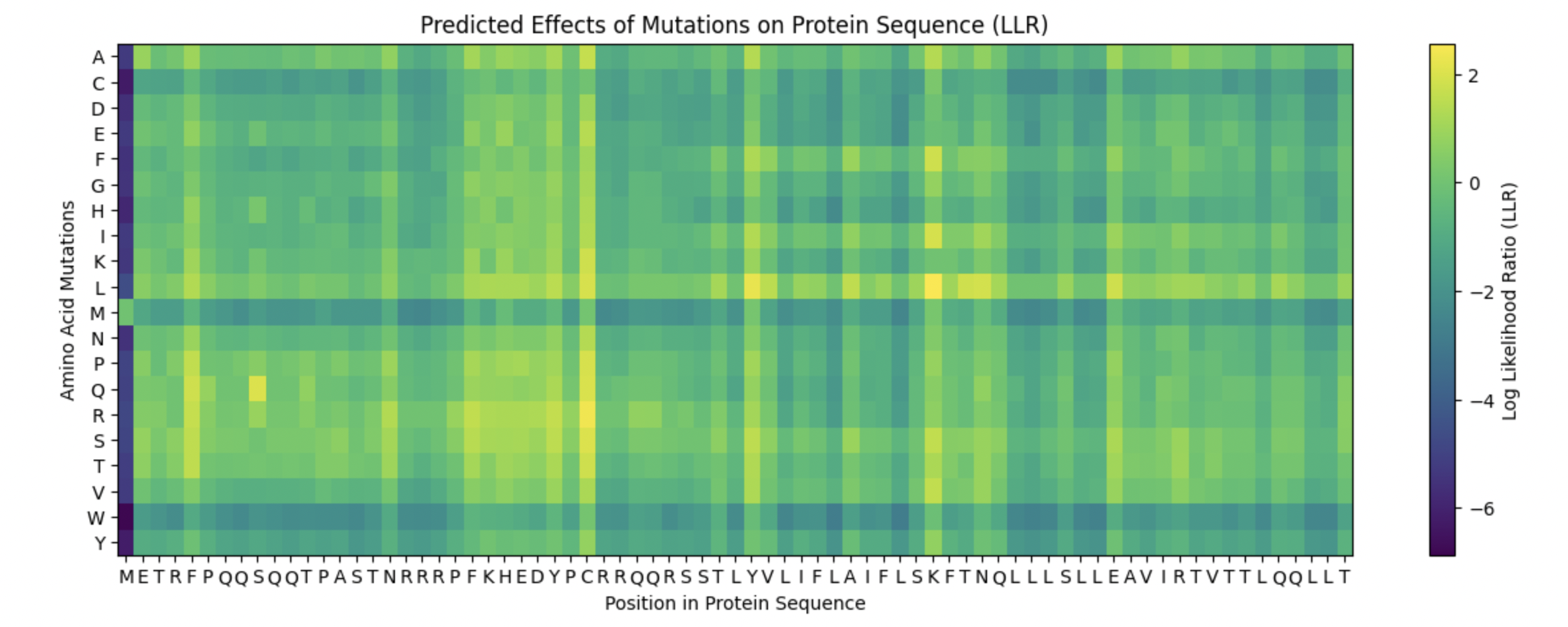
5 Mutations
I looked at the Protein Sequence Heatmap. And chose spots for mutations depending on the most yellow matrix squares.
- C29L: METRFPQQSQQTPASTNRRRPFKHEDYPLRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- K50I: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSIFTNQLLLSLLEAVIRTVTTLQQLLT (Protein Level 1)
- S9Q: METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- K50F: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSFFTNQLLLSLLEAVIRTVTTLQQLLT
- C29R: METRFPQQSQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Solube Region is around 1 - 39: So the S and C Mutations
Transmembrane Region is afterwards 40 - 83: So the K Mutations
Among the proposed mutations, K50I and K50F are strong candidates in the transmembrane region, as they replace a charged lysine with hydrophobic residues, which is more compatible with the membrane environment and likely stabilizes the helix. In the soluble region, S9Q is a safe, conservative mutation that maintains polarity and could enhance hydrogen bonding, while C29L and C29R are riskier, since C29 may form disulfide bonds or contribute to structural stability; replacing it with hydrophobic or charged residues could destabilize the protein. Overall, K50I, K50F, and S9Q are likely effective mutations, while the C29 variants could be informative but carry higher structural risk.