Hi, I’m Fanni, an engineer finding her way in the creative industry while trying to actively promote sustainable change. Last summer, I completed my BSc in Environmental Engineering in Vienna, and two months later I moved to London to begin my Master’s in Biodesign at Central Saint Martins.
This past autumn has been incredibly exciting, offering many new perspectives on sustainable innovation. In early November, I attended a talk by David S. Kong in London, where he introduced the HTGAA course. I was fascinated by the field of Synthetic Biology, which felt like the perfect bridge back to my engineering background - something that had taken a quieter role during my more creatively focused recent studies.
I am still figuring out where exactly I fit within these overlapping fields, but I’m confident that gaining knowledge in synthetic biology will guide me closer to where I want to go.
Contact info
here you can find my two instagram accounts and dm me if you want to discuss/collab:
Part I 1. First, describe a biological engineering application or tool you want to develop and why.
I am interested in the development of engineered bee gut bacteria or similar that help bees resist viral infections, pesticide stress but especially harmful varroa mites. The presence of varroa mites in bee colonies place an important pressure on bee health since they attack and feed on them in a parasitism relationship. 1 Instead of genetically modifying bees themselves, I aim to modify their symbiotic bacteria to strengthen colony resilience while minimizing ecological risks. Bees are increasingly threatened by habitat loss, unsustainable agricultural practices, climate change and pollution. Their decline jeopardizes food production, increases costs and exacerbates food insecurity, particularly for rural communities. I am convinced that supporting pollinators will get more and more critical for global food systems and biodiversity and this approach could offer a scalable and ecologically sensitive alternative to chemical treatments currently used in agriculture. Even if it needs human intervention into nature to keep our ecosystem in balance, I think supporting these small often unnoticed pollinators could make a real difference.
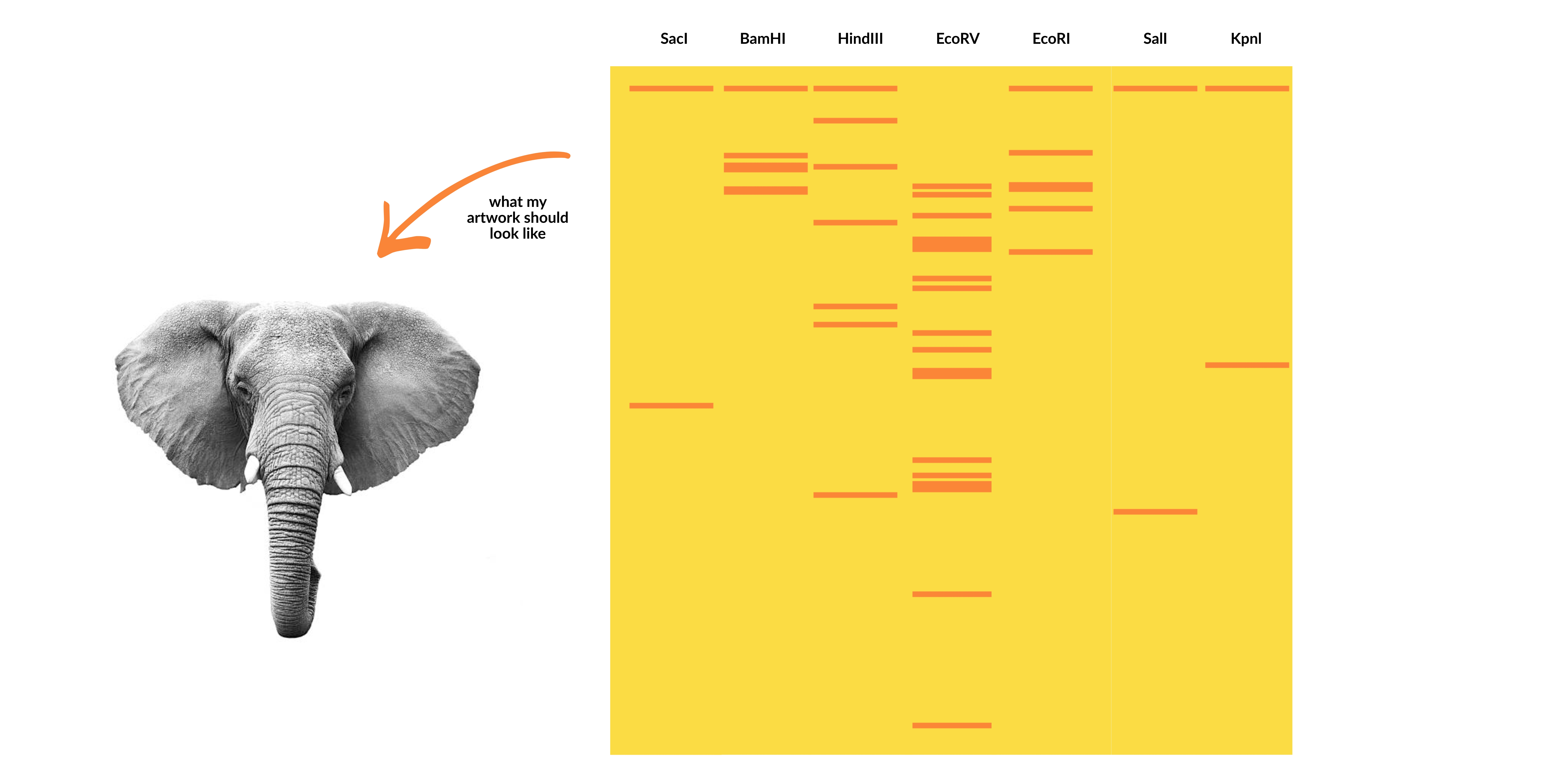
Part I: Benchling & In-silico Gel Art Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Part III: DNA Design Challenge 1. Choose your protein.
Since my project proposal from last week focuses on honeybee health, I searched for relevant proteins in Apis mellifera. During this process, I identified three candidates that seemed particularly interesting: Defensin-1, Hymenoptaecin and Vitellogenin. Working with Twist Bioscience’s codon optimization tool, I learned that the tool only accepts sequences within a specific length range — proteins that are too short or too long cannot be optimized. After several iterations, vitellogenin was the only protein for which I could successfully perform codon optimization. Vg, a phospholipoglycoprotein synthesized and stored in the honey bee fat body, is an ancient reproduction-associated protein that provides nutrients to eggs in most oviparous animals. Honey bee queens, who produce hundreds of eggs each day, have high levels of Vg gene expression. It is involved in nutrient storage, immune regulation and longevity in honeybees. Its expression is closely linked to colony health and higher vitellogenin levels are associated with improved immune responses and tolerance to Varroa destructor infestation. 1
Part I: Python Script for Opentrons Artwork Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Firstly, I used Ronan’s Automation Art Interface to translate my logo into a pixelated biological artwork. The software converted the image into a set of coordinate outputs, where each tuple (x, y) represents the precise millimeter offset from the calibrated center of the agar plate. Each of these coordinate pairs defines the placement of a single 1 µL droplet, allowing the robot to reconstruct the digital logo physically on the plate.
Part I: Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
$1 Dalton = 1 g/mol$
The protein content of meat is about 20%.1 That means 500g of meat contain 100g protein.
$100g/100g/mol=1mol$
$1mol = Avogadro_constant = 6*10^{23}$
Part A: SOD1 Binder Peptide Design Part I: Generate Binders with PepMLM 1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V Mutation -> means one needs to change the A at position 4 to an V. This protein sequence only had one at the fifth position so I changed this Alanine.
DNA Assembly Answer these questions about the protocol in this week’s lab: 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Typical components include:
Phusion DNA Polymerase A high-fidelity enzyme that synthesizes new DNA strands with very low error rates (proofreading activity → fewer mutations). dNTPs (deoxynucleotide triphosphates) Building blocks (A, T, G, C) used to create new DNA strands. Reaction Buffer (HF buffer) Maintains optimal pH and salt conditions for enzyme activity and fidelity. Mg²⁺ ions (magnesium chloride) Essential cofactor for polymerase activity; affects enzyme efficiency and specificity. Stabilizers (sometimes included) Help maintain enzyme stability during thermal cycling. 2. What are some factors that determine primer annealing temperature during PCR?
Part I: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Continuous Signal Processing
Boolean circuits output only 0 or 1 (OFF/ON) states. IANNs operate on continuous, graded gene expression levels. Ability to Model Complex Relationships
Boolean logic is limited to simple combinations of AND/OR/NOT gates. IANNs can approximate complex, nonlinear input–output functions. Efficient Integration of Multiple Inputs
Part A: General and Lecturer-Specific Questions General 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Quantification of pigment concentration through color intensity measurements Analysis of pigment degradation (as a proxy for biochemical stability) under environmental conditions Material–pigment interaction effects on color retention 2. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Somehow I didn’t receive an email, so I couldn’t contribute.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents 1. Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Subsections of Homework
Week 1 HW: Principles and Practices
Part I
1. First, describe a biological engineering application or tool you want to develop and why.
I am interested in the development of engineered bee gut bacteria or similar that help bees resist viral infections, pesticide stress but especially harmful varroa mites. The presence of varroa mites in bee colonies place an important pressure on bee health since they attack and feed on them in a parasitism relationship. 1
Instead of genetically modifying bees themselves, I aim to modify their symbiotic bacteria to strengthen colony resilience while minimizing ecological risks.
Bees are increasingly threatened by habitat loss, unsustainable agricultural practices, climate change and pollution. Their decline jeopardizes food production, increases costs and exacerbates food insecurity, particularly for rural communities. I am convinced that supporting pollinators will get more and more critical for global food systems and biodiversity and this approach could offer a scalable and ecologically sensitive alternative to chemical treatments currently used in agriculture. Even if it needs human intervention into nature to keep our ecosystem in balance, I think supporting these small often unnoticed pollinators could make a real difference.
Inspiration: Leonard, S. P., Perutka, J., Powell, J. E., Geng, P., Richhart, D. D., Byrom, M., Kar, S., Davies, B. W., Ellington, A. D., Moran, N. A., & Barrick, J. E. (2018). Genetic engineering of bee gut microbiome bacteria with a toolkit for modular assembly of broad-host-range plasmids. ACS Synthetic Biology, 7(5), 1279–1290. https://doi.org/10.1021/acssynbio.7b00399
1: Le Conte, Y., Ellis, M. & Ritter, W. (2010). Varroa mites and honey bee health: can Varroa explain part of the colony losses?. Apidologie, 41, 353–363. https://doi.org/10.1051/apido/2010017
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
1. Prevent ecological harm
Require controlled field trials and ecological risk assessments before environmental release.
Develop containment or reversibility strategies, such as microbes that cannot survive outside bee hosts.
Monitor impacts on wild pollinators and microbial communities long-term.
2. Avoid technology dependancy in nature
Ensure solutions complement ecological practices instead of replacing them.
Link deployment to reduction of harmful pesticide use, rather than allowing continued pollution.
3. Ensure fair access and prevent corporate control
Prevent exclusive patents that make beekeepers dependent on private companies. (monsanto scandal)
Encourage open-access or public research partnerships.
Ensure affordable access for small-scale and community beekeepers.
4. Transparency and public participation
Include beekeepers and environmental groups in decision-making.
Maintain international cooperation since pollinators cross borders.
Raise awareness about the relevance of bees around May 20th and beyond.
5. Ensure safe and responsible deployment of engineered microbes
Require biosafety training and certification for researchers.
Establish traceability and monitoring systems so released microbial strains can be tracked and evaluated over time.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Transparency Standards for DNA Synthesis Providers
Purpose
I believe that many DNA synthesis companies voluntarily screen orders against limited threat models to block synthesis of harmful sequences, but standards vary widely. That’s why, we need an universal regulatory requirement for all commercial DNA synthesis providers (domestic and international selling into regulated markets) to implement robust sequence screening, standardized reporting and data sharing with trusted authorities to build transparency.
Design
What is needed to make it work?
A regulatory body defines minimum screening criteria and risk thresholds.
All providers must opt-in by compliance through certification, non-compliant firms cannot legally sell into the regulated market.
Public funding or tax credits to support smaller providers implementing screening software.
Actors involved:
Federal regulators (standard setting and enforcement)
DNA synthesis companies (compliance)´
Independent auditors (certify implementation)
Assumptions
That most synthesis providers will respond to regulatory pressure and that screening software is reliable.
That standards can keep pace with rapid advances in gene editing and novel organisms.
That international firms will comply or that governments will enforce import controls tied to compliance.
That smaller companies can hold against the competitiveness of certification costs.
Risks of Failure & “Success”
Failure risks
Providers find loopholes or perform minimal compliance without effective safety.
Adversaries migrate to unregulated markets or underground vendors, worsening risk.
High compliance costs drive small innovators out of business, reducing competition.
Risks of “success”
Genuine research slows due to increased cost and time to order DNA.
Fragmented global adoption creates asymmetries: robust safety in some countries, weak in others.
Involvment of local stakeholders & community
Purpose
From what I have read, bee-related synbio solutions are mostly developed in labs and tested with limited involvement of local beekeepers or communities who depend on pollinators. The proposed change is to actively involve beekeepers, farmers and local communities (most practical knowledge because of living with/around them) before deploying biotech solutions affecting bee populations.
Design
Projects deploying engineered microbes or treatments in hives must include local beekeeper collaboration.
Workshops and pilot projects with beekeeping associations allow practical feedback.
Farmers, urban beekeepers, and conservation groups participate in decision-making.
Actors involved
Researchers & biotech companies
Local governments & environmental authorities
Farmers & beekeper associations
Assumptions
Beekeepers are willing and able to participate.
Public engagement improves trust and project outcomes.
Communication between scientists and practitioners works effectively.
Risks of Failure & “Success”
Failure risks
Engagement becomes symbolic rather than meaningful.
Misinformation or fear blocks beneficial projects.
Participation dominated by a few voices, not representative groups.
Risks of “success”
Projects become slowed by lengthy consultation processes.
Communities may expect veto power over projects beyond reasonable risk concerns.
Secure Testing & Containment Framework for Bee Biotechnology
Purpose
Currently, biotechnology innovations may move from lab testing to real hives without fully coordinated safeguards if unexpected ecological effects occur. This action proposes a controlled testing environment (sandbox ecosystem) combined with mandatory containment and emergency response plans before wider deployment.
Design
New bee biotech solutions are first tested in regulated pilot environments with selected partner beekeepers and oversight from authorities.
Engineered microbes or treatments must include biological containment mechanisms (e.g., limited survival outside managed hives).
Continuous monitoring tracks spread and bee health impacts.
Emergency protocols allow rapid withdrawal or containment if problems appear.
Actors involved
Researchers
Biotech companies
Beekeeper networks (monitoring)
Environmental and agricultural authorities
Assumptions
Small-scale sandbox ecosystems manage to imitate natural ecosystems.
Containment mechanisms work reliably in real ecosystems.
Monitoring detects problems early enough to intervene.
Beekeepers cooperate in reporting unexpected outcomes.
Risks of Failure & “Success”
Failure Risks
Containment fails or spread occurs before detection.
Response measures may be too slow.
Risk of “success”
Confidence in safe testing could encourage faster or riskier deployments.
Strict requirements might limit participation by small innovators.
Bügl, H., Danner, J. H., Molinari, R. J., Mulligan, J. T., Park, H., Reichert, B., Roth, D. A., Wagner, R., Budowle, B., Scripp, R. M., Smith, J. A. L., Steele, S. J., Church, G. & Endy, D. (2007). DNA synthesis and biological security. Nature Biotechnology, 25(6), 627–629
Leckenby, E., Dawoud, D., Bouvy, J. & Jónsson, P. (2021). The Sandbox Approach and its Potential for Use in Health Technology Assessment: A Literature Review. Applied Health Economies Health Policy, 19, 857–869. https://doi.org/10.1007/s40258-021-00665-1
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the evaluation of the governance options, I would prioritize Transparency Standards, combined with a Secure Testing and Containment Framework.
Transparency is essential for building trust in new biotechnological tools and for ensuring accountability. If projects, testing procedures and releases are openly documented and traceable, it becomes possible to identify where problems arise and address them early. Similar to other industries - for example, the fashion industry, where lack of supply chain transparency hides environmental and social impacts - insufficient transparency in biotechnology makes it difficult to understand risks or intervene effectively when things go wrong.
However, transparency alone is not sufficient. Even if processes are visible, interventions must also be safe in practice. Therefore, I would combine transparency with a Secure Testing and Containment Framework that ensures technologies are tested in controlled environments and include emergency response mechanisms before large-scale deployment. In the case of bee-related biotech applications, unintended spread or ecological effects could impact entire ecosystems. A containment and rapid-response system would help minimize damage if interventions do not behave as expected.
The main trade-off considered is that stronger transparency and safety requirements may slow innovation or increase costs for smaller research groups. There is also uncertainty about whether containment mechanisms will always function reliably in complex natural environments. Nevertheless, given the ecological importance of pollinators and the potential scale of unintended consequences, prioritizing safety and accountability over rapid deployment seems justified.
Part II
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error Rate of Polymerase: 1:108
Length of Human Genome: 3.2 Gbp = 3.2 * 109 base pairs
If the error rate is 1 in 10⁸, copying the whole genome would lead to roughly:
3*109 / 108 ~ 30
That means there are about 30 errors per cell division without additional repair. To deal with this decrepancy biology developed a error correcting polymerase including proofreading mechanisms and mismatch repair systems.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Formula from the Slides:
The complexion for the total number of different ways to arrange N blocks of Q different types (where each type has the same number) is given by:
((20!)/(((20/4)!)(4))) = 11732745024 ~ 11.7 * 109
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite solid-phase synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis?
The main problem is stepwise synthesis errors. Each nucleotide addition is not perfect. Typical coupling efficiency: ~99–99.5% per step.
0.995200 ~ 0.37
Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct oligonucleotide synthesis adds nucleotides step by step, and each step has a small error rate (≈99–99.5% efficiency). Over thousands of steps, these small errors accumulate, leading to very low yields of full-length, correct DNA. As a result, direct chemical synthesis becomes impractical beyond ~150–200 nucleotides. So companies like Twist Bioscience instead assemble long genes (up to 7kbp) from short oligos and then clone and sequence-verify them.
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
There are 9 essential amino acids: phenylalanine, valine, tryptophan, threonine, isoleucine, methionine, histidine, leucine, and lysine.
However, amino acids such as arginine and histidine may be considered conditionally essential because the body cannot synthesize them sufficiently during specific physiological periods of growth, including pregnancy, adolescent growth or recovery from trauma.1
As I understand it, the “Lysine Contingency” is derived from Jurassic Park and is fictional. I believe it raises important ethical questions about human intervention in nature. In the movie, the dinosaurs depend on lysine supplements provided by the park’s staff, so they cannot survive or escape without them. This system was intended to prevent the dinosaurs from disrupting the global ecosystem. Although the idea aimed to protect the environment, it also involved engineering organisms to depend on a single nutrient for survival which is questionable. All in all, it is striking to me that the absence of just one essential amino acid could determine life or death.[^2]
1: Lopez, M.J. and Mohiuddin, S.S. (2024) Biochemistry, essential amino acids, National Library of Medicine. Available at: https://www.ncbi.nlm.nih.gov/sites/books/NBK557845/ (Accessed: 10 February 2026).
Other References from Part II: Slides from Lecture 2
Week 2 HW: DNA read/write/edit
Part I: Benchling & In-silico Gel Art
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Part III: DNA Design Challenge
1. Choose your protein.
Since my project proposal from last week focuses on honeybee health, I searched for relevant proteins in Apis mellifera. During this process, I identified three candidates that seemed particularly interesting: Defensin-1, Hymenoptaecin and Vitellogenin. Working with Twist Bioscience’s codon optimization tool, I learned that the tool only accepts sequences within a specific length range — proteins that are too short or too long cannot be optimized. After several iterations, vitellogenin was the only protein for which I could successfully perform codon optimization. Vg, a phospholipoglycoprotein synthesized and stored in the honey bee fat body, is an ancient reproduction-associated protein that provides nutrients to eggs in most oviparous animals. Honey bee queens, who produce hundreds of eggs each day, have high levels of Vg gene expression. It is involved in nutrient storage, immune regulation and longevity in honeybees. Its expression is closely linked to colony health and higher vitellogenin levels are associated with improved immune responses and tolerance to Varroa destructor infestation. 1
1: Amdam, G.V., Fennern, E., Havukainen, H. (2012). Vitellogenin in Honey Bee Behavior and Lifespan. In: Galizia, C., Eisenhardt, D., Giurfa, M. (eds) Honeybee Neurobiology and Behavior. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-2099-2_2
2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Once the codon-optimized DNA sequence is obtained, it can be used to produce the protein through transcription and translation. In a cell-dependent system, the DNA is cloned into an expression vector, such as pET-21, and introduced into E. coli, where the bacterial machinery transcribes the DNA into mRNA and translates it into the vitellogenin protein. Alternatively, cell-free systems can carry out transcription and translation in vitro, using extracted enzymes and ribosomes without living cells. In both cases, the DNA sequence serves as a template that determines the amino acid sequence of the resulting protein. 2
I would use the cell-free mechanism “PUREexpress”. Vitellogenin is a very large protein, which can be difficult to express in living cells because of size, folding and potential toxicity. A reconstituted, cell‑free system like “PURExpress” provides a clean, RNase‑ and protease‑poor environment, so long mRNAs and large proteins are less likely to be degraded during expression.3
2: Claassens, N. J., Burgener, S., Vögeli, B., Erb, T. J., Bar-Even, A. (2019) A critical comparison of cellular and cell-free bioproduction systems, Current Opinion in Biotechnology, 60 (221-229)
3: Tuckey, C., Asahara, H., Zhou, Y., Chong, S. (2014) Protein synthesis using a reconstituted cell-free system. Curr Protoc Mol Biol, 108, doi: 10.1002/0471142727.mb1631s108
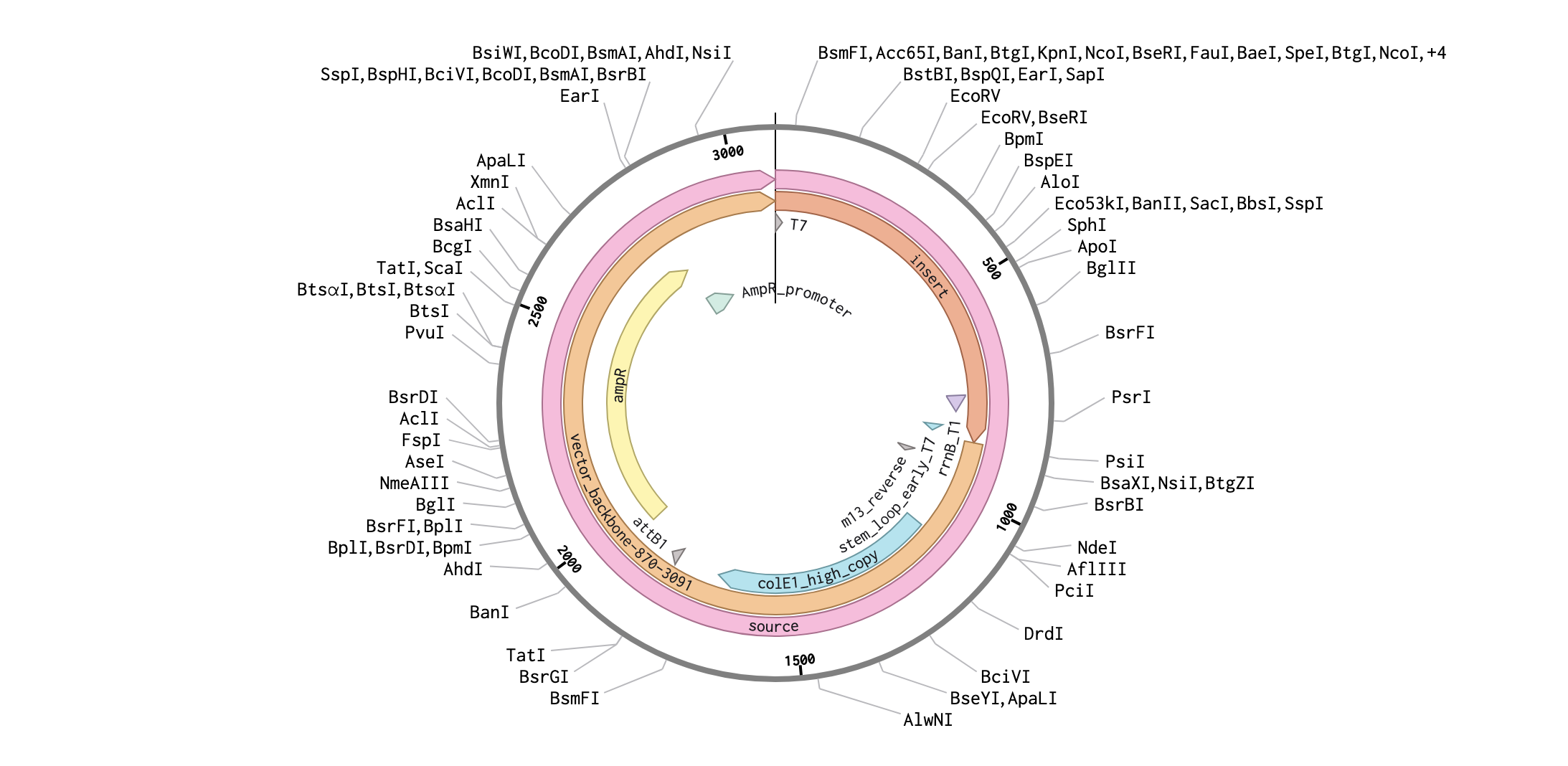
Part IV: Prepare a Twist DNA Synthesis Order
Annotation
supportet by AI - “If you have a DNA strang how do you know which is what to annotate like: task instruction”
Plasmid
Part V: DNA Read/Write/Edit
1. What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would like to sequence Varroa mite DNA because Varroa destructor is a key global parasite of honey bees and a major cause of colony losses. Sequencing its genome and mitochondrial markers would help identify treatment‑resistance mutations, track the spread of different mite lineages between regions, and link mite genotypes to disease outcomes in colonies. This information can directly support better Varroa monitoring, more targeted control strategies, and breeding of honey bees that are more resilient to the specific Varroa populations in their environment, ultimately benefiting pollination, food security, and ecosystem health.4
4: Grindrod, I., Martin, SJ. (2021) Parallel evolution of Varroa resistance in honey bees: a common mechanism across continents? Proc Biol Sci, 288(1956), doi: 10.1098/rspb.2021.1375.
2. In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Chosen sequencing technology
I would use Illumina next-generation sequencing to analyze honeybee genes associated with Varroa mite resistance because it provides highly accurate and cost-efficient sequencing for comparing many samples or studying specific gene regions.5
5: Hu, T., Chitnis, N. , Monos, D., Dinh, A. (2021) Next-generation sequencing technologies: An overview,
Human Immunology, 82(11), 801-811, https://doi.org/10.1016/j.humimm.2021.02.012.
Generation of technology
This method belongs to the second generation of sequencing technologies because it sequences millions of short DNA fragments in parallel, unlike first-generation Sanger sequencing or third-generation long-read single-molecule sequencing.
Input and preparation steps
The input is genomic DNA extracted from honeybees or mites. Preparation involves:
DNA extraction from samples
DNA fragmentation into short pieces
Adapter ligation to fragment ends
PCR amplification of fragments
Loading fragments onto a sequencing flow cell
How bases are decoded (sequencing principle)
Fragments bind to the flow cell and are amplified into clusters. During sequencing, fluorescently labeled nucleotides are incorporated one base at a time. A camera records the color signal after each cycle, and software converts these signals into DNA base sequences — this process is called base calling.
Output of sequencing
The output consists of millions of short DNA reads, typically stored in FASTQ files containing:
DNA sequences
quality scores for each base
These reads are then assembled or mapped to a reference genome to analyze genetic variation related to disease resistance.
3. What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs!
For my project, I would like to synthesize DNA that enables the production of a honeybee protein relevant to resistance against Varroa mite infection, specifically a codon-optimized fragment of the vitellogenin gene for expression in E. coli. Producing this protein in a laboratory system would allow further investigation of its structure and function and could support future research on improving honeybee resilience, which is crucial for pollination, biodiversity, and food production.
4. What technology or technologies would you use to perform this DNA synthesis and why? What are the essential steps of your chosen sequencing methods? What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Modern DNA synthesis relies on chemical oligonucleotide synthesis combined with enzymatic assembly. Short DNA fragments are chemically synthesized and then assembled into longer genes. This method is efficient, scalable, and allows full customization of DNA sequences, including codon optimization and removal of unwanted restriction sites.
Essential steps of DNA synthesis
Digital sequence design of the gene or construct.
Chemical synthesis of short DNA oligonucleotides.
Assembly of oligos into longer DNA fragments using enzymatic methods.
Error correction and amplification of assembled fragments.
Cloning into plasmids and propagation in bacteria.
Sequence verification to confirm correctness before delivery.
Limitations of this method
Speed: Gene synthesis can take days to weeks depending on sequence length and complexity.
Accuracy: Errors can occur during synthesis or assembly, especially in repetitive or GC-rich sequences, requiring verification and correction.
Scalability: Although modern platforms are highly scalable, very long DNA constructs or entire genomes remain costly and technically challenging.
Sequence constraints: Certain sequences (e.g., strong repeats or toxic genes) can be difficult to synthesize or maintain in host organisms.
5. What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Once again I would want to edit DNA related to honeybee health, specifically genes that contribute to resistance against Varroa mite infestation. Potential edits could focus on genes involved in immune response, grooming behavior, or parasite detection, enhancing bees’ natural ability to remove mites or better tolerate infections transmitted by them. Instead of introducing entirely new traits, the goal would be to support or amplify naturally occurring resistance traits, similar to selective breeding but with greater precision.
More broadly, responsible DNA editing could also be applied in agriculture and conservation to help organisms adapt to climate change, reduce pesticide use, and improve resilience in vulnerable ecosystems.
6. What technology or technologies would you use to perform these DNA edits and why? How does your technology of choice edit DNA? What are the essential steps? What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? What are the limitations of your editing methods (if any) in terms of efficiency or precision?
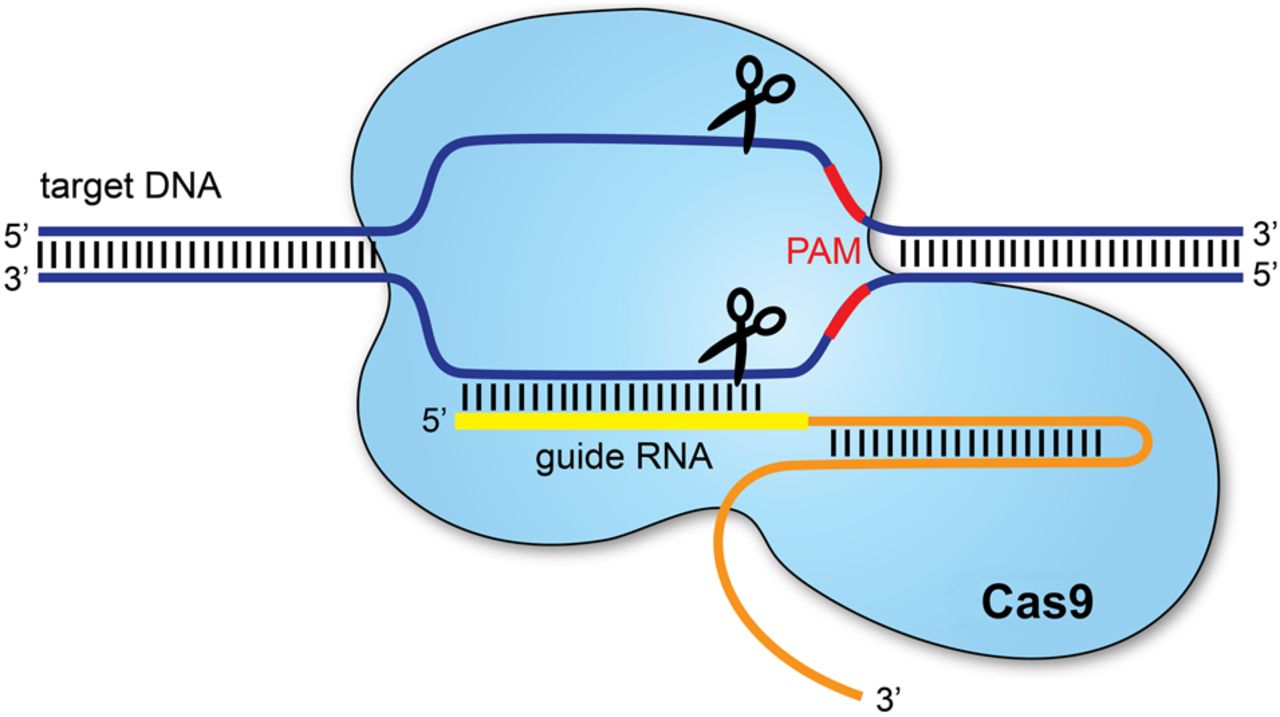
I would use CRISPR–Cas9, which allows precise modification of genes within an organism’s genome. This technology is widely used because it is relatively simple, efficient and adaptable to many organisms.
CRISPR–Cas9 edits DNA by using a guide RNA (gRNA) that directs the Cas9 enzyme to a specific DNA sequence. Cas9 then creates a cut at the targeted location. The cell’s natural DNA repair mechanisms repair this cut, and during repair, scientists can either disable a gene or insert a modified DNA sequence.
Essential editing steps include:
Designing a guide RNA targeting the gene of interest.
Delivering the guide RNA and Cas9 enzyme into cells.
Cas9 cutting the DNA at the chosen site.
Cellular repair mechanisms introducing deletions, modifications, or inserting new DNA.
Screening cells or organisms to confirm successful edits.
Preparation and required inputs
Before editing, we must design the genetic modification and ensure the target gene is well characterized. Required inputs typically include:
Guide RNA sequences targeting the gene
Cas9 enzyme or Cas9-encoding plasmid
A donor DNA template if inserting new sequences
Delivery system (e.g., plasmids, viral vectors, or microinjection)
Target cells or embryos to be edited
Limitations
The main limitations of CRISPR/Cas9 relate to delivery, accuracy and ethical concerns. A major challenge is safely delivering the editing system into the correct cells in living organisms, as current delivery vectors have size or efficiency limitations. Another concern is off-target effects, where unintended parts of the genome may be edited, potentially causing harmful consequences such as cancer. Editing efficiency can also vary, meaning not all cells receive the desired modification. Additionally, editing germline cells or embryos raises significant ethical and long-term safety concerns, since changes would be inherited by future generations and their consequences are uncertain. 6
6: Redman, M., King, A., Watson, C., King, D. (2016) What is CRISPR/Cas9?, Archives of Diseases in Childhood, 101, 213–215. doi:10.1136/archdischild-2016-310459
Week 3 HW: Lab Automation
Part I: Python Script for Opentrons Artwork
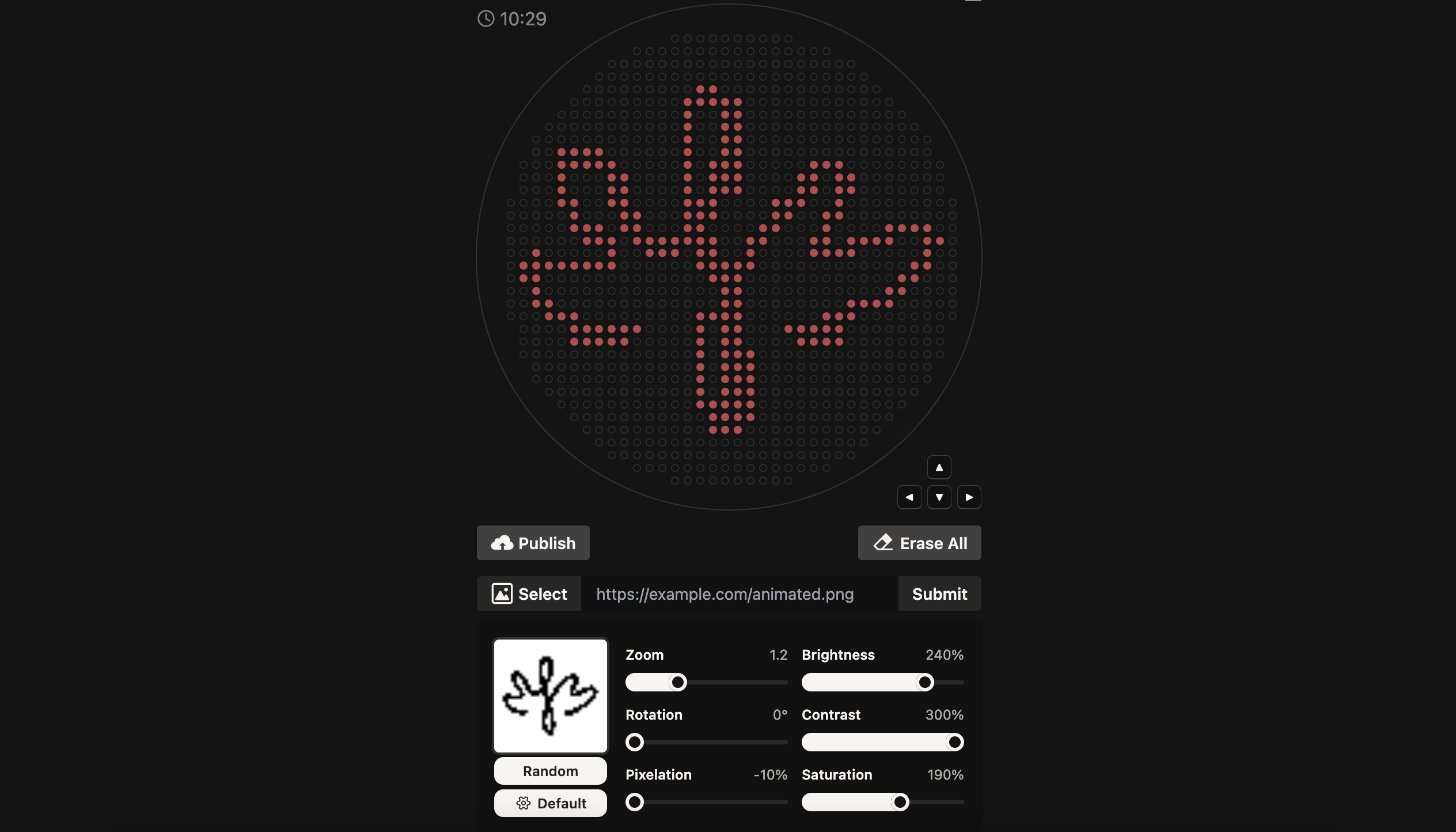
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Firstly, I used Ronan’s Automation Art Interface to translate my logo into a pixelated biological artwork. The software converted the image into a set of coordinate outputs, where each tuple (x, y) represents the precise millimeter offset from the calibrated center of the agar plate. Each of these coordinate pairs defines the placement of a single 1 µL droplet, allowing the robot to reconstruct the digital logo physically on the plate.
I then transferred these coordinates into Google Colab, where I programmed the Opentrons protocol. Before beginning the patterning process, the robot picks up a single 20 µL pipette tip. Since the entire artwork is executed in one color, only one tip is required for the whole procedure, minimizing material use.
The main challenge was preventing the pipette from exceeding its 20 µL capacity. Because each droplet dispenses 1 µL and the artwork consists of many coordinate points, the system must repeatedly refill the pipette. However, simply aspirating 20 µL at fixed intervals can lead to overfilling if residual liquid remains inside the tip.
To solve this, I implemented a volume-tracking mechanism in the code with support of AI (ChatGPT). A variable continuously monitors the remaining liquid in the pipette. The robot only aspirates when the remaining volume reaches zero, and it calculates the exact amount needed—either the full 20 µL capacity or just the remaining volume required to complete the artwork. After each 1 µL dispense, the tracked volume is reduced accordingly. This ensures that the pipette never exceeds its capacity while allowing the artwork to be executed seamlessly and efficiently.
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
A suitable published paper that utilizes an automation tool for a novel biological application is:
DeRoo, J. B., Jones, A. A., Caroline K. Slaughter, C. K., Ahr, T. W., Stroup, S. M., Thompson, G. B., Snow, C. D. (2025) Automation of protein crystallization scaleup via Opentrons-2 liquid handling. SLAS Technology, 32, https://doi.org/10.1016/j.slast.2025.100268
This study employs the Opentrons OT-2 automated liquid handling robot to optimize protein crystallization workflows. Protein crystallization is a critical step in structural biology, particularly for determining protein structures via X-ray crystallography. Traditionally, crystallization screening requires extensive manual pipetting, which is time-consuming, error-prone, and difficult to standardize.
The novelty of the paper lies in:
Low-cost automation of crystallization screening
The researchers adapted the open-source OT-2 robot to perform precise nanoliter- to microliter-scale liquid handling for setting up crystallization trials. This democratizes access to automation, as most conventional crystallization robots are prohibitively expensive.
Workflow optimization and reproducibility
The study demonstrates how automated pipetting improves reproducibility and throughput compared to manual methods. It allows systematic variation of crystallization conditions (e.g., precipitant concentration, pH, additives) in a controlled and programmable manner.
Open-source customization
A key contribution is the development of customizable protocols that other laboratories can replicate and modify. The paper highlights how open-source hardware and software can accelerate biological research innovation without reliance on proprietary systems.
Biological Impact
By automating crystallization optimization:
Screening becomes more efficient and scalable.
Experimental variability is reduced.
Smaller laboratories gain access to structural biology techniques that were previously limited to well-funded institutions.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
Rather than relying on manual trial-and-error biology, I aim to build programmable experimental pipelines using robotic liquid handling, custom labware, and computational control for my final project. I intend to use modular lab automation tools to develop reproducible, scalable biological systems.
How automation would be used
High-Throughput Construct Assembly
Using a modular liquid handler such as the Opentrons OT-2:
Automated Golden Gate or Gibson assemblies
Parallel plasmid construction
Controlled transformation setups
Replicate culture inoculations
Example pseudocode:
for construct in plasmid_library: pipette.transfer(2, promoter_plate[construct], assembly_well) pipette.transfer(2, gene_plate[construct], assembly_well) pipette.transfer(6, master_mix, assembly_well)
This would allow rapid screening of different antimicrobial peptide constructs, dsRNA delivery systems, or immune-modulating pathways.
Custom 3D-Printed Bee Microbiome Holders
I would design:
3D-printed micro-incubation chambers
Gut-simulating microfluidic inserts
Controlled feeding modules
Ginkgo Nebula Integration
Through Ginkgo Bioworks’s Nebula platform, I could:
Analyze microbiome sequencing data
Identify candidate symbionts
Screen gene clusters linked to antimicrobial production
Compare engineered vs natural strain performance
To put it in a nutshell, lab automation transforms these projects from speculative ideas into experimentally rigorous, iterative engineering systems.
Part III: Final Project Ideas
As explained in this week’s recitation, add 1-3 slides in your Node’s section of this slide deck with 3 ideas you have for an Individual Final Project. Be sure to put your name, city, and country on your slide!
Week 4 HW: Protein Design Part I
Part I: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
$1 Dalton = 1 g/mol$
The protein content of meat is about 20%.1 That means 500g of meat contain 100g protein.
$100g/100g/mol=1mol$
$1mol = Avogadro_constant = 6*10^{23}$
Under the given assumption your intake of molecules of amino acids equals the value of the avogadro constant.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
You don’t become what you eat because your body breaks food down into basic building blocks, then rebuilds them according to your own DNA instructions.
3. Why are there only 20 natural amino acids?
There aren’t only 20 natural amino acids. Two additional amino acids exist in nature:
Selenocysteine (the 21st)
Pyrrolysine (the 22nd)
For humans the 20 basic amino acids are essential for surviving. There are 20 canonical ones because evolution selected a set that is chemically sufficient, efficient, and stable.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes. Although natural proteins are built from only 20 amino acids, nature has expanded this repertoire by genetically encoding Selenocysteine and Pyrrolysine through stop codon reassignment. Inspired by this flexibility, scientists have engineered the genetic code to incorporate many non-natural amino acids, greatly expanding the chemical and functional diversity available in protein design. 2
5. Where did amino acids come from before enzymes that make them, and before life started?
Before life and enzymes existed, amino acids were formed through abiotic chemical processes—that is, purely chemical reactions in the environment. There are a few major sources scientists think contributed:
Atmospheric and lightning synthesis: Simple gases like CH₄, NH₃, H₂, and H₂O could react under energy input (like lightning or UV light) to form amino acids. This was famously demonstrated in the Miller–Urey experiment.3
Hydrothermal vents: Hot, mineral-rich water at the ocean floor could drive chemical reactions that produce amino acids from simple carbon and nitrogen compounds.4
Extraterrestrial delivery: Amino acids have been found in meteorites, suggesting that some were formed in space and brought to Earth.4
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
It would be left‑handed.
If you make an α-helix using D-amino acids instead of the natural L-amino acids, the helix will have the opposite handedness. Natural α-helices made of L-amino acids are right-handed. α-helices made of D-amino acids will be left-handed.
This happens because the chirality at the α-carbon determines the backbone geometry, so inverting the stereochemistry flips the helix’s screw direction. 5
7. Can you discover additional helices in proteins?
Yes, additional helices beyond the classical α-helix can exist and have been discovered or designed. Besides the common α-helix, proteins can form structures like the 3₁₀ helix and the π-helix, which differ in hydrogen bonding patterns and backbone geometry. Moreover, by incorporating non-natural or β-amino acids, researchers have created entirely new helical structures (“foldamers”) that do not occur in natural proteins.
So both nature and protein engineers can access additional helical architectures by altering backbone chemistry or hydrogen-bonding patterns.
8. Why are most molecular helices right-handed
Because most helices consist of only L-amino acids. Most molecular helices, including α-helices in proteins and the DNA double helix, are right-handed because of the chirality of their building blocks. 5
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their extended backbone and repetitive hydrogen-bonding pattern allow multiple strands to stack together in a very regular way.
Why β-sheets aggregate: The exposed edges of a β-sheet present backbone C=O and N–H groups already arranged in the correct geometry to hydrogen bond with any compatible β-strand they encounter, so two sheets or strands can zip together without major rearrangement
What is the driving force: Backbone hydrogen bonding, Hydrophobic interactions, Van der Waals forces 6
Part II: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I selected the protein with UniProt accession A0A0F6TNJ6_DWV, which is part of the genome polyprotein of Deformed wing virus (DWV), a positive-strand RNA virus that infects honey bees and contributes to wing deformities and colony collapse. DWV expresses its proteins as one large polyprotein that is later cleaved into functional units including capsid and enzymatic proteins, and I chose it because viral polyproteins illustrate how a single chain can generate multiple sub-proteins with distinct 3D structures and functions that you can visualize in molecular viewers.
2. Identify the amino acid sequence of your protein.
The most common amino acid is: V, which appears 26 times.
homologs: There are 24 protein sequence homologs for this protein
family: The protein A0A0F6TNJ6_DWV belongs to the Iflavirus polyprotein family, which is part of the larger Picornavirus-like superfamily of positive-sense single-stranded RNA viruses.
3. Identify the structure page of your protein in RCSB.
Structure of deformed wing virus, a honeybee pathogen: 5L7Q | pdb_00005l7q
2017
Resolution 3.50 Å
vp1, vp2, vp3
Classification: VIRAL PROTEIN
4. Open the structure of your protein in any 3D molecule visualization software:
Part III: Using ML-Based Protein Design Tools
Deep Mutational Scans
1a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
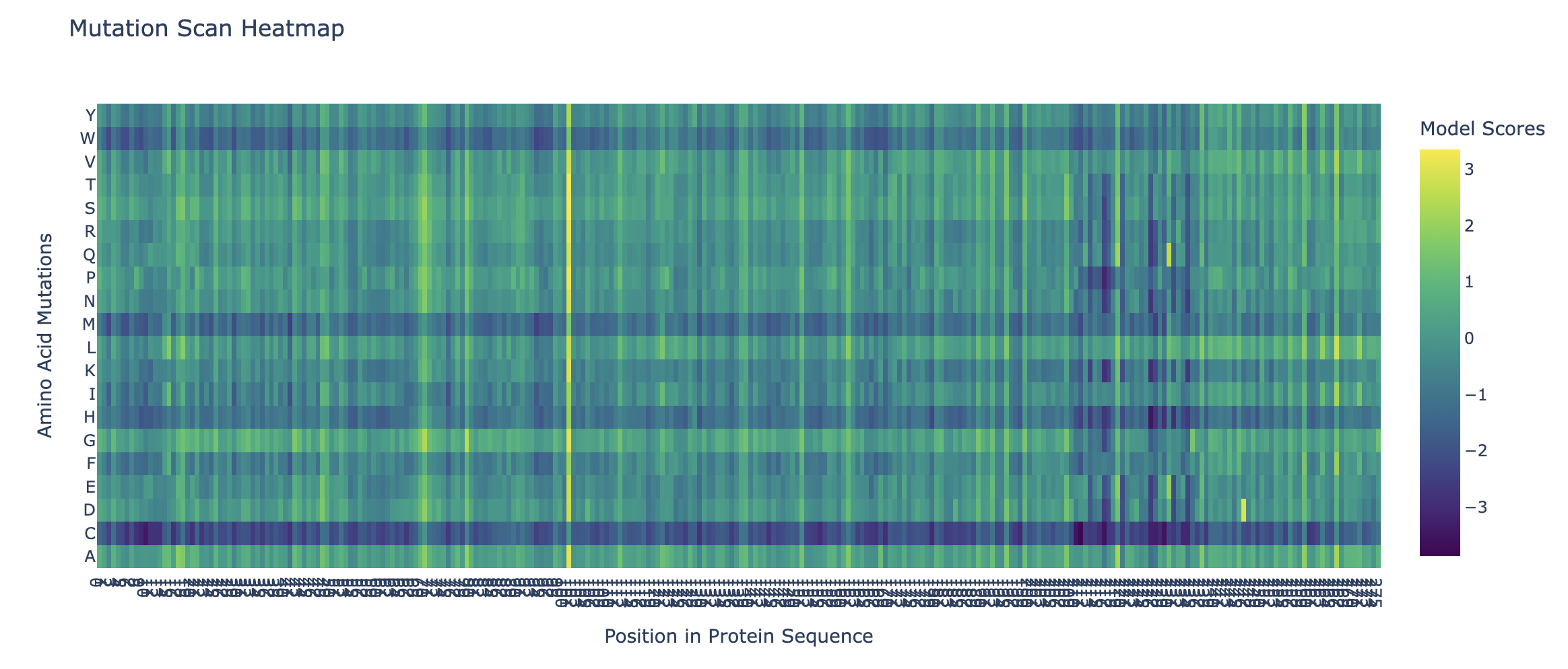
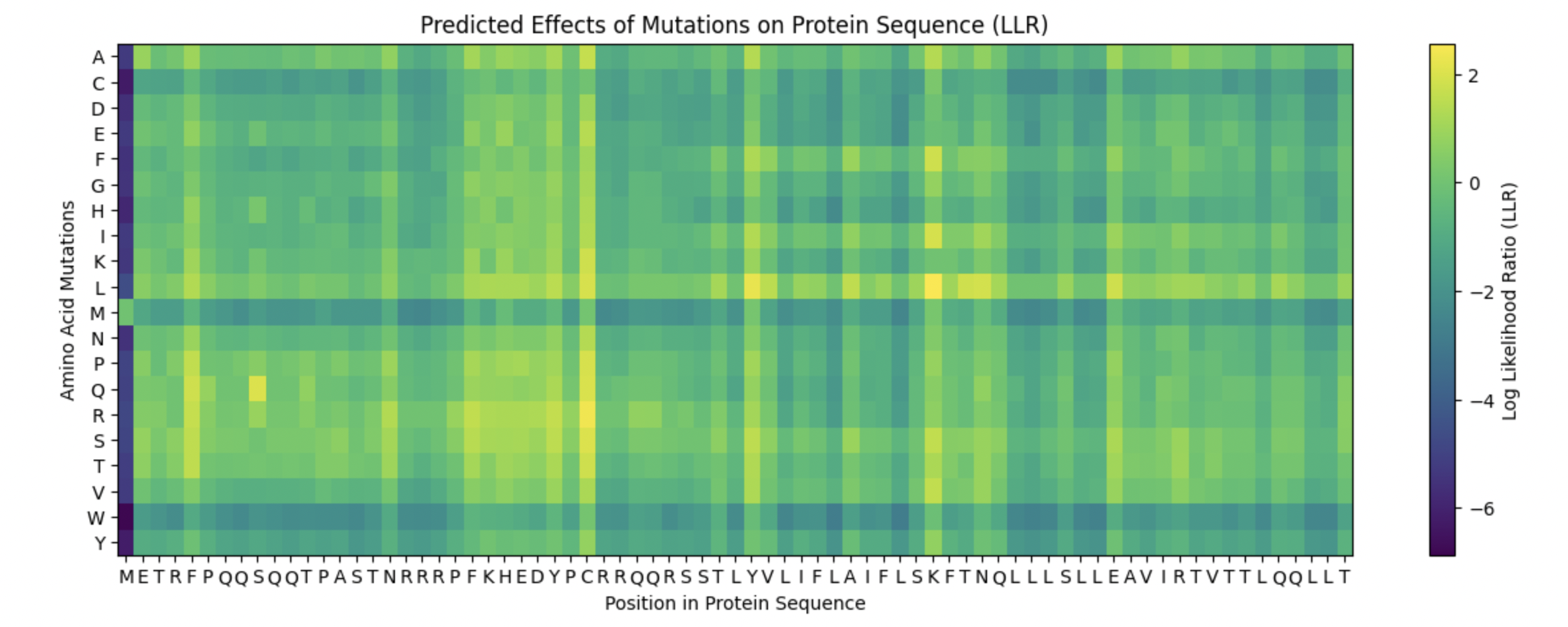
The heatmap of my capsid protein:
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
You can clearly observe:
Some vertical stripes (columns) are very purple
These positions are mutation-intolerant.
Likely structurally or functionally critical residues.
Some regions are mostly green
These are mutation-tolerant regions.
Likely surface-exposed loops or flexible regions.
The Cysteine row is strongly purple across many positions as well as Methionine and Tryptophan. This means that these three amino acids disfavor the occurence of mutations.
Looking at the colums there is one that is specifically interesting but i can’t see the x-axis right.
Latent Space Analysis
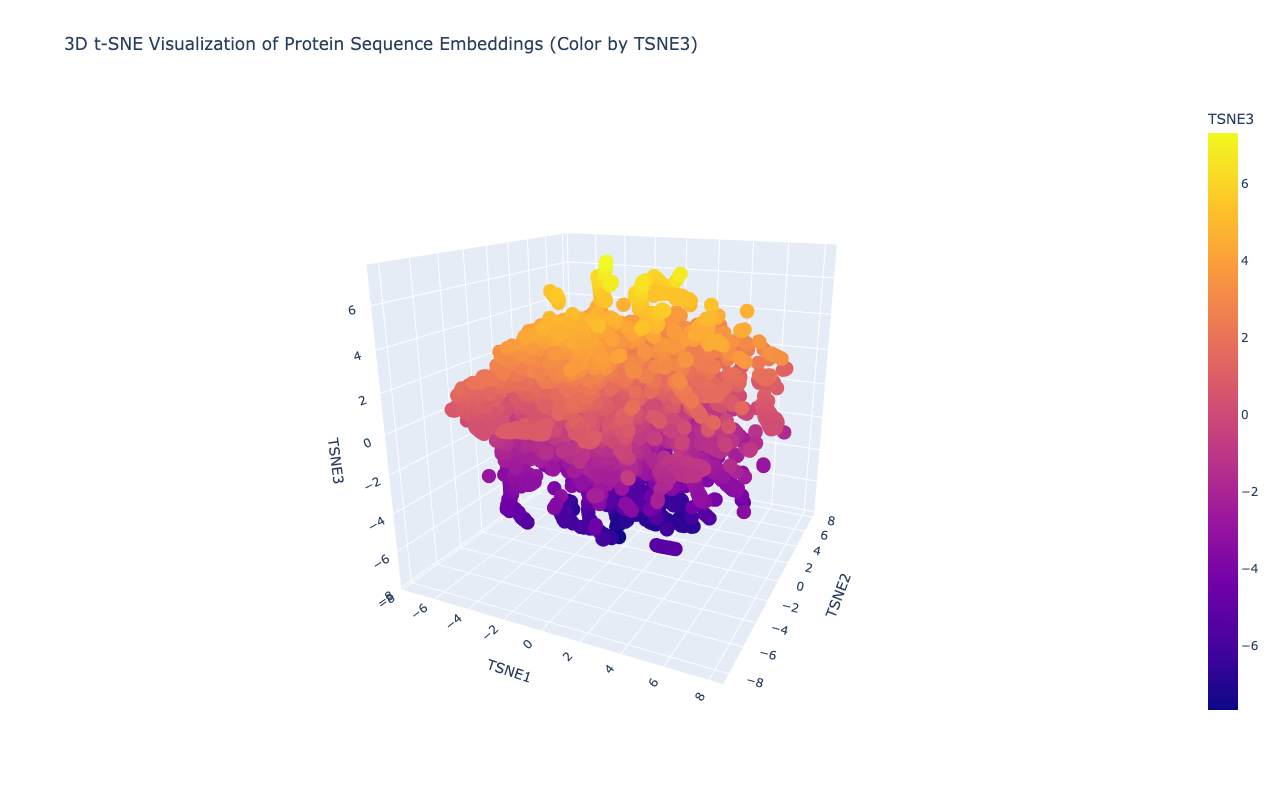
2a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
Shape of embeddings array after 3D t-SNE: (15177, 3)
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes,typically they do. In protein language model embeddings:
Proteins from the same family cluster together
Proteins with similar functions cluster
Proteins with similar folds cluster
e.g. Viral polyproteins cluster with other viral polyproteins
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
After embedding the protein sequences into a reduced latent space, distinct neighborhoods emerged that correspond to proteins with similar evolutionary and functional characteristics. Proteins belonging to related viral families cluster closely, indicating that the language model captures conserved sequence motifs and domain architecture. My selected protein, the Deformed Wing Virus polyprotein (A0A0F6TNJ6_DWV), clusters with other positive-sense RNA viral polyproteins, reflecting shared conserved domains such as RNA-dependent RNA polymerase and viral protease regions.
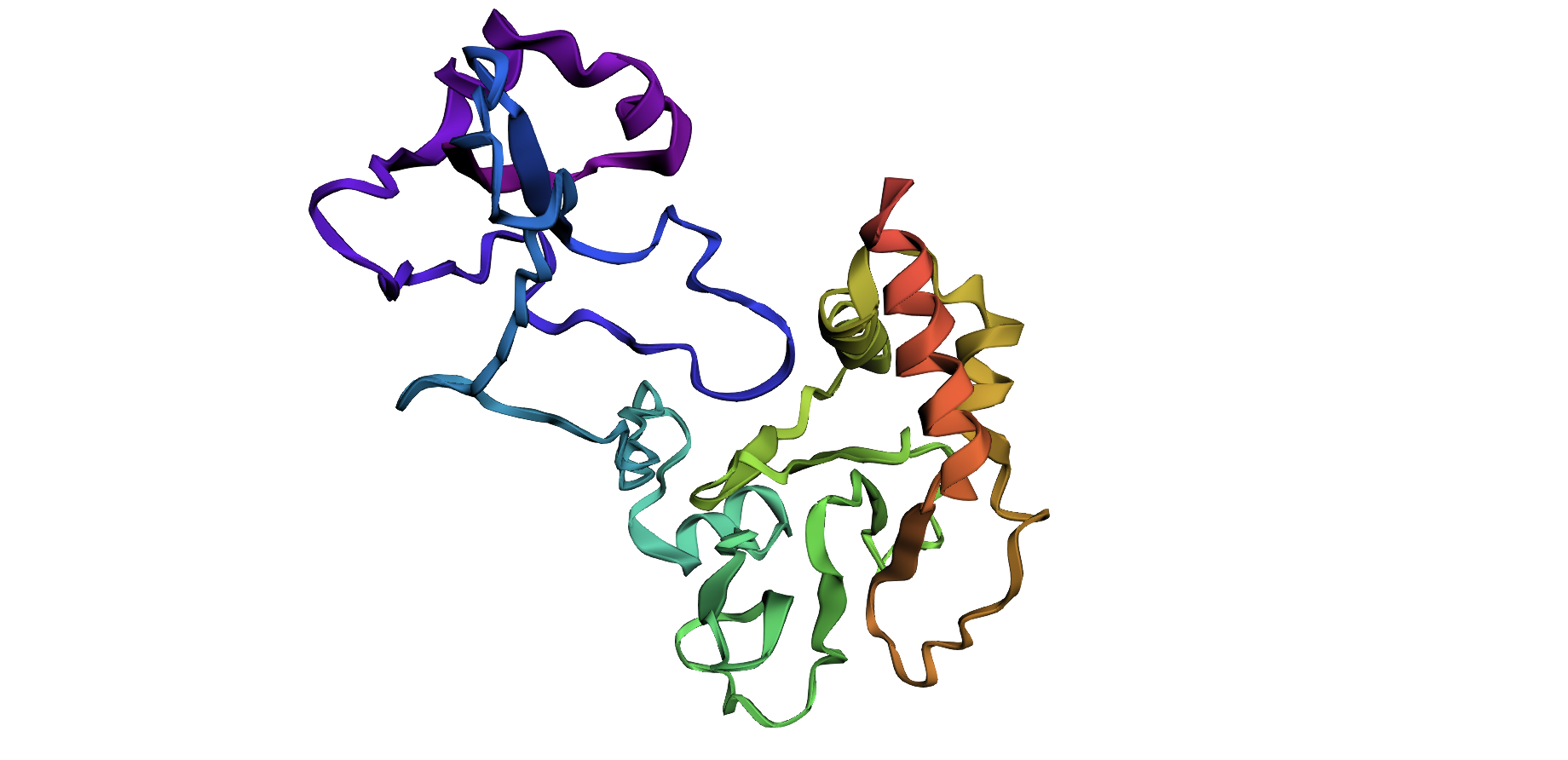
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Total sequence length: 278
Running ESMFold inference for sequence with length 278…
Prediction complete. ptm: 0.329 plddt: 46.496
Results saved to test_877d8/
CPU times: user 27.2 s, sys: 8.79 s, total: 36 s
Wall time: 1min
Structure display:
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I changed the sampling temperature to 0.5 and the number of sequences to 4. I
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
Zeng, Y., Chen, E., Zhang, X., Li, D., Wang, Q., & Sun, Y. (2022). Nutritional Value and Physicochemical Characteristics of Alternative Protein for Meat and Dairy—A Review. Foods, 11(21), 3326. https://doi.org/10.3390/foods11213326↩︎
Zitti, A., Jones, D. (2023). Expanding the genetic code: a non-natural amino acid story. The Biochemist, 45(1), 2–6. https://doi.org/10.1042/bio_2023_102↩︎
Ring D, Wolman Y, Friedmann N, Miller S.L. (1972) Prebiotic synthesis of hydrophobic and protein amino acids. PNAS, 69(3), 765-8. https://doi.org/10.1073/pnas.69.3.765↩︎
Higgs P.G., Pudritz R.E. (2009). A Thermodynamic Basis for Prebiotic Amino Acid Synthesis and the Nature of the First Genetic Code. Astrobiology, 9(5), 483-490. https://doi.org/10.1089/ast.2008.0280↩︎↩︎
Hoang, H. N., Abbenante, G., Hill, T. A., Ruiz-Gómez, G., Fairlie, D. P. (2012). Folding pentapeptides into left and right handed alpha helices. Tetrahedron, 68(23), 4513-4516, https://doi.org/10.1016/j.tet.2011.10.108↩︎↩︎
Richardson, J. S., Richardson, D.C. (2002). Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. PNAS, 99(5), 2754-9. https://doi.org/10.1073/pnas.052706099↩︎
Week 5 HW: protein design part ii
Part A: SOD1 Binder Peptide Design
Part I: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
A4V Mutation -> means one needs to change the A at position 4 to an V. This protein sequence only had one at the fifth position so I changed this Alanine.
2. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
1 KRVYVVVVEHKE 43.699289
2 WRYPVVAAALGX 7.252220
3 WLYYPAAVELGX 10.791046
4 HRYYPTAVRHWK 13.771030
5 FLYRWLPSRRGG
The aim of PepMLM is to finetune the ESM-2 protein language model to fully reconstruct the binder region, achieving low perplexities matching or improving upon validated peptide–protein sequence pairs. The lower the perplexity, the better the model.
Perplexity in protein models measures how well a language model predicts the next amino acid in a sequence, acting as a proxy for how “natural” or physically plausible a protein sequence is. Lower perplexity indicates the model understands the protein structure constraints better, frequently used to estimate sequence fitness and stability.1
1.: Kantroo, P., Wagner, G. P., Machta, B. B. (2025) Pseudo-perplexity in one fell swoop for protein fitness estimation, PRX Life 3, https://doi.org/10.1103/zhx7-hcmm
Part II: Evaluate Binders with AlphaFold3
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
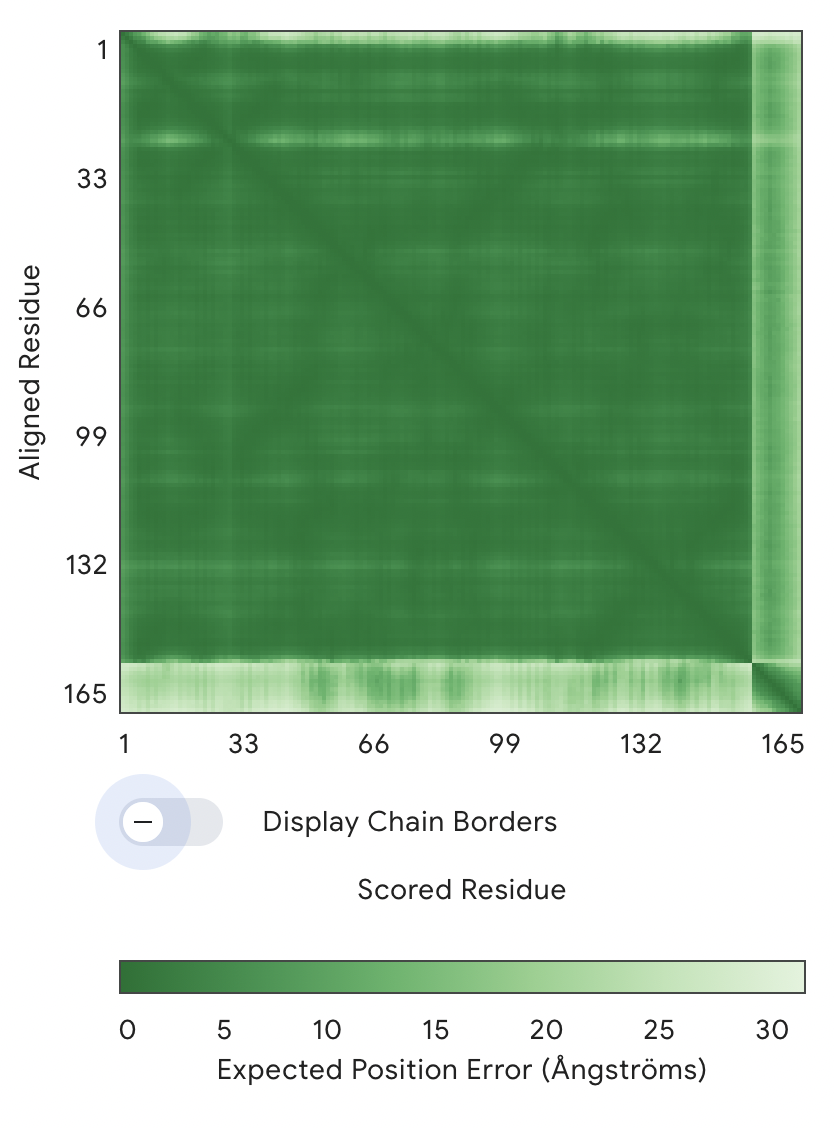
1: ipTM = 0.34, pTM = 0.88
PAE Graph
The main large green square (1–154 residues) = SOD1 protein
The thin strip on the right/bottom (~154–165) = peptide
Key observation:
The SOD1–SOD1 region is dark green → very low error → high confidence structure
The peptide vs SOD1 region is much lighter (higher error) → low confidence in relative positioning
→ AlphaFold is uncertain where the peptide sits
→ The interaction is likely weak, flexible, or non-specific
As a result, no specific binding site can be confidently assigned. The peptide does not appear to localize near the N-terminal region where the A4V mutation is located, nor is there clear evidence that it engages the β-barrel core or the dimer interface. Instead, the peptide appears to be loosely associated with the protein surface, likely in a flexible and non-specific manner rather than forming a stable, partially buried interaction.
For the next two peptides, I replaced the Xs with As for 2 and 3. A X means there is not amino acid defined.
2: ipTM = 0.37, pTM = 0.83
3: ipTM = 0.36, pTM = 0.83
4: ipTM = 0.26, pTM = 0.84
5: ipTM = 0.39, pTM = 0.83
Overall, all the ipTM scores I received weren’t convincing. ipTM (interface predicted TM-score) measures how confidently AlphaFold predicts the interaction between chains -> the lower the worse the prediction.
Part III: Evaluate Properties of Generated Peptides in the PeptiVerse
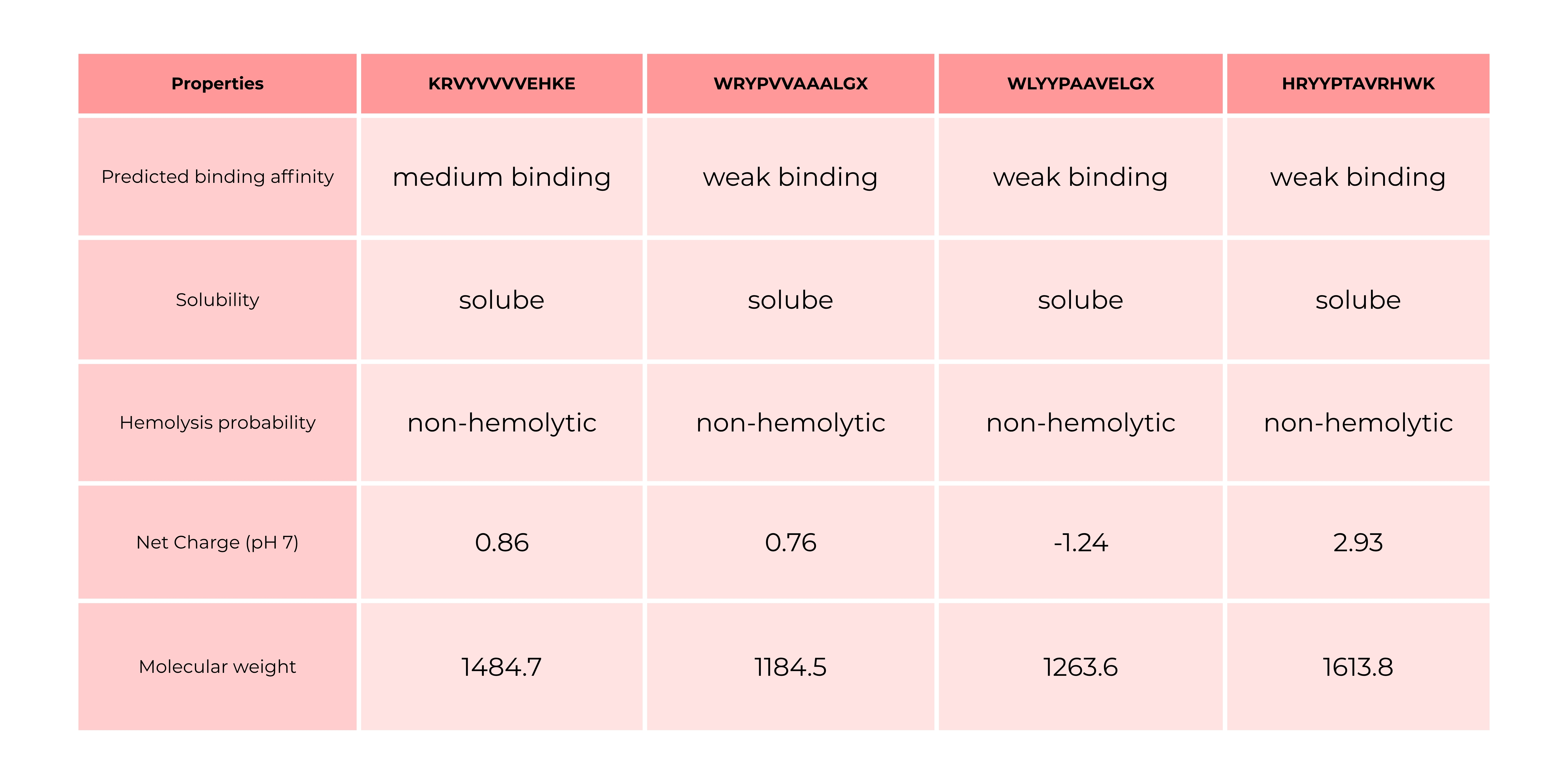
PeptiVerse is a universal therapeutic peptide property prediction platform.
The comparison between AlphaFold3 predictions and PeptiVerse results shows that structural confidence (ipTM) does not necessarily correlate with predicted binding affinity. Peptides with higher ipTM scores did not consistently exhibit stronger predicted affinity, highlighting differences between structure-based and sequence-based approaches. Interestingly, the only peptide predicted to have medium binding affinity was the one with an ipTM of 0.34 and a perplexity of 43, which were the highest values among the tested candidates. All other peptides were predicted to bind only weakly, even in cases where they showed comparable or slightly better ipTM and perplexity scores. This suggests that neither ipTM nor perplexity alone is sufficient to reliably predict binding strength, and that combining multiple evaluation methods is necessary for a more comprehensive assessment.
In that case, I would go with peptide 1 because it binds the best.
Part IV: Generate Optimized Peptides with moPPIt
1: KKTKTYKETRGD
2: RTGSETGTEEKY
3: TKTKRERGYNKQ
4: QATKKKKETNKE
The moPPit peptides differ significantly from the PepMLM-generated peptides in both composition and physicochemical properties. While the PepMLM peptides contain a mix of hydrophobic and aromatic residues (e.g., W, Y, V, L), suggesting potential for structured binding and interaction with hydrophobic regions of SOD1, the moPPit peptides are dominated by charged and polar residues, particularly lysine (K), arginine (R), and glutamic acid (E). This makes the moPPit peptides more hydrophilic and likely more soluble, but also suggests that they may interact more nonspecifically through electrostatic interactions rather than forming well-defined binding interfaces.
In contrast, the PepMLM peptides appear more “protein-like” and may be better suited for specific binding, although some sequences include undefined residues (X), introducing uncertainty. The moPPit peptides, with their high positive charge, may resemble cell-penetrating or antimicrobial peptides, which can increase membrane interaction but also raise concerns about cytotoxicity or hemolysis.
Before advancing any of these peptides toward clinical studies, a comprehensive evaluation would be required. This would include computational validation such as structure prediction (e.g., via AlphaFold) and binding assessment, as well as sequence-based predictions of solubility, toxicity, and hemolysis (e.g., using PeptiVerse). Promising candidates should then be tested experimentally through in vitro assays to measure binding affinity, stability, and aggregation effects on SOD1. Additionally, toxicity assays, including hemolysis and cell viability tests, would be essential to assess safety.
Solube Region is around 1 - 39: So the S and C Mutations
Transmembrane Region is afterwards 40 - 83: So the K Mutations
Among the proposed mutations, K50I and K50F are strong candidates in the transmembrane region, as they replace a charged lysine with hydrophobic residues, which is more compatible with the membrane environment and likely stabilizes the helix. In the soluble region, S9Q is a safe, conservative mutation that maintains polarity and could enhance hydrogen bonding, while C29L and C29R are riskier, since C29 may form disulfide bonds or contribute to structural stability; replacing it with hydrophobic or charged residues could destabilize the protein. Overall, K50I, K50F, and S9Q are likely effective mutations, while the C29 variants could be informative but carry higher structural risk.
Week 6 HW: Genetic Circuits Part I
DNA Assembly
Answer these questions about the protocol in this week’s lab:1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Typical components include:
Phusion DNA Polymerase
A high-fidelity enzyme that synthesizes new DNA strands with very low error rates (proofreading activity → fewer mutations).
dNTPs (deoxynucleotide triphosphates)
Building blocks (A, T, G, C) used to create new DNA strands.
Reaction Buffer (HF buffer)
Maintains optimal pH and salt conditions for enzyme activity and fidelity.
Mg²⁺ ions (magnesium chloride)
Essential cofactor for polymerase activity; affects enzyme efficiency and specificity.
Stabilizers (sometimes included)
Help maintain enzyme stability during thermal cycling.
2. What are some factors that determine primer annealing temperature during PCR?
Annealing temperature is critical for specificity. It depends on:
Primer melting temperature (Tm)
Main determinant; annealing temperature is usually ~2–5°C below the lowest Tm.
*Primer length
Longer primers → higher Tm.
GC content (40–60% ideal)
More G/C = stronger binding → higher Tm.
Sequence composition
GC-rich regions bind more tightly than AT-rich ones.
Salt concentration in buffer
Higher salt stabilizes DNA duplexes → increases effective Tm.
Primer mismatches
Intentional mismatches (like your mutation) can slightly lower binding strength.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR
Protocol:
Uses primers + polymerase to amplify a specific DNA region
Can introduce mutations (e.g., your chromophore changes)
No need for restriction sites
Very flexible and precise
Limitations:
Can introduce errors (though minimized with high-fidelity enzymes)
Requires careful primer design
Restriction Enzyme Digest
Protocol:
Uses enzymes that cut DNA at specific recognition sites
Produces sticky or blunt ends
Advantages:
Highly specific and reproducible
No amplification errors
Limitations:
Requires existing restriction sites
Leaves “scars” (extra sequences)
Less flexible for mutations
When one wants to mutate Dna and doesn’t have suitable restriction sites, one uses PCR. However, if one wants a simple, clean cloning and suitanle restriction sites already exist, one uses Restriction Enzyme Digest.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure compatibility:
Design overlapping regions (20–40 bp): Adjacent fragments must share identical sequences.
Correct orientation (5′ → 3′): Overlaps must align properly for assembly.
Accurate primer design: Overhangs must match the adjacent fragment exactly.
High-quality DNA fragments: Clean PCR products (no contaminants or leftover template).
Verify fragment sizes (gel electrophoresis): Confirms correct amplification.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Cells are made chemically competent (membrane destabilized).
DNA is added and incubated on ice → DNA associates with membrane.
Heat shock (42°C) creates temporary pores in the membrane.
DNA enters the cell through these pores (diffusion-driven).
Cells recover in SOC media and begin expressing the plasmid (e.g., antibiotic resistance).
6. Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly uses Type IIS restriction enzymes (e.g., BsaI) that cut DNA outside of their recognition sequence, generating custom overhangs. These overhangs allow multiple DNA fragments to be assembled in a specific order in a single reaction. Unlike Gibson Assembly, Golden Gate uses a cut-and-ligate mechanism, where restriction enzyme digestion and ligation occur simultaneously in a thermocycling reaction. Because the recognition sites are removed during assembly, the final construct is scarless. This method is highly efficient for assembling multiple fragments (e.g., synthetic biology constructs). It is especially useful for modular cloning systems. However, it requires that internal restriction sites be removed beforehand.
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembbly Method needs to be added
b. Model this assembly method with Benchling or Asimov Kernel!
N/A because we don’t have access.
Week 7 HW: Genetic Circuits Part II
Part I: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Continuous Signal Processing
Boolean circuits output only 0 or 1 (OFF/ON) states.
IANNs operate on continuous, graded gene expression levels.
Ability to Model Complex Relationships
Boolean logic is limited to simple combinations of AND/OR/NOT gates.
IANNs can approximate complex, nonlinear input–output functions.
Efficient Integration of Multiple Inputs
Boolean circuits: combining many inputs requires many layers of logic gates, which increases circuit size and burden.
IANNs: Combininbg inputs through weighted interactions means processing many signals in parallel.
IANNs outperform Boolean genetic circuits by enabling continuous, tunable, and scalable processing of complex, multi-input signals, making them more robust and biologically realistic.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Idea: An intracellular artificial neural network (IANN) can be engineered into bacteria to detect and degrade textile dye pollutants in wastewater, while adapting its response to varying environmental conditions.
Inputs (continuous signals):
The IANN processes multiple environmental inputs simultaneously, for example:
Dye concentration (e.g., azo dyes)
Toxic byproducts (aromatic amines)
pH levels
Oxygen availability
Presence of heavy metals or inhibitors
Each input is graded (not just present/absent), allowing the system to distinguish between low, medium, and highly polluted water.
Processing (IANN behavior):
Each input is assigned a weight (via promoter strength, TF affinity, etc.).
The network integrates signals and computes a nonlinear response:
Optional: reporter signals (color/fluorescence to indicate pollution level)
The output is graded:
Low pollution → minimal enzyme production
High pollution → strong enzyme expression
IANNs are particularly useful for wastewater bioremediation because real wastewater is highly variable and noisy, meaning traditional Boolean circuits would often fail or overreact. Instead, IANNs can integrate multiple weak and continuous signals into a meaningful decision, enabling adaptive and energy-efficient responses. For example, they can activate dye-degrading enzymes only under suitable conditions, such as optimal pH, preventing unnecessary metabolic cost. However, there are limitations: environmental variability may exceed the network’s operating range, and the system can impose a metabolic burden on the host cell. Additionally, crosstalk with native pathways, evolutionary instability over time, challenges in scaling from lab to real-world systems, and biosafety concerns related to releasing engineered organisms must all be considered.
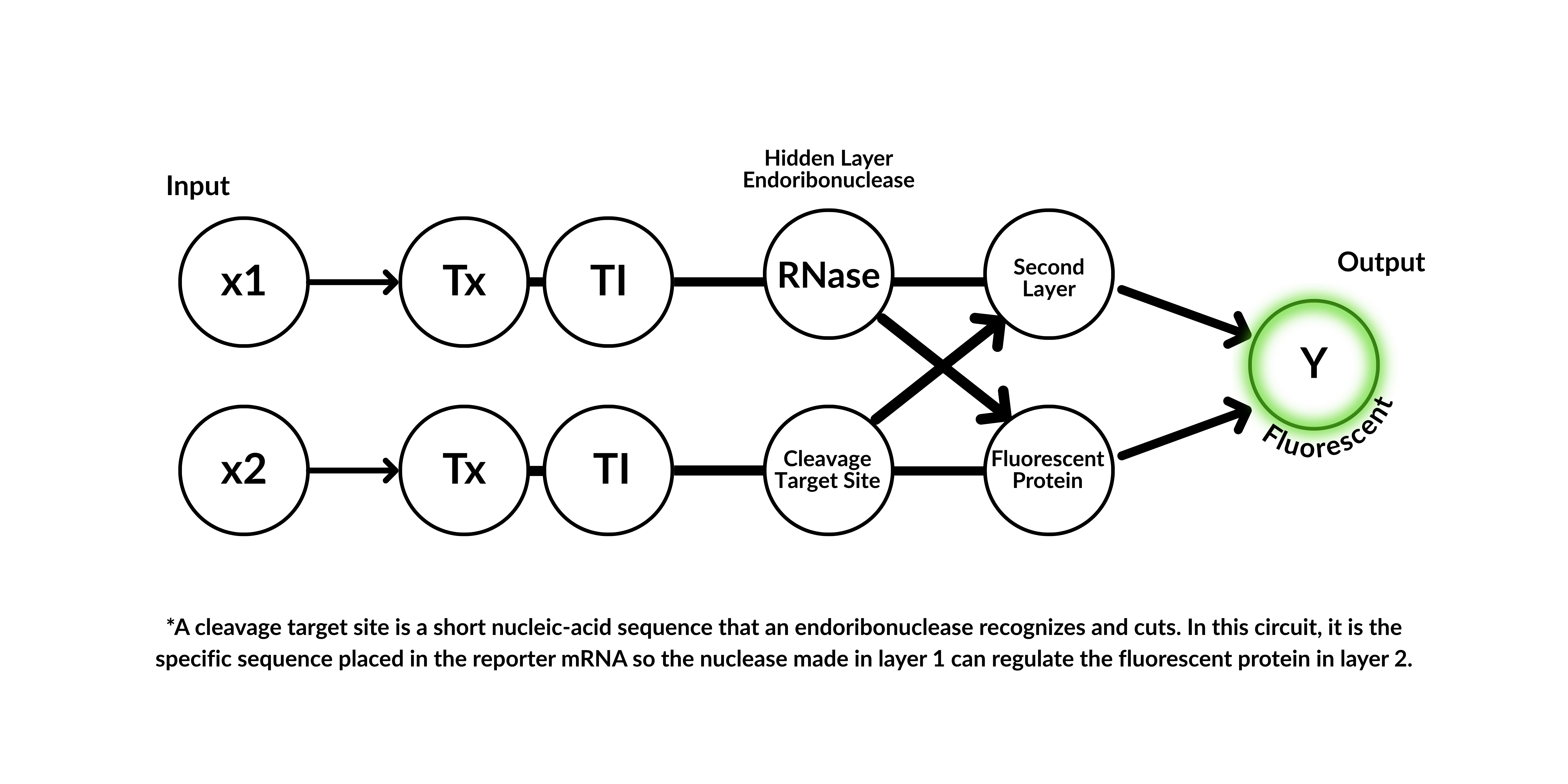
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Definition Csy4 endoribonuclease: Csy4 is a highly specific bacterial CRISPR-associated endoribonuclease from Pseudomonas aeruginosa that processes precursor CRISPR RNA (crRNA) by recognizing and cleaving a 16-nucleotide hairpin stem-loop. It is widely used in biotechnology for controlling gene expression, RNA imaging, and creating inducible gene switches due to its high-affinity RNA binding, including a catalytic H29A mutant that binds but does not cleave.1
Diagram:
Part II: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
mycelium bricks/biocement:
used for: Sustainable construction materials (e.g., insulation panels, lightweight bricks), packaging as an alternative to polystyrene
advantages:
Biodegradable and compostable
Low energy production (grown, not manufactured)
Uses agricultural waste as feedstock
Good insulation properties (thermal and acoustic)
disadvantages:
Lower mechanical strength than conventional bricks or concrete
Sensitive to moisture if not properly treated
Limited structural applications (not yet suitable for load-bearing walls in most cases)
Variability in material properties
mycelium leather:
used for: Fashion products (bags, shoes, clothing), Upholstery and accessories, Alternative to animal leather and synthetic (PU/PVC) leather - because even the common vegan leather is bad for the planet
advantages:
Animal-free and more ethical than traditional leather
Lower environmental impact (less water, no livestock emissions)
Can be grown into desired shapes → reduces waste
Potentially biodegradable (depending on finishing)
disadvantages:
Durability and longevity may be lower than high-quality animal leather
Often still requires chemical treatments or coatings
Not always fully biodegradable in commercial forms
Higher cost and limited large-scale production (still emerging technology)
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
To make them grow in speciic directions. (for building without waste)
To make them grow more structural mycelium. (for creating objects e.g. vases, furniture, …)
To make them degrade environmental pollutants e.g., dyes, plastics, pesticides (fungi naturally secrete powerful enzymes, so engineering them could enhance bioremediation)
To make them improve agricultural systems (for enhancing plant growth, nutrient uptake, or protect against pathogens)
Fungi offer several advantages over bacteria in synthetic biology, particularly due to their ability to form macroscopic, three-dimensional structures, such as mycelium networks, which makes them ideal for applications in construction and textile-like materials. They naturally secrete large amounts of extracellular enzymes, enabling efficient breakdown of complex substrates like lignin or synthetic dyes, which is highly valuable for bioremediation. As eukaryotic organisms, fungi can also perform post-translational modifications, allowing them to produce more complex and functional proteins than bacteria - an important feature for pharmaceutical applications. Additionally, fungi can grow on low-value waste streams, such as agricultural residues, making them especially attractive for sustainable production systems. Their mycelial networks also provide intrinsic material properties, allowing functional materials to be grown directly. However, compared to bacteria, fungi typically grow more slowly, are more difficult to genetically engineer, and currently have fewer standardized synthetic biology tools available, although this field is rapidly advancing.
Part III: First DNA Twist Order
N/A for Lifefabs node because we haven’t had feedback for final project.
Borchardt, E. K., Meganck, R. M., Vincent, H. A., Ball, C. B., Ramos, S. B. V., Moorman, N. J., Marzluff, W. F., Asokan, A. (2017) Inducing circular RNA formation using the CRISPR endoribonuclease Csy4 23(5):619-627. doi: 10.1261/rna.056838.116. ↩︎
Week 9 HW: Cell Free Systems
Part A: General and Lecturer-Specific Questions
General
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
In vitro transcription-translation (TX-TL) can enable faster engineering of biological systems. This speed-up can be significant, especially in difficult-to-transform chassis. It is much easier to modify conditions and fine-tune levels of concentration. 1
1. Toxic Protein Production
Proteins that are toxic to living cells (e.g., membrane-disrupting proteins or toxins) can be safely produced
In in vivo systems, these would kill the host cells before sufficient protein is made
2. Rapid Prototyping / Synthetic Biology
Useful for quickly testing genetic constructs (e.g., promoters, circuits) without cloning and culturing
Ideal for iterative design workflows
Describe the main components of a cell-free expression system and explain the role of each component.
Cell-free expression works through the coupling of transcription (TX) and translation (TL) inside of a test tube.
Transcription from a DNA template to a mRNA -> RNA polymerase
Translation from RNA into protein(s) -> ribosome complex + tRNA
Main Components
Cell extract (lyse): It is composed of the molecular machinery and co-factors need for reactions
ribosome ribonucleic complex
RNA polymerase
other desired proteins
tRNA: Transfers specific amino acids to the ribosome during protein synthesis by matching codons with anticodons.
polymerase: An enzyme (e.g., RNA polymerase) that synthesizes RNA from a DNA template during transcription.
nucleotides: Building blocks of nucleic acids that are used to construct RNA (and DNA).
folic acid: Acts as a cofactor in one-carbon metabolism, supporting nucleotide synthesis and overall metabolic activity.
coenzyme A: Functions as an acyl group carrier, playing a key role in energy metabolism and biochemical reactions.
3-PGA: Serves as an energy source in some cell-free systems by helping regenerate ATP.
RNA template: Provides the direct sequence information for protein synthesis during translation.
hepes buffer: Due to its high solubility, low membrane permeability, and negligible metal ion binding, it is considered a “Good’s” buffer ideal for optimizing biochemical reactions.
spermidine: Stabilizes nucleic acids and enhances transcription and translation efficiency.
sodium oxalate: Acts as a chelating agent that can influence ion balance in the reaction mixture.
NAD: A cofactor involved in redox reactions, supporting metabolic processes and energy balance.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Cell-free systems lack metabolism, so they cannot naturally regenerate ATP. However, protein synthesis is highly energy-demanding.
One common strategy is to use 3-PGA as an energy source: 3-PGA is metabolized by enzymes present in the cell extract. This generates ATP from ADP through endogenous metabolic pathways. Instead of adding a fixed amount of ATP (which is quickly depleted), energy regeneration systems like 3-PGA ensure a continuous ATP supply, enabling longer and more efficient protein synthesis in cell-free systems.
benefits
Provides a slow, sustained release of energy
Reduces accumulation of inhibitory byproducts compared to direct ATP addition
Extends the reaction time and increases protein yield
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems (e.g., Escherichia coli lysate)
Advantages:
Fast protein synthesis
High yields
Cost-effective and simple to use
Limitations:
Lack of post-translational modifications (PTMs) like glycosylation
Limited ability to correctly fold complex eukaryotic proteins
Green Fluorescent Protein (GFP): It doesn’t require complex PTMs
Eukaryotic systems (e.g., wheat germ, insect, or mammalian lysates)
Advantages:
Capable of post-translational modifications (e.g., glycosylation, disulfide bonds)
Better folding of complex proteins
Limitations:
More expensive
Slower protein production
Typically lower yields
Human Insulin: It requires correct disulfide bond formation
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Challenges
Hydrophobic regions → cause aggregation or precipitation
Lack of membrane → improper folding and loss of function
Low solubility → reduced yield
Complex structure → requires correct insertion and orientation
To optimize membrane protein expression in a cell-free system, I would design an experiment that recreates a membrane-like environment while systematically controlling reaction conditions. A key challenge is that membrane proteins contain hydrophobic regions, which can lead to aggregation and misfolding in the absence of a lipid bilayer. To address this, I would supplement the system (e.g., based on Escherichia coli lysate) with membrane mimetics such as detergents, liposomes, or nanodiscs to enable proper folding and insertion. Additionally, I would optimize parameters like ion concentrations, temperature, and DNA levels, and include chaperones to improve protein stability. By running parallel reactions with different conditions, I could identify the setup that maximizes soluble and functional protein yield.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
One possible reason is insufficient or degraded DNA template, which limits transcription. This can be addressed by checking DNA quality (e.g., avoiding degradation), increasing template concentration, or using a stronger promoter to enhance transcription efficiency.
A second reason could be inefficient energy supply, leading to early termination of protein synthesis. Since cell-free systems cannot regenerate ATP naturally, adding or optimizing an energy regeneration system (e.g., using 3-PGA or PEP) can help sustain longer and more productive reactions.
A third issue may be protein misfolding or aggregation, especially for complex or hydrophobic proteins. This can be improved by lowering the reaction temperature, adding chaperones, or including membrane mimetics (for membrane proteins) to promote proper folding and increase the amount of soluble, functional protein.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1. Pick a function and describe it.
The synthetic cell produces natural pigments that could be used to dye cotton fibers sustainably directly on fibers.
2. hat would your synthetic cell do? What is the input and what is the output?
Function: Biosynthesis of plant-based pigments (e.g., anthocyanins)
Input: Simple nutrients (glucose) + an inducer molecule (e.g., light or small molecule)
Output: Visible color (e.g., red/purple pigment)
3. Could this function be realized by cell-free Tx/Tl alone, without encapsulation? Could this function be realized by genetically modified natural cell?
Cell-free Tx/Tl alone?
Partially yes — pigment enzymes can be expressed in a cell-free system, but:
Yield and stability are limited
No compartmentalization → less control
Genetically modified natural cell?
Yes, very feasible
Plants or microbes already produce pigments naturally
But less controllable and less “designable” than synthetic cells
4. Describe the desired outcome of your synthetic cell operation.
A self-contained synthetic vesicle that:
Produces visible pigment when triggered
Could be applied to cotton fibers
Enables localized, low-waste dyeing
5. Design all components that would need to be part of your synthetic cell. What would be the membrane made of? What would you encapsulate inside? Enzymes, small molecules. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Genes for Pigment Production: PAL, CHS, F3H, DFR, ANR, and UFGT 2Small Molecules & Cofactors
Glucose → energy source
ATP regeneration system (e.g., 3-PGA)
NADPH → required for biosynthesis
Salts & nucleotides
6. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Challenge: Liposomes are semi-permeable but limited
-> Solutions:
Passive diffusion of small molecules (e.g., glucose)
OR express membrane channels:
Example:
α-hemolysin (αHL) pore protein
→ allows small molecules to enter/exit
7. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
8. How will you measure the function of your system?
Color Output
Visual observation (color change)
Spectrophotometry (absorbance ~520–550 nm for anthocyanins)
Protein Expression
SDS-PAGE or fluorescence tagging
Efficiency
Pigment concentration over time
Application Test
Dye uptake on cotton fibers
Wash stability
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
1. Write a one-sentence summary pitch sentence describing your concept.
A swimwear garment embedded with freeze-dried cell-free biosensors that activate upon water exposure and produce a visible signal indicating unsafe water quality.
2. How will the idea work, in more detail? Write 3-4 sentences or more.
The bikini is made from textiles integrated with microencapsulated, freeze-dried BioBits® cell-free systems. These systems contain DNA constructs encoding fluorescent or color-producing reporter proteins under the control of contaminant-responsive genetic circuits. When the bikini is immersed in water, the embedded systems rehydrate and become biologically active. If contaminants such as bacteria, toxins, or chemical pollutants are present, the genetic circuit is activated and triggers the expression of the reporter protein. This results in a visible color change or fluorescence in specific areas of the fabric, indicating that the water quality may be unsafe. Different regions of the garment could be engineered to respond to different types of contaminants, enabling a multi-signal readout.
3. What societal challenge or market need will this address?
This concept addresses the growing need for personal, real-time environmental monitoring in recreational and natural water bodies such as lakes, rivers, and oceans. Water pollution can pose health risks that are not always visible, and current testing methods are often not accessible to individuals at the point of use. A wearable biosensor provides immediate feedback, empowering users to make informed decisions about water safety. It also aligns with increasing demand for smart, functional, and sustainable textiles that integrate biological sensing without electronics.
4. How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Activation with water: The system is intentionally designed to activate upon immersion, using water as the trigger to rehydrate the freeze-dried components.
Stability: Freeze-drying preserves the cell-free reactions during storage, while encapsulation within protective polymer or hydrogel microdomains embedded in the fabric prevents premature activation and degradation.
One-time use: The biosensor could be designed as a semi-consumable feature, where certain zones activate per exposure, or the garment could include replaceable sensing patches.
Environmental robustness: Encapsulation also helps protect the system from mechanical stress, UV exposure, and repeated washing until activation is desired.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
In space missions, astronauts depend on closed-loop water recycling systems, making water quality monitoring critical for survival. Contaminants such as microbial byproducts, toxins, or chemical impurities can accumulate without easy detection. Traditional laboratory testing is impractical in space due to limited equipment, time, and resources. A portable, cell-free biosensor based on BioBits® offers a lightweight, stable, and on-demand solution for detecting water contamination. This approach is significant for ensuring astronaut health, enabling safe long-duration missions, and advancing compact diagnostic technologies that are also relevant for water quality monitoring on Earth in remote or resource-limited environments.
2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
A contaminant-responsive genetic circuit encoding a fluorescent reporter protein under the control of a promoter activated by bacterial toxins or stress-inducible regulatory elements.
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The molecular target is directly linked to detecting harmful substances in recycled spacecraft water. In a cell-free BioBits® system, the genetic circuit is designed so that the presence of contaminants activates transcription and translation of a fluorescent reporter gene. This fluorescence serves as a measurable output indicating contamination levels. By coupling contaminant-sensitive regulatory elements to reporter expression, the system translates otherwise invisible molecular signals into a detectable readout. This allows rapid, in situ assessment of water safety without the need for complex instrumentation, addressing the critical need for reliable environmental monitoring in space habitats.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
We hypothesize that a BioBits® cell-free system containing a contaminant-responsive genetic circuit will produce a measurable fluorescent signal in response to contaminated water samples, while remaining inactive in clean water. Specifically, regulatory elements sensitive to bacterial components or stress-related molecules will activate expression of a fluorescent reporter protein when contaminants are present. The intensity of fluorescence will correlate with contaminant concentration, enabling semi-quantitative assessment of water quality. The reasoning behind this hypothesis is that cell-free transcription and translation machinery can be programmed with DNA constructs to function as biosensors, converting specific molecular inputs into optical outputs. Demonstrating this capability would show that freeze-dried cell-free systems can serve as reliable, portable diagnostic tools for monitoring water safety in space environments where conventional testing methods are not feasible.
5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
BioBits® freeze-dried cell-free reactions will be prepared with DNA constructs encoding a fluorescent reporter under a contaminant-responsive promoter. Test samples will include clean water (negative control), water spiked with simulated contaminants (e.g., bacterial lysate or chemical analogs), and reactions without DNA (background control). Water samples will be added to activate the reactions, followed by incubation. Fluorescence will be measured using the P51 Molecular Fluorescence Viewer. Fluorescence intensity will be compared across conditions to evaluate sensitivity and specificity of the biosensor in detecting contamination levels.
Meyerowitz, J. T., Larsson, E.M., Murray, R.M. (2024) Development of Cell-Free Transcription-Translation Systems in Three Soil Pseudomonads. ACS Synth Biol, 13(2):530-537. doi: 10.1021/acssynbio.3c00468 ↩︎
Shi, S., Tang, R., Hao, X., Tang, S., Chen, W., Jiang, C., Long, M., Chen, K., Hu, X., Xie, Q., Xie, S., Meng, Z., Ismayil, A., Jin, X., Wang, F., Liu, H., & Li, H. (2024). Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants, 13(15), 2028. https://doi.org/10.3390/plants13152028↩︎
Week 10 HW: Imaging and Measurement
Part I: Final Project
For your final project:
1. Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Quantification of pigment concentration through color intensity measurements
Analysis of pigment degradation (as a proxy for biochemical stability) under environmental conditions
Material–pigment interaction effects on color retention
2. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Pigment production (via color intensity as proxy)
I will assess whether and to what extent pigments are successfully expressed in fibers by quantifying color intensity. Since direct biochemical quantification may not be feasible, color measurements (RGB or CIELAB values) obtained through standardized imaging will serve as a proxy for pigment concentration. This allows comparison between different pigment systems (e.g. anthocyanins vs. betalains) or expression strategies.
Pigment stability (biochemical and environmental stability)
To evaluate whether biologically produced pigments are viable for textile use, I will analyze their stability under environmental stressors such as light exposure, washing, and pH variation. Changes in color intensity and hue over time will indicate degradation rates and overall pigment robustness.
Material integration (fiber-specific color retention)
A key aspect of the project is whether pigments remain stable and visible within fiber structures. I will therefore evaluate how pigments perform within different fiber types (or simulated substrates), focusing on color retention, uniformity, and resistance to fading. This reflects how effectively pigments are integrated into the material during or after fiber formation.
3. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Bioinformatics and pathway design tools
To identify suitable pigment systems, I will use bioinformatics databases and literature-based analysis to select and map biosynthetic pathways (e.g. anthocyanin or betalain pathways). This includes identifying key enzymes and exploring strategies for fiber-specific expression.
Digital color quantification
Color intensity will be measured using a standardized photographic setup and analyzed with software such as ImageJ. Extracted RGB or CIELAB values will provide quantitative data for comparing pigment expression and stability.
Spectrophotometric analysis
Spectrophotometry will be used to measure absorbance or reflectance of fibers, allowing more precise quantification of pigment presence and optical properties.
Environmental testing protocols
To simulate real-world conditions, samples will be exposed to controlled stress factors such as UV/light exposure, washing cycles, and pH variation. These tests will help evaluate pigment durability and material performance.
Waters Part I — Molecular Weight
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Theoretical isoelectric point (pI): 5.90
Theoretical Molecular weight (Mw): 28006.60 Da
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
2.1 Determine z for each adjacent pair of peaks (n, n+1):2.2 Determine the MW of the protein:2.3 Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, since the protein is in a larger charged state than the mass spectrometer can catch. It isn’t visible.
Waters Part II — Secondary/Tertiary structure
not required
Waters Part III — Peptide Mapping - primary structure
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
2. How many peptides will be generated from tryptic digestion of eGFP? Navigate to https://web.expasy.org/peptide_mass/ Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides. Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP. Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
It reports 19 peptides.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
According to Figure 5a, there are 22 peaks: 0.43, 0.61, 0.79, 1.20, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70, 4.30, 4.48, 4.64, 4.87, 5.06, 5.43
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
There are more peaks in the chromatogram. -> 3
5. Identify the mass-to-charge of the peptide shown in Figure 5b. What is the charge () of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide
526,25918 - 525,76712 = 0.49206 ~ 0.5
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
FEGDTLVNR
7. What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
90.7% of sequence covered
Waters Part IV — Oligomers
1. We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Regarding the peaks in Figure 7, I see peaks at 3.4 MDa, 8.33 MDa, 12.67 MDa and a smaller one close to 16-17 MDa.
Waters Part V — Did I make GFP?
1. Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Week 11 HW: Building Genomes
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Somehow I didn’t receive an email, so I couldn’t contribute.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): Contains high levels of T7 RNA polymerase, which specifically recognizes the T7 promoter on the DNA template and drives strong transcription of the target gene. The “Star” variant reduces RNA degradation, improving mRNA stability and increasing overall protein yield in the cell-free reaction.
Salts/Buffer
Potassium glutamate: Maintains ionic strength and mimics intracellular conditions, helping stabilize ribosomes and enzymes for efficient translation.
HEPES-KOH pH 7.5: Buffers the reaction to keep a stable pH, which is critical for enzyme activity and protein synthesis.
Magnesium glutamate: Provides Mg²⁺ ions, essential cofactors for ribosome structure, tRNA binding, and enzymatic reactions in transcription/translation.
Potassium phosphate (dibasic/monobasic): Contributes to buffering capacity and supplies phosphate ions needed for energy metabolism and nucleotide balance.
Energy / Nucleotide System
Ribose: Serves as a precursor for nucleotide synthesis, enabling regeneration of NTPs required for transcription and energy transfer.
Glucose: Acts as an energy source, feeding metabolic pathways that regenerate ATP and drive the reaction.
AMP, CMP, UMP: Provide nucleotide building blocks for RNA synthesis and can be converted into triphosphates (ATP, CTP, UTP) for transcription.
GMP (0 µM): Its absence suggests reliance on salvage pathways (e.g., from guanine) to generate GTP, a key molecule for translation.
Guanine: Precursor for GMP/GTP synthesis via salvage pathways, supporting RNA synthesis and ribosomal function.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies the building blocks for protein synthesis (excluding specific ones added separately for stability or solubility reasons).
Tyrosine: Added separately due to solubility issues; required as a protein building block once adjusted to a usable form.
Cysteine: Included separately because it is prone to oxidation; essential for forming disulfide bonds in proteins.
Additives
Nicotinamide: Precursor for NAD⁺/NADH, supporting redox balance and metabolic reactions involved in energy regeneration.
Backfill
Nuclease Free Water: Serves as the solvent to bring all components to the desired final volume while preventing degradation of DNA/RNA by nucleases, ensuring the stability of the transcription–translation machinery.
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour optimized PEP–NTP master mix relies on directly supplied high-energy molecules (PEP) and fully formed nucleotides (ATP, GTP, CTP, UTP), enabling rapid and immediate protein synthesis but limiting the reaction’s duration. In contrast, the 20-hour NMP–ribose–glucose system uses metabolic precursors such as NMPs, ribose, and glucose, which are enzymatically converted within the system to regenerate energy and nucleotides over time. This results in a slower initial rate but allows for more sustained, long-term protein production with a simpler and more resource-efficient composition.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: Superfolder GFP is engineered for robust folding, allowing it to fold efficiently even in challenging conditions like cell-free systems, which leads to strong and reliable fluorescence output.
mRFP1: has a relatively slow maturation time, meaning fluorescence develops more slowly after translation, which can delay signal detection in short experiments. -> low acid sensitivity.
mKO2: is relatively acid-sensitive, so its fluorescence can decrease under lower pH conditions that may arise during longer cell-free reactions.
mTurquoise2: has a high quantum yield and brightness, making it very efficient for fluorescence readout even at lower expression levels. It’s also a rapidly-maturing monomer with very low acid sensitivity.
mScarlet_I: is optimized for fast maturation and high brightness, enabling strong fluorescence signals relatively quickly compared to older red fluorescent proteins.
Electra2: is designed for enhanced brightness and stability, but like many fluorescent proteins, it is oxygen-dependent for chromophore formation, which can limit fluorescence if oxygen is depleted in the system
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mRFP1, which is limited by slow chromophore maturation, increasing oxygen availability in the cell-free mastermix (e.g., by reducing reaction volume-to-surface ratio or incorporating oxygen-rich buffers) and supplementing with energy-regeneration components (e.g., higher glucose or ribose concentrations) will enhance chromophore formation and sustain ATP levels. This will accelerate maturation and prolong protein synthesis, ultimately leading to higher cumulative fluorescence over a 36-hour incubation.
For mKO2, whose fluorescence is acid-sensitive, the 36-hour mastermix could be improved by increasing buffering capacity (e.g., higher HEPES concentration and optimized phosphate ratio) and slightly reducing glucose concentration. In the 20-hour system, glucose metabolism can lead to acidification over time, which would quench mKO2 fluorescence. By strengthening the buffer and limiting excess glucose-driven acid production, the pH can be kept more stable, resulting in higher and more stable fluorescence over a 36-hour incubation.
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
N/A
4. The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
1 2
Flavonoids are abundant and widely distributed plant secondary metabolites. They are the primary compounds of plant pigments, provide signals for pollinators and symbiotic bacteria, protect plants from UV-B and environmentally induced oxidative stress. 3
The ability to manipulate plant pigmentation has been extensively studied, particularly in ornamental flowers such as tulips or petunia. Flower color is primarily determined by anthocyanins, a class of flavonoid pigments responsible for red, purple and blue hues. In species like tulipa, variation in color is achieved through differences in anthocyanin composition, concentration and cellular localization. Genetic engineering approaches have successfully modified flower color by altering key enzymes in the anthocyanin biosynthesis pathway or by introducing transcription factors that regulate pigment production. 4
Flavonoids are abundant and widely distributed plant secondary metabolites. They are the primary compounds of plant pigments, provide signals for pollinators and symbiotic bacteria, protect plants from UV-B and environmentally induced oxidative stress. 3
The ability to manipulate plant pigmentation has been extensively studied, particularly in ornamental flowers such as tulips or petunia. Flower color is primarily determined by anthocyanins, a class of flavonoid pigments responsible for red, purple and blue hues. In species like tulipa, variation in color is achieved through differences in anthocyanin composition, concentration and cellular localization. Genetic engineering approaches have successfully modified flower color by altering key enzymes in the anthocyanin biosynthesis pathway or by introducing transcription factors that regulate pigment production. 4
For example, previous studies have demonstrated that overexpression or suppression of enzymes such as dihydroflavonol reductase (DFR) or anthocyanidin synthase (ANS) can shift pigmentation outcomes. Additionally, regulatory genes such as MYB transcription factors have been used to activate entire pigment pathways, enabling predictable and stable color changes in petals. These advances illustrate that plant pigmentation can be rationally engineered when the biosynthetic pathway and its regulation are well understood. 5
In contrast to ornamental flowers, pigmentation in fiber-producing crops such as Gossypium hirsutum remains largely unexplored as a designable trait. Cotton fibers are single elongated epidermal cells composed primarily of cellulose and are typically white or off-white. While naturally colored cotton varieties exist (e.g. brown or green), their color range is limited and not easily tunable. 6
However, existing research shows that cotton is capable of producing anthocyanins under specific conditions. In response to infection by the bacterial pathogen Xanthomonas campestris pv. malvacearum, cotton accumulates red pigmentation at infection sites. This coloration is caused by anthocyanins, particularly cyanidin-3-glucoside, which play a protective role by absorbing light and mitigating damage from reactive oxygen species and light-activated phytoalexins. Importantly, studies have shown that these pigmented cells can absorb 3–4 times more photo-activating light, protecting surrounding healthy tissue. 78
This demonstrates that:
Cotton possesses a functional anthocyanin biosynthesis pathway
Pigment production is inducible and spatially regulated
Pigmentation is linked to stress and defense responses, not material development
Knowledge Gap
This project addresses this gap by proposing to reprogram the regulatory control of anthocyanin biosynthesis in cotton, shifting it from a stress-induced response to a developmentally controlled trait.
By drawing on established strategies from flower color engineering (e.g. transcription factor activation and pathway modification) and applying them to cotton, this work explores the possibility of creating intrinsically colored fibers. This represents a novel intersection of synthetic biology, plant science and material design, with potential applications in sustainable textile production.
Despite advances in engineering pigmentation in flowers, there is a clear gap in applying these strategies to fiber-producing plant tissues such as cotton. Specifically:
Anthocyanin production in cotton is restricted to stress conditions and does not occur during normal fiber development
There is currently no established method to program pigment production directly into cotton fibers
The integration of pigmentation into fibers during growth remains unexplored 9
Innovation
This project could be innovative in that it proposes a shift from post-production dyeing to biologically embedded color, reframing pigmentation as a material property that is designed during growth rather than applied afterward. While anthocyanin pathways have been extensively engineered in ornamental plants for aesthetic purposes, their application to fiber-producing crops like Gossypium hirsutum remains largely unexplored. By transferring and adapting these biological concepts, the project introduces a new use of existing genetic tools to address environmental challenges in the textile industry.Furthermore, the work challenges the prevailing assumption that pollution must be managed through downstream solutions such as filtration, instead proposing a preventative, design-based approach rooted in synthetic biology. It expands the boundaries of the field by positioning plants not only as organisms to be engineered for yield or resistance, but as programmable material systems capable of producing functional and aesthetic properties simultaneously.
Research by Stephen Chandler and Yoshikazu Tanaka (2007) provides a comprehensive review of genetic modification in floriculture, focusing particularly on the engineering of flower color. The study explains that traditional breeding is limited by the natural gene pool of a species, whereas genetic modification enables the introduction of new genes to create novel traits, especially through manipulation of the anthocyanin biosynthesis pathway. A key achievement highlighted is the development of genetically modified carnations with new colors (e.g. violet/blue hues), demonstrating that pigment pathways can be successfully reprogrammed to produce traits not naturally present in the plant. However, the authors note that despite significant scientific progress, commercial applications remain limited due to regulatory costs, intellectual property constraints, and perceived public acceptance issues.
Shi, S.; Tang, R.; Hao, X.; Tang, S.; Chen, W.; Jiang, C.; Long, M.; Chen, K.; Hu, X.; Xie, Q.; et al. (2024) Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants, 13, 2028. https:// doi.org/10.3390/plants131520288
Research on pigmentation in Gossypium hirsutum demonstrates that naturally colored cotton fibers derive their pigmentation primarily from the flavonoid biosynthesis pathway. A recent transcriptomic and metabolomic study (Shi et al., 2024) showed that key genes such as CHS, DFR, F3H, and UFGT are significantly upregulated in brown cotton fibers, particularly during later developmental stages. The study also identified metabolites including cyanidin-3-O-glucoside as major contributors to fiber coloration, and highlighted the role of MYB transcription factors in regulating pigment production. However, despite identifying these pathways and regulatory networks, the study concludes that the mechanism of pigment deposition in fibers remains poorly understood, limiting the ability to engineer new or controllable colors in cotton.
In contrast, research on ornamental plants such as Tulipa has demonstrated that anthocyanin-based pigmentation can be precisely engineered through genetic modification. Studies have shown that altering the expression of biosynthetic enzymes (e.g. DFR, ANS) or regulatory transcription factors (e.g. MYB proteins) enables predictable changes in flower color. These systems illustrate that pigmentation pathways can be externally controlled and fine-tuned, resulting in a wide spectrum of stable colors. Together, these studies highlight a key gap: while pigmentation in flowers is highly programmable, cotton fibers possess similar biochemical pathways but lack the regulatory control needed for designed and scalable color production.
Why this project matters?
It addresses a fundamental limitation in the textile industry: the reliance on post-production dyeing, which leads to significant environmental pollution. In my Bachelor thesis, I conducted a life cycle assessment of a cotton T-shirt grown in India, manufactured in Bangladesh, and sold in Austria. The results challenged common assumptions: contrary to expectations, transportation contributed only minimally to the overall impact, accounting for approximately 0.1 kg CO₂-eq, whereas the production phase—particularly textile finishing — was responsible for nearly 4 kg CO₂-eq for a 200 g cotton T-shirt. Additionally, I found that dyeing alone can require around 5 liters of water per T-shirt, highlighting the disproportionate environmental burden of this stage. These findings emphasize that addressing the finishing process is critical for achieving meaningful reductions in the environmental footprint of clothing.
The wastewater generated from textile dyeing presents substantial environmental and social challenges, it threatens access to safe water for drinking, sanitation and hygiene, while also damaging aquatic ecosystems. (SDG Goals 6 & 14) Common dye classes—including naphthalene-based, heterocyclic, anthraquinone, and indigo dyes—are associated with serious health risks such as skin irritation, respiratory issues, carcinogenic effects, and liver and kidney damage. On a global scale, it is estimated that approximately 280,000 tons of textile dyes are discharged into wastewater annually, with up to 80% released untreated into the environment, exacerbating ecological and human health impacts.
As highlighted in recent research on Gossypium hirsutum, pigment production in cotton fibers is already biologically possible through the flavonoid biosynthesis pathway. However, this mechanism is not yet controllable or scalable for industrial use, representing a key barrier to progress. While naturally colored cotton offers a promising alternative, its limited color range, instability and inconsistent expression prevent widespread adoption and maintain dependence on chemical dyeing. 10
Building on this gap, the project proposes to move beyond observation toward engineering controllable pigmentation directly within the fiber, eliminating the need for external dyes. The potential impact is significant: reducing water consumption, chemical inputs and energy use across the textile lifecycle. Beyond environmental benefits, this approach could fundamentally transform how materials are designed, enabling fibers to possess both functional and aesthetic properties from the moment they are grown.
Bioethics & Biosafety
This project raises several ethical considerations related to the use of genetic modification in agriculture, particularly in a widely cultivated species such as Gossypium hirsutum. The deliberate alteration of plant metabolic pathways to produce colored fibers introduces questions about biosafety, ecological impact and long-term consequences. For example, engineered pigment pathways could unintentionally affect plant fitness, interactions with pests or surrounding ecosystems if gene flow occurs. Working with GMOs requires careful design, testing and containment strategies to be certain that innovations in synthetic biology are applied safely.
In addition, the cultivation of cotton - as one of the most relevant crops - engages with broader ethical questions around justice and accessibility. Textile pollution disproportionately affects communities in major production regions, where untreated wastewater impacts local water resources and public health. By proposing a preventative alternative, this work has the potential to contribute to a more equitable distribution of environmental burdens. However, it is also important to consider who benefits from such innovations: access to genetically modified seeds, intellectual property rights and economic implications for hydroponic farmers must be addressed to avoid reinforcing existing inequalities. Transparent communication, inclusive decision-making and consideration of local contexts are therefore essential to ensure that the benefits of this technology are shared fairly, aligning the project with principles of justice and global responsibility.
Measures
To ensure that this project is conducted ethically, it will be implemented exclusively within hydroponic systems in closed, monitored greenhouse environments. Restricting cultivation to controlled settings significantly reduces the risk of unintended environmental exposure, including gene flow to wild relatives, ecological escape, or disruption of surrounding ecosystems. Nevertheless, even when plants are grown hydroponically without contact with soil, it remains essential to conduct controlled greenhouse trials, apply gene-flow mitigation strategies, and perform comprehensive ecological risk assessments. Continuous monitoring is necessary to evaluate potential unintended consequences, such as impacts on plant metabolism, ecosystem interactions, or the spread of modified traits beyond the intended system. Transparency and open communication with stakeholders — including farmers, policymakers, and the public — should be integrated throughout the research process to foster trust and support informed decision-making.
Potential unintended consequences may include unforeseen ecological effects, limitations in color diversity or durability, and socioeconomic challenges such as unequal access to modified seeds or dependency on proprietary technologies. These uncertainties underline the importance of iterative testing and interdisciplinary collaboration.
To put it in a nutshell, colorful grown cotton aims to contribute to society by indirectly improving equitable access to safe drinking water, reducing potential health risks for workers in conventional dyeing facilities, and relocating cultivation closer to the areas where the products are sold, thereby potentially decreasing transport-related environmental impacts.
I will use databases and Benchling to organize and develop the genetic constructs needed for engineering naturally colorful cotton. Scientific databases such as NCBI GenBank and UniProt would help me research pigment-producing genes, regulatory elements and existing studies related to plant coloration and cotton genetics. In Benchling, I will design my expression casette, annotate it and export it to Twist Bioscience.
Additionally, designing a Twist order will be an important step in transforming the digital genetic designs into physical DNA constructs. Through Twist Bioscience, I would order synthesized DNA fragments containing the selected pigment genes and promoters optimized for cotton expression. This process would allow me to efficiently move from computational design to experimental implementation in developing naturally colorful cotton fibers.
Experimental plan (7-9 weeks)
1. Literature Research into flavonoid pathway Literature Research on the Flavonoid Biosynthesis Pathway (1–2 weeks)
The project will begin with an extensive literature review on the flavonoid biosynthesis pathway, with a particular focus on anthocyanin and pigment production in plants. Scientific databases such as Google Scholar and NCBI will be used to identify key enzymes, regulatory genes, and previous genetic engineering approaches related to pigmentation in plants.
Expected result: Identification of the most relevant enzymes and pathways involved in stable pigment formation.
2. Selection of and Specialized Research into a Gene of Interest (1 week)
After comparing multiple candidate genes, one gene involved in anthocyanin biosynthesis will be selected based on criteria such as pigment intensity, stability, previous use in plant engineering studies, and compatibility with cotton. Particular attention will be given to genes reported to strongly influence visible coloration. Once a candidate gene has been chosen, a more detailed literature review will investigate its biological function, regulation, enzymatic activity, and performance in previous transformation experiments.
Expected result: Selection of the most promising pigmentation-related gene and further investigation.
3. Retrieval of Gene Sequences from Multiple Species (3–4 days)
DNA and amino acid sequences of the selected gene will be retrieved from databases such as NCBI GenBank and UniProt. Sequences from multiple plant species will be collected to compare natural variation and identify the most suitable version for expression in cotton.
Expected result: A collection of candidate sequences from different species for comparative analysis.
4. Reverse Translation of Amino Acid Sequences and Codon Optimization for Gossypium hirsutum (2 days)
Amino acid sequences of the selected protein will be reverse translated into nucleotide sequences using the Reverse Translate Tool from Bioinformatics.org, subsequently the nucleotide sequence will be codon optimized for cotton using the VectorBuilder Codon Optimization Tool
to improve predicted protein expression efficiency. Parameters such as GC content and codon adaptation index will be evaluated.
Expected result: An optimized gene sequence predicted to express efficiently in cotton cells.
5. Evaluation of Expression Efficiency (3–4 days)
Computational analysis will compare optimized and non-optimized sequences to assess possible differences in translational efficiency, sequence stability, and mRNA structure.
Expected result: Evidence that codon optimization improves theoretical expression potential.
6. Research into Regulatory DNA Elements (1 week)
Suitable promoters, terminators, and regulatory elements for plant transformation will be identified through literature research.
Expected result: Selection of appropriate regulatory sequences for stable gene expression.
7. Design of the Expression Cassette (3–5 days)
An expression cassette containing the promoter, RBS, start codon, optimized gene sequence, his tag, terminator and stop codon will be digitally assembled.
Expected result: A complete DNA construct for hypothetical plant transformation.
8. Selection, Assembly and Validation of the Plasmid Construct (4–6 days)
An appropriate plasmid vector for plant transformation will be selected based on cloning compatibility, selectable markers and compatibility with Agrobacterium tumefaciens systems. The designed expression cassette will then be integrated into the selected vector and uploaded to Twist Bioscience to evaluate whether the sequence is technically synthesizable and free of problematic sequence characteristics such as repetitive regions or unstable motifs.
Expected result: Identification of a suitable plasmid vector and confirmation that the construct is feasible for synthetic DNA production.
9. Visualization, Annotation, and Optimization of the Construct (1 day)
The completed plasmid construct will be imported into Benchling to generate a detailed annotated plasmid map. Functional regions including promoters, coding sequences, terminators, and selectable markers will be clearly labeled. The construct will then be analyzed for unwanted restriction sites, sequence instabilities, or cloning incompatibilities, and refinements will be made where necessary to optimize the design for stable expression.
Expected result: A fully annotated, refined and optimized plasmid construct suitable for hypothetical plant transformation experiments.
10. Creation of Visual Workflow and Plasmid Graphs (1 week)
Diagrams and graphs will be produced to visualize the project workflow, plasmid composition, and sequence proportions. These figures will support communication of the experimental design.
Expected result: Clear visual materials illustrating the genetic engineering workflow and construct design.
11. Final Analysis and Ethical Assessment (1 week)
The completed project design will be evaluated regarding feasibility, sustainability, biosafety, and societal implications. Potential ecological risks, greenhouse containment strategies, and ethical considerations surrounding genetically modified crops will be discussed.
Expected result: A comprehensive assessment showing how the project addresses both scientific feasibility and responsible innovation.
After extensive research, I decided to focus on Dihydroflavonol 4-reductase, or DFR. DFR is a key enzyme in the flavonoid biosynthetic pathway and plays an essential role in the production of anthocyanin pigments - the compounds responsible for red, purple, and blue coloration in plants. Beyond coloration, these pigments contribute to UV protection and can support plant stress responses.
Instead of introducing an entirely foreign protein into the organism, I wanted to work with a naturally existing pathway and explore how its expression could be intensified. Therefore, I experimented with codon optimization of the DFR gene itself in order to potentially increase expression efficiency while maintaining biological compatibility. This approach reflects the idea that laboratory experimentation is necessary to evaluate feasibility, but in this case, optimizing a native system appeared to be the most plausible and sustainable strategy.
As shown in the graphic, I compared DFR gene sequences from multiple plant species. First, I generated the nucleotide sequences using the Reverse Translate tool from Bioinformatics.org. Afterwards, the sequences were codon-optimized specifically for Gossypium hirsutum, or upland cotton, using the official VectorBuilder codon optimization tool.
The optimization results did not reveal major differences between the variants. Because of this, I ultimately selected the cotton-derived DFR sequence, as it showed a relatively high Codon Adaptation Index, or CAI, value of 0.93 and offered the advantage of native compatibility with the host organism. But as a next step, and if I had access to a lab I would love for my aim 2 to experiment with DFR from multiple species - especially ones that have an intense red/purple/blue coloration, like strawberries or grapes. It seems there must be a difference because cotton has the same gene but is still not appearing red or blue.
In the following points, you can see the individual steps of my working process and how I developed this experimental approach.
Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
reverse translation of tr|C8YQV2|C8YQV2_GOSHI Dihydroflavonol 4-reductase OS=Gossypium hirsutum OX=3635 GN=LOC107957957 PE=2 SV=1 to a 1020 base sequence of most likely codons.
By comparing the original and optimized sequences, I increased the codon adaptation index (CAI) from 0.66 to 0.93, meaning the redesigned DNA now uses codons that better match those found in highly expressed cotton genes. This was especially interesting since I learned that even native genes in the same organism often have suboptimal codon usage, regulatory constraints and evolutionary tradeoffs. This suggests the optimized sequence would likely be translated more efficiently, potentially increasing enzyme production in the flavonoid pathway.
ATGGGTTCATCTGTTACAGATGGTGAGATTGTTTGTGTCACTGGAGGTTCTGGATTCATCGGATCATGGCTTATTAAACTTCTTCTTGAAAGGGGTTATGTTGTTAGAGCTACAGTTAGAGATCCAGGAAACTCAAAGAAGGTTAAACACCTTTTGGAGCTTCCTAAGGCCGAGACTCATCTCACCCTTTGGAAGGCCGATTTGGCTGAAGAGGGTTCATTTGATGATGCTATTCAAGCATGTACCGGTGTTTTTCATGTGGCTACTCCTATGGATTTCGAGTCTGAGGATCCTGAAAACGAGGTTATCAAGCCTACAATCAACGGTGTTTTGTCCATCATGAAGGCATGCGCTAAAGCTAAGACTGTTAGGAGATTGGTTTTCACTTCCTCAGCTGGTACTATCGATGTTGCTGAACAACAAAAGCCTTGCTACGATGAGACATGTTGGTCTGATTTGGAATTCATTCAAGCTAAGAAGATGACCGGATGGATGTATTTCGTTTCAAAGACAATGGCCGAACAAGCAGCTTGGAAGTTTGCTAAGGAAAACAATATAGATTTCGTTTCAATTATCCCACCACTTGTTGTAGGACCTTTCATTATGCAATCCATGCCTCCTTCTCTTATTACTGCTTTGAGTCCCATTACTGGTAATGAGGCTCATTATTCTATTATTAAACAAGGACAATTTATTCATCTTGATGATCTTTGTAGAGCCCATATTTTTTTGTTCGAAAACCCTAAAGCAGAGGGTAGGCATATTTGTGCTTCCCATCATGCTACTATTATCGATCTTGCTAAGATGCTTTCAGAGAAGTATCCTGAATATAACGTTCCTACTAAGTTCAAGGATGTCGATGAAAACTTGAAATCTGTTGAATTCTCCTCAAAGAAACTCTTGGATCTTGGATTTGAATTCAAGTACTCTCTTGAAGATATGTTCGTTGGGGCTGTTGAGACTTGCAGGGAGAAGGGACTTCTGCCTCTTTCAAACGAGAAGAAAATTAAGAACATTGAT
Promoter
The SCFP promoter fragment was extracted from the GenBank entry (GQ411495.1) of Gossypium hirsutum cultivar Handan 5833 fiber-specific protein (SCFP) gene, promoter region and 5’ UTR, which contains a 1005 bp sequence annotated as promoter region plus 5’ UTR/mRNA. The promoter region corresponds to nucleotides 1–633, while nucleotides 634–1005 correspond to the 5’ UTR/mRNA. 14131516
I chose a 6×His tag because it is a small and widely established affinity tag that enables efficient detection and purification of the recombinant protein without significantly increasing the size of the expressed sequence. Compared to longer variants such as 7×His, the 6×His tag is more commonly used in standard molecular biology workflows and generally provides sufficient binding affinity. In the context of DFR expression, using a small tag is advantageous because it minimizes the risk of interfering with protein folding or enzymatic activity.18
Stop codon
The TAA (UAA in mRNA) stop codon was selected for the cotton DFR construct because its usage pattern in plants suggests it is the most efficient and reliable termination signal, particularly in dicotyledonous species. In dicots, UAA is the most frequently used stop codon, occurring in approximately 46% of genes, whereas UGA is used in about 36% and UAG in only 18%. This distribution indicates a clear preference for UAA in dicot genomes, suggesting better compatibility with the endogenous translation termination machinery. In contrast, UAG is comparatively underrepresented and has been associated with stronger context dependence, which may increase the likelihood of inefficient termination or readthrough. Therefore, selecting UAA provides a biologically supported choice that aligns with natural codon usage bias in dicots and supports efficient translation termination in plant expression systems. 19
nos terminator
The NOS terminator was selected because it is a well-established and reliable regulatory sequence commonly used in plant genetic engineering. The terminator ensures proper transcription termination and supports consistent and stable expression of the introduced construct. Its compatibility with plant transformation systems and extensive use in previous studies also made it a suitable and low-risk choice for achieving stronger color development in the cotton fibers.
I imported all the DNA fragments into Benchling one after another and annotated the sequence for the first time. After exporting the annotated sequence as a FASTA file, I imported it into Twist Bioscience and selected the vector pTwist Amp High Copy. The order validation worked successfully, confirming that the construct was functional.
Key Synthetic Biology Techniques for Realization
I utilized several core synthetic biology techniques, including DNA sequencing, DNA construct design, databases, and designing a Twist order.
DNA sequencing was important for analyzing and comparing DFR gene sequences from multiple plant species in order to identify suitable variants for anthocyanin production. I used publicly available genetic databases such as NCBI GenBank and protein information from UniProt to obtain the original sequences and study their biological function within the flavonoid biosynthesis pathway.
Another important technique was DNA construct design. In Benchling, I assembled and annotated a complete cotton expression construct containing the SCFP fiber-specific promoter, start codon, optimized DFR coding sequence, 5’ UTR, 6×His tag, stop codon and NOS terminator.
Finally, I utilized the process of designing a Twist order to validate whether the construct was technically synthesizable. After exporting the annotated plasmid sequence from Benchling, I uploaded it to Twist Bioscience and selected the pTwist Amp High Copy vector. The successful order validation confirmed that the construct met synthesis requirements and could theoretically be manufactured for laboratory testing.
Challenges
One unexpected challenge during the validation process was realizing that increasing DFR expression alone may not automatically result in visibly red, purple or blue cotton fibers. Although Gossypium hirsutum naturally contains the DFR gene, cotton fibers do not normally have any anthocyanin pigmentation, which suggests that additional regulatory factors or pathway limitations are involved. This made it difficult to predict whether codon optimization by itself would be sufficient to significantly enhance pigment production. To address this limitation, I compared DFR sequences from multiple highly pigmented plant species and considered the possibility of testing alternative DFR variants from plants such as grapes or strawberries in future experiments.
A further limitation was that the project remained computational and theoretical due to the lack of access to a wet laboratory. Although the construct was successfully validated through Twist Bioscience and assembled digitally in Benchling, I could not experimentally test transformation efficiency, pigment intensity, or unintended physiological effects in real cotton plants.
Analysis
Conclusion
This project explored the possibility of rethinking textile coloration by shifting pigment production from an external industrial process to an intrinsic biological property of the material itself.
By focusing on the anthocyanin biosynthesis pathway in Gossypium hirsutum, the project demonstrated that cotton already possesses the biological machinery required for pigment production, but that this capability is currently restricted to stress responses rather than fiber development. Through literature research, sequence analysis, codon optimization and plasmid design, a theoretically functional genetic construct for DFR expression in cotton was developed and digitally validated. The successful validation of the construct through Twist Bioscience confirmed that the designed sequence is technically synthesizable and compatible with standard molecular cloning systems.
The project further highlighted the importance of regulatory design in plant synthetic biology. The use of a fiber-specific promoter, codon optimization strategies and established regulatory elements illustrated how gene expression can be computationally optimized to potentially improve anthocyanin production in cotton fibers. At the same time, the work demonstrated that even naturally occurring genes can be redesigned to achieve higher predicted expression efficiency, emphasizing the role of codon usage and regulatory architecture in synthetic construct design.
Beyond the technical aspects, the project positioned genetically engineered pigmentation within a broader environmental and ethical context. Rather than treating wastewater after pollution has occurred, the concept proposes a preventative strategy that could reduce reliance on chemical dyes, water-intensive finishing processes and toxic wastewater generation. The use of hydroponic greenhouse systems additionally addressed biosafety concerns by providing a controlled cultivation environment that minimizes ecological exposure and supports responsible experimentation with genetically modified plants.
Although the project remains conceptual and no biological transformation experiments were conducted, it demonstrates the possibility of intrinsically colored cotton fibers as a more sustainable alternative to conventional textile dyeing. Hopefully, one day it can be tested more thorougly within a lab environment.
Supply List and Budget
Since I am a non in-person comitted listener, my project is only conceptual and no budget is needed. My Twist Bioscience plasmid order would be $288.22.
References
Dhakal, K., Julkowska, M. M., Shekoofa, A. (2026) A hydroponic approach to assess the morpho-physiological responses of cotton cultivars under varying vapor pressure deficit conditions. Frontiers in Plant Science, 17:1751642 ↩︎
Natalio, F., Tahir M. N., Friedrich, N. et al. (2016) Structural analysis of Gossypium hirsutum fibers grown under greenhouse and hydroponic conditions. Journal of Structural Biology, http://dx.doi.org/10.1016/j.jsb.2016.03.005↩︎
Tan, J., Tu, L., Deng, F. et al. (2013) A Genetic and Metabolic Analysis Revealed that Cotton Fiber Cell Development Was Retarded by Flavonoid Naringenin, Plant Physiology, 162, 86-95 ↩︎
Katsumoto, Y., Fukuchi-Mizutani, M., Fukui, Y. et al. (2007) Engineering of the Rose Flavonoid Biosynthetic Pathway Successfully Generated Blue-Hued Flowers Accumulating Delphinidin, Plant and Cell Physiology, 48:11, 1589–1600. https://doi.org/10.1093/pcp/pcm131↩︎
Parmar, M. S., Sharma, R. P. (2002) Development of various colours and shades in naturally coloured cotton fabrics. Indian Journal of Fibre & Textile Research, 27, 397-407 ↩︎
Gě, Q., Cūi, Y., Lǐ, J. et al. (2020) Disequilibrium evolution of the Fructose-1,6-bisphosphatase gene family leads to their functional biodiversity in Gossypium species. BMC Genomics 21, 379. https://doi.org/10.1186/s12864-020-6773-z↩︎
Shi, S., Tang, R., Hao, X. et al. (2024) Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants, 13, 2028. https://doi.org/10.3390/plants13152028↩︎↩︎
Pinnika, G., Thyssen, G. N., Madison, C. A. et al. (2025) The brown fiber phenotype in cotton line SA-40 is linked to a missing Ty3-like retrotransposon upstream of the GhTT2_A07. Frontiers in Plant Science, 16:1668965. doi: 10.3389/fpls.2025.1668965 ↩︎
Periyasamy, A. P. (2025) Textile Dyes in Wastewater and its Impact on Human and Environment: Focus on Bioremediation, Water Air Soil Pollut, 236:562 https://doi.org/10.1007/s11270-025-08204-7↩︎
Xie, D., Jackson, L. A., Cooper, J. D. (2004) Molecular and Biochemical Analysis of Two cDNA Clones Encoding Dihydroflavonol-4-Reductase from Medicago truncatula. Plant Physiology, 134, 979–994 ↩︎
Lewis, J. A., Zhang, B., Harza, R. et al. (2023) Structural Similarities and Overlapping Activities among Dihydroflavonol 4-Reductase, Flavanone 4-Reductase, and Anthocyanidin Reductase Offer Metabolic Flexibility in the Flavonoid Pathway. International Journal of Molecular Sciences, 24, 13901. https://doi.org/10.3390/ijms241813901↩︎
National Library of Medicine (2025). National center for biotechnology information. Nih.gov. Available at: https://www.ncbi.nlm.nih.gov/. ↩︎↩︎
Atella, A. L., Grossi-de-Sá, M. F., Alves-Ferreira, M. (2023) Cotton promoters for controlled gene expression. Electronic Journal of Biotechnology, 26, 1-12, https://doi.org/10.1016/j.ejbt.2022.12.002↩︎
Liliana, V. U.,Tatiana, C. N., Ricardo, P. C. et al. (2023) Plant Promoters: Their Identification, Characterization, and Role in Gene Regulation. Genes, 14, 1226. https://doi.org/10.3390/genes14061226↩︎
Yaqoob, A., Ali Shahid, A. , Salisu, I. B. et al. (2020) Comparative analysis of Constitutive and fiber-specific promoters under the expression pattern of Expansin gene in transgenic Cotton. PLOS ONE, 15(3):e0230519. https://doi.org/10.1371/journal.pone.0230519↩︎
Mayr, C. (2019) What Are 3′ UTRs Doing? Cold Spring Harbor Perspectives in Biology, 11:a034728 doi: 10.1101/cshperspect.a034728 ↩︎
Kimple, M. E., Brill, A. L., Pasker, R.L. (2013) Overview of affinity tags for protein purification. Curr Protoc Protein Sci, ;73:9.9.1-9.9.23. doi: 10.1002/0471140864.ps0909s73 ↩︎
Angenon, G., Van Montagu, M., Depicker, A. (1990). Analysis of the stop codon context in plant nuclear genes. , 271(1-2), 144–146. doi:10.1016/0014-5793(90)80392-v ↩︎