First, describe a biological engineering application or tool you want to develop and why. I want to develop a biological engineering system for smarter nutrient liberation in agricultural soils, especially for sugarcane. The current nutrient management system depends heavily on chemical fertilizers that release nutrients in poorly timed pulses, leading to massive nutrient losses, soil degradation, and environmental damage. I do not want to sugarcoat it: the situation is bad. I am Brazilian, which also means I come from the country that exports the most sugar in the world, largely through intensive sugarcane monoculture. My goal is to explore how engineered soil microorganisms could sense plant-derived signals and release nutrients only when they are biologically needed, turning the soil from a passive substrate into an active, responsive system.
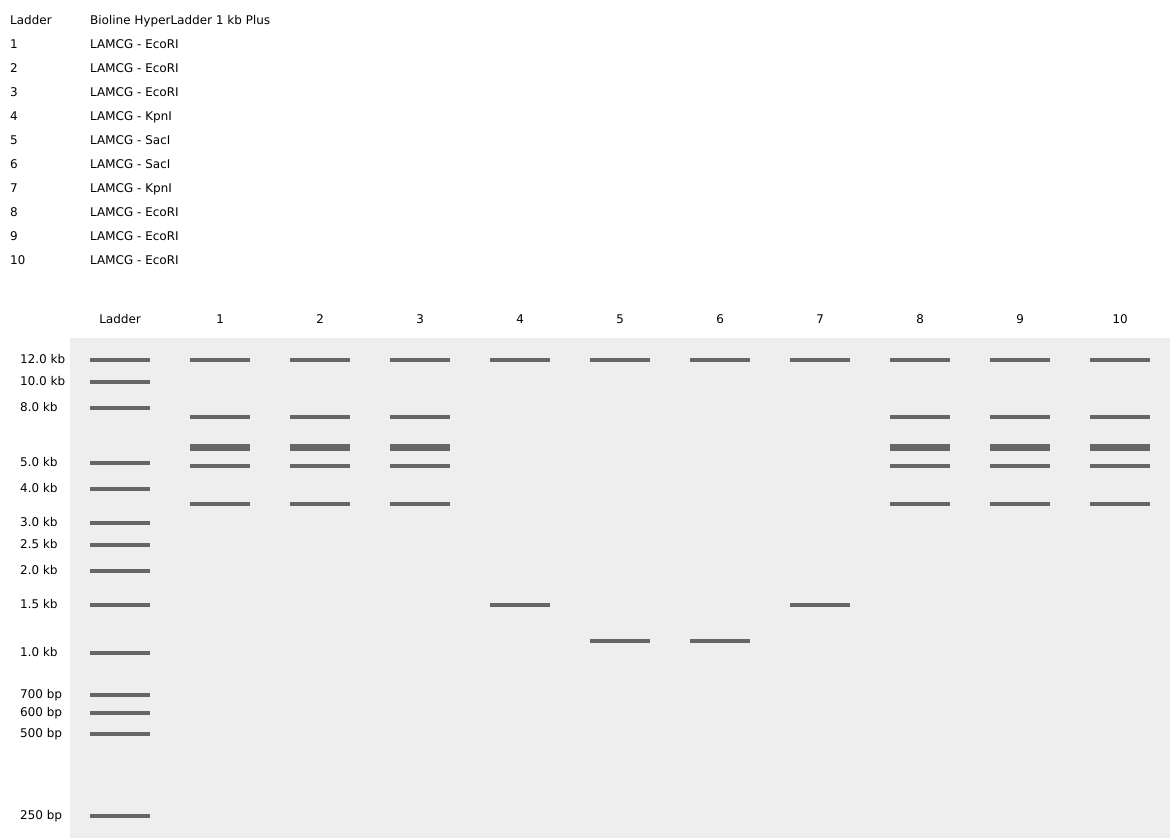
Part 1: Benchling & In-silico Gel Art: Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. It’s a smiley face!! :) Part 3: DNA Design Challenge: 3.1 Chose your protein Alkaline phosphatase. I chose this protein because it plays because it is important for nutrient sensing, as it is naturally activated under conditions of phosphate limitation. In Escherichia coli, alkaline phosphatase (PhoA) hydrolyzes organic phosphate compounds to release inorganic phosphate, directly linking environmental signals to nutrient availability. Additionally, it is a well-characterized enzyme with a resolved structure and extensively documented sequence information, making it an ideal model protein for computational and experimental analysis.
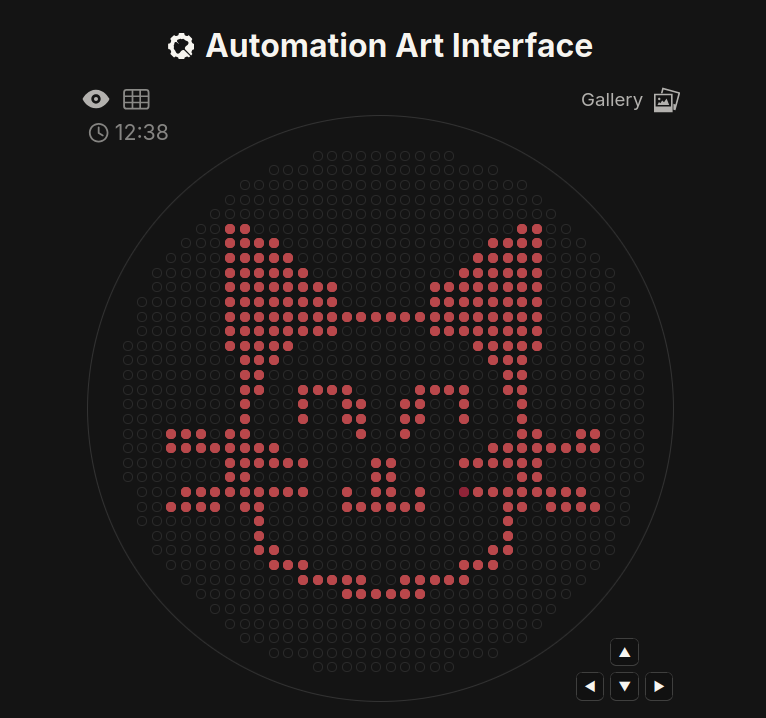
Part 1: Python Script for Opentrons Artwork I used the amazing Donovan’s tool to create this, then used the coordinates… It’s a homage to my cat Nico. He is a crazy cute orange cat. UPDATE (THE RESULT!) And… here is the result! I’m so happy, this is so cool! Thank you so much, USFQ node :) Oh, and here is the real Nico haha
Part A. Conceptual Questions All of the questions are so interesting, so c :D 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) To answer this question, we first need to know how much of meat is made of proteins (therefore, aminoacids). A quick search on Google returned that in 100g of meat we have ~26g of protein (this, of course, changes based on what type of meat we’re talking about, but let’s use this number). Then we have 130g of protein in 500g of meat. If on average an aminioacid is ~100 Daltons (equivalent to 100g/mol) then we have 1.3 mol in 500 grams of meat. Therefore 1.3 x 6 x 10^23 molecules.
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM I first generated the mutated sequence: SOD1 A4V MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I then used the PepMLM-650M notebook to generate peptide binders conditioned on the mutant SOD1 sequence. I set the peptide length to 12 amino acids and generated candidate binders from the model output. The resulting peptides and their pseudo-perplexity values are shown below.
Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix contains Phusion DNA polymerase, a enzyme that synthesizes DNA (this one also has proofreading activity). It also includes HF buffer, which provides the optimal pH and salt concentration (MgCl2) required for enzyme activity, as well as dNTPs, which are the nucleotide building blocks used to synthesize the new DNA strands. Glycerol helps stabilize the enzyme, and DMSO may be added to improve amplification of GC-rich regions by reducing secondary structure formation.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? An advantage of IANNs is that gene expression can vary continuously, meaning that—unlike Boolean functions, whose outputs are limited to ON or OFF states—IANNs can weigh inputs and produce graded responses based on their relative strengths, enabling more nuanced, flexible, and biologically realistic decision-making within the cell.
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis (CFPS) offers much greater flexibility and experimental control than in vivo expression because protein production occurs outside living cells, typically in a test tube using cellular extracts. This means variables such as DNA concentration, ion composition, temperature, cofactors, chaperones, and energy substrates can be precisely adjusted without worrying about cell viability, metabolism, or toxicity. Another notable example is time, as to perform CFPS takes 1 –2 days, whereas in vivo protein expression may take 1–2 weeks. Here are two examples of when CFPS can be efficiently used:
Final Project For my final project I would like to measure a few things:
whether the biosensor can detect carbon monoxide (CO) in a reliable and quantifiable way. The main readout will be the signal generated after CO binding, such as fluorescence, or a visible color change, depending on the final design. By exposing the system to known CO concentrations, I can build a calibration curve and evaluate sensitivity, detection range, and response time. also if the protein sensor itself is being produced correctly and remains functional. For this, I would use SDS-PAGE to confirm protein size and purity, and UV-visible spectroscopy to verify heme incorporation and observe spectral changes upon CO binding. To test specificity, I would compare the response to CO with other gases or environmental factors such as CO2 and humidity. Waters Part I — Molecular Weight Question 1 Using the online calculator, the calculated molecular weight (unmodified) is 28006.60
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Unfortunately, I didn’t have a chance to contribute because I never received the personalized URL. I guess I’ll have to make up for it by becoming a TA this fall :)
That said, I really loved the idea of collaborative bioart. My favorite part was the upper right section with the gene expression, it looks so cool. For next year, I think it could be even more engaging if there were multiple canvases or themes (for example, one per node or topic), so more people could contribute and explore different ideas within the same project.
1. First, describe a biological engineering application or tool you want to develop and why.
I want to develop a biological engineering system for smarter nutrient liberation in agricultural soils, especially for sugarcane. The current nutrient management system depends heavily on chemical fertilizers that release nutrients in poorly timed pulses, leading to massive nutrient losses, soil degradation, and environmental damage. I do not want to sugarcoat it: the situation is bad. I am Brazilian, which also means I come from the country that exports the most sugar in the world, largely through intensive sugarcane monoculture. My goal is to explore how engineered soil microorganisms could sense plant-derived signals and release nutrients only when they are biologically needed, turning the soil from a passive substrate into an active, responsive system.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
The sugar industry is a highly profitable one: the global sugar market was estimated to be valued at $46.4 billion in 2023. That being said, profitability also creates incentives for misuse. If a biological system significantly improves nutrient efficiency, it could be exploited to further intensify monoculture, expand cultivation into fragile ecosystems, or concentrate power in the hands of large agribusinesses rather than improving sustainability. A central governance goal, therefore, is to prevent technological gains from amplifying extractive or environmentally harmful practices. To do that, we can break down into sub-goals such as regulating deployment to avoid land-use expansion and establishing public or open-access frameworks that limit exclusive corporate ownership of engineered soil systems. This is important because it is very important that small and medium-scale farmers also have access to this technology.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Establishment of a biosafety and ecological oversight committee;
Purpose: Ensure that engineered soil microorganisms are biologically safe, ecologically contained, and function as intended. This includes preventing unintended persistence in the environment, horizontal gene transfer, or disruption of native soil microbiomes.
Design: Development of standardized biosafety evaluation protocols, including genetic safeguards (such as kill-switches or nutrient dependencies), controlled soil microcosm experiments, and long-term monitoring of microbial survival and gene stability. Ecological impact assessments would be required before any field deployment.
Risk of failure and “success”: Failure: engineered microorganisms persist uncontrollably in the environment, transfer genetic material to native species, or alter soil ecosystems in harmful ways. Success: the system demonstrates predictable behavior, effective containment, and minimal ecological disruption under real soil conditions.
Action 2: Phased field trials and agronomic validation
Purpose: Test whether the engineered microorganisms actually improve nutrient synchronization with plant demand without causing harm, ensuring that efficiency gains are real, reproducible, and context-dependent rather than theoretical.
Design: Multi-phase field trials beginning with small, controlled plots and gradually expanding to larger agricultural settings. Always havinf a control group, including runnning comparative studies against conventional fertilization practices, measuring nutrient use efficiency, crop yield, soil health indicators, and environmental runoff.
Risk of failure and “success”: Failure: the system fails to improve nutrient efficiency, behaves inconsistently across environments, or introduces new agronomic risks. Success: nutrient release becomes better synchronized with plant needs, fertilizer input is reduced, and soil quality is maintained or improved over multiple growing cycles.
Action 3: Governance frameworks to prevent extractive or monopolistic use
Purpose: Prevent efficiency gains from being exploited to intensify monoculture, expand cultivation into ecologically sensitive areas, or concentrate control within large agribusiness corporations.
Design: Creation of policy frameworks that tie deployment of engineered soil systems to sustainability metrics, land-use regulations, and open or publicly governed licensing models. This includes transparency requirements, public-sector involvement, and safeguards against exclusive proprietary ownership of essential soil biotechnologies.
Risk of failure and “success”: Failure: the technology is captured by profit-driven actors and used to accelerate environmental degradation or deepen agricultural inequality. Success: the system is deployed in ways that promote long-term soil resilience, equitable access for small and medium-scale farmers, and environmentally responsible agricultural practices.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option help to:
Option 1
Option 2
Option 3
Ensure biological and ecological safety
• Prevent unintended environmental harm
1
2
3
• Limit uncontrolled spread or persistence
1
2
3
Validate real-world effectiveness and reliability
• Demonstrate consistent performance across contexts
2
1
n/a
• Identify failures before large-scale deployment
2
1
n/a
Prevent extractive or profit-driven misuse
• Limit intensification of monoculture
3
2
1
• Prevent concentration of control by large agribusiness
3
2
1
Promote equitable access to the technology
• Enable access for small and medium-scale farmers
2
2
1
Maintain feasibility and avoid blocking research
• Feasible to implement in practice
2
2
3
• Does not unnecessarily slow scientific progress
2
3
2
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties
I would prioritize a combination of Option 1 (biosafety and ecological oversight) and Option 2 (phased field trials and agronomic validation) as the foundation for responsible governance. Because before thinking about the politics of how and by whom the technology will be used (although these questions should be considered from the beginning) we need to make sure it is actually safe to deploy. Establishing biological safety and ecological predictability should be (ALWAYS!) a prerequisite for any ethical discussion about scale, access, or commercialization.
FINAL REFLECTION
One ethical question that really made me stop and think during this project was whether making biological systems more efficient automatically makes them more sustainable. Engineering soil microorganisms to release nutrients in a smarter way could reduce fertilizer use and environmental damage, but it could also be used to push monoculture systems even further or expand agriculture into already fragile ecosystems (as I see it so frequently happening in Brazil :Brazil:). And honestly, I’m not completely sure where the safety limits of genetic modification are, especially in complex environments like soil. That uncertainty is part of what excites me - and I’m eager to explore, question, and learn more about this during HTGA :D
Week 2 lecture preparation
Homework Questions from Professor Jacobson:
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase makes about one mistake every one million bases copied (an error rate of ~10⁻⁶) after its built-in proofreading activity. The human genome is about three billion base pairs long, so if DNA were copied using only polymerase proofreading, each cell division would introduce thousands of errors 🤯. Biology solves this problem by adding extra layers of quality control after replication, especially mismatch repair and other DNA repair pathways, which reduce the final error rate to roughly one mistake per billion to ten billion bases.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein (~1036 bp, ~345 amino acids) can be encoded in astronomically many ways because most amino acids are specified by multiple synonymous codons; in theory this corresponds to roughly to 1 followed by 150 zeros (🤯) different DNA sequences that encode the same protein.
In practice, most of these sequences fail because of codon bias, effects on mRNA stability and secondary structure (as shown by studies linking codon usage to mRNA half-life and folding), translation speed and co-translational folding (as revealed by ribosome-profiling and folding-kinetics experiments), GC-content constraints, and embedded regulatory or splicing signals within coding regions.
We can think of this problem as
Homework Questions from Dr. LeProust:
1. What’s the most commonly used method for oligo synthesis currently?
The typical method for Oligonucleotide synthesis is the phosphoramidite method
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Chemical oligo synthesis accumulates errors because each nucleotide coupling step is slightly imperfect. Altoug there exists papers on
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2000 bp gene would require thousands of sequential coupling steps, causing error rates and truncation products to completely dominate over correct full-length molecules. The yield of an error-free sequence would be essentially zero, so long genes must instead be assembled from shorter, high-quality oligos using enzymatic methods like PCR or Gibson assembly (ChatGPT helped me aswer. My prompt: The question + so what is usually done when a 2000bp+ gene is needed?.
Homework Questions from George Church:
1. What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The ten essential amino acids in animals are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine. Lysine Contigency is a safety mechanism from Jurassic Park that moodifies dinosaurs so that they aren’t able to produce the essential amino acid lysine, and therefore need the assistance of the staff from the Park to acquire this necessary molecule. This, in theory (in the story), prevents the dinosaurs from living outside the island. But this “Lysine Contingency” doesn’t make much sense to me, as lysine is already an essential amino acid, meaning that our bodies do not produce it and we must obtain it from diet.
Week 2 HW: DNA r/w/e
Part 1: Benchling & In-silico Gel Art:
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. It’s a smiley face!! :)
Part 3: DNA Design Challenge:
3.1 Chose your protein Alkaline phosphatase. I chose this protein because it plays because it is important for nutrient sensing, as it is naturally activated under conditions of phosphate limitation. In Escherichia coli, alkaline phosphatase (PhoA) hydrolyzes organic phosphate compounds to release inorganic phosphate, directly linking environmental signals to nutrient availability. Additionally, it is a well-characterized enzyme with a resolved structure and extensively documented sequence information, making it an ideal model protein for computational and experimental analysis.
3.3 Codon optimization Codon optimization is important because organisms differ on the way the use certain codons. We know the genetic code is universal and degenerate, meaning that one aminoacid can be encoded by multiple codons, but different organisms do not use these synonymous codons with the same frequency. This phenomenon, known as codon usage bias, affects how efficiently a gene is translated. If a DNA sequence contains codons that are rare in the host organism, the corresponding tRNAs may be scarce, which can slow down translation, reduce protein yield, or even affect proper folding. Therefore codon optimization adjust the sequence to match the codons preferred by the organism, therefore improving the yield and overall expression. For this project, I chose to optimize the codon sequence for Escherichia coli because it is one of the most widely used host organisms for recombinant protein expression. It grows rapidly, is inexpensive to culture, has well-characterized genetics, and many expression vectors are available. Since alkaline phosphatase (PhoA) is naturally found in E. coli, optimizing for this host ensures efficient production while maintaining compatibility with standard molecular biology workflows. I used *VectorBuilder’s Codon Optimization Tool. Result: Pasted Sequence: GC=61.29%, CAI=1.00 // Improved DNA[1]: GC=59.38%, CAI=0.93 Sequence: ATGAAACAGAGCACCATTGCGCTGGCGCTGCTGCCGCTGCTGTTTACCCCGGTCACGAAAGCGCGCACCCCGGAAATGCCGGTGCTGGAAAACCGCGCAGCACAGGGTGATATCACCGCGCCGGGCGGTGCGCGCCGCCTGACCGGCGATCAGACCGCGGCCCTGCGCGATAGCCTGAGCGATAAACCGGCGAAAAATATTATTCTGCTGATTGGCGACGGTATGGGCGATAGCGAAATCACCGCCGCGCGCAATTATGCGGAAGGCGCCGGTGGCTTTTTTAAAGGTATCGATGCGCTGCCGCTGACCGGCCAGTACACCCACTACGCGCTGAATAAAAAAACTGGTAAACCGGATTATGTCACCGATAGTGCGGCCAGCGCAACCGCGTGGAGCACCGGCGTGAAAACCTACAATGGCGCGCTGGGCGTGGATATTCATGAAAAAGATCACCCGACGATTCTGGAAATGGCGAAAGCGGCCGGCCTGGCGACCGGCAATGTGAGCACCGCGGAACTGCAGGATGCCACCCCGGCGGCGCTGGTGGCCCATGTGACCAGCCGTAAATGCTATGGTCCGAGCGCGACCAGCGAAAAATGTCCGGGCAACGCGCTGGAAAAAGGTGGCAAAGGCAGCATTACCGAACAGCTGCTGAACGCGCGTGCCGATGTGACCCTGGGCGGAGGCGCAAAAACCTTTGCCGAAACCGCGACCGCGGGCGAATGGCAGGGCAAAACCCTGCGCGAACAGGCGCAGGCCCGCGGTTATCAGCTGGTTAGCGATGCGGCCAGCCTGAATAGCGTGACCGAAGCGAACCAGCAGAAACCGCTGCTGGGCCTGTTTGCGGATGGTAATATGCCGGTGCGCTGGCTGGGTCCGAAAGCGACCTATCACGGTAACATTGATAAACCGGCGGTGACCTGCACCCCGAACCCGCAGCGCAACGATAGCGTGCCGACCCTGGCACAGATGACCGATAAAGCCATTGAACTGCTGAGCAAAAATGAAAAAGGCTTTTTCCTGCAGGTGGAAGGCGCGTCCATTGATAAACAGGATCACGCAGCCAACCCGTGTGGCCAGATTGGCGAAACCGTGGATCTGGATGAAGCGGTGCAGCGTGCCCTGGAATTTGCGAAAAAAGAAGGTAACACCCTGGTGATTGTGACCGCAGACCATGCGCATGCGAGCCAGATCGTGGCGCCGGATACCAAAGCGCCGGGTCTGACCCAAGCGTTGAATACCAAAGATGGTGCGGTGATGGTGATGAGCTATGGCAACAGCGAGGAAGATAGCCAGGAACACACCGGCAGTCAGCTGCGTATTGCCGCATACGGCCCGCATGCGGCGAACGTGGTGGGCCTGACCGATCAGACCGACCTGTTTTACACCATGAAAGCGGCACTGGGCCTGAAA
3.4. You have a sequence! Now what? Once the optimized DNA sequence is obtained, it can be cloned into an expression plasmid containing a promoter, ribosome binding site, and selection marker, and then introduced into a host such as Escherichia coli through transformation. Inside the cell, the promoter drives transcription of the DNA into mRNA by RNA polymerase, and ribosomes bind to the mRNA to translate its codons into the corresponding amino acid sequence, producing the alkaline phosphatase protein, which then folds into its functional structure. This is the cell-dependent method.
Part 4: Prepare a Twist DNA Synthesis Order:
I attempted to place the Twist DNA synthesis order, but the platform did not function properly. Although I was able to log in, a message stating “Contact your Distributor” repeatedly appeared, and the system automatically logged me out, preventing me from proceeding with the order. I am aware that other students in my region (Brazil/Latin America) experienced similar difficulties accessing the platform.
Part 5: DNA Read/Write/Edit:
DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? I would sequence soil metagenomic DNA from the sugarcane rhizosphere (and bulk soil as a control) to understand which microbial communities and genes are present that control nutrient cycling. In particular, I’d look for taxa and functional genes involved in phosphorus solubilization/mineralization (e.g., phosphatases), nitrogen transformations (nitrification/denitrification), and other pathways that determine whether nutrients are retained, lost, or made bioavailable. This would let me link nutrient status (low/high P or N) to shifts in microbial composition and metabolic potential.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Illumina shotgun metagenomic sequencing, which is a second-generation technology because it performs massively parallel sequencing of millions of short DNA fragments using sequencing-by-synthesis chemistry. The input would be total DNA extracted from soil samples, which would be fragmented, end-repaired, ligated to sequencing adapters (with barcodes if multiplexing), PCR-amplified if necessary, and loaded onto a flow cell to generate clusters. During sequencing, fluorescently labeled nucleotides are incorporated one at a time, and imaging after each cycle detects the emitted fluorescence to determine the identity of each base (base calling). The output consists of millions of short reads in FASTQ format, containing both nucleotide sequences and quality scores, which can then be analyzed to identify microbial taxa and nutrient-cycling genes.
DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? I would like to synthesize a nutrient-responsive genetic circuit for soil bacteria that turns nutrient mobilization “on” only when the environment is nutrient-limited—for example, a phosphate-starvation–responsive promoter (Pho regulon) driving a codon-optimized alkaline phosphatase (PhoA) module to help mineralize organic phosphate into bioavailable phosphate. Concretely, the construct would look like [P_Pho]–RBS–phoA–terminator, where the PhoA protein sequence can be sourced from UniProt P00634 and the Pho regulon is sourced from the well-characterized PhoB/PhoR phosphate-sensing regulatory system in bacteria.
(ii) What technology or technologies would you use to perform this DNA synthesis and why? I would use commercial gene synthesis based On sdlid-phase phosphoramidite oligonucleotide synthesis followed by DNA assembly (like Gibson Assembly,as I think we are going to learn in HTGAA) because this is the most reliable way to “write” a custom genetic circuit with a good sequence control. In practice, short oligos are chemically synthesized cycle-by-cycle, then cleaved/deprotected and purified, and multiple fragments are assembled into the full construct (they are oftten sequneced afterward, to check if everything is ok). The main limitations are that error rates and cost increase with sequence length, as long constructs require multi-fragment assembly and verification.
DNA Edit
(i) What DNA would you want to edit and why? I would want to edit the genomes of plant-associated soil bacteria to enhance their ability to sense nutrient limitation and respond in a controlled, beneficial way. Specifically, I would modify regulatory regions of genes involved in phosphate metabolismto fine-tune their sensitivity and dynamic range.
(ii) What technology or technologies would you use to perform these DNA edits and why? I would use CRISPR-Cas9 genome editing because it is the standard technique and it allows precise modification of specific DNA sequences in bacteria. CRISPR edits DNA by using a designed guide RNA (gRNA) that directs the Cas9 nuclease to a complementary target sequence, where Cas9 introduces a double-strand break; the cell then repairs this break either through non-homologous end joining (introducing small insertions or deletions) or through homology-directed repair if a donor DNA template is provided, enabling precise edits. Preparation involves designing the guide RNA sequence, constructing or obtaining a plasmid encoding Cas9 and the gRNA, optionally designing a repair template with homology arms, and delivering these components into the target bacterial cells (e.g., via transformation or electroporation). Limitations include variable editing efficiency depending on the organism, potential off-target effects if guide design is imperfect, lower efficiency of homology-directed repair in some bacteria, and challenges in delivering editing machinery into non-model environmental strains.
Week 3 HW: Lab Automation
Part 1: Python Script for Opentrons Artwork
I used the amazing Donovan’s tool to create this, then used the coordinates… It’s a homage to my cat Nico. He is a crazy cute orange cat.
UPDATE (THE RESULT!)
And… here is the result! I’m so happy, this is so cool! Thank you so much, USFQ node :)
Oh, and here is the real Nico haha
Part 2: Post-Lab Questions
Question 1 I chose this paper: Automation of protein crystallization scaleup via Opentrons-2 liquid handling by DeRoo et al.
I’m really into structural biology, and protein crystallization is a critical step in it, because it enables structure determination via X-ray crystallography. However, traditional crystallization trials are highly manual, repetitive, and sensitive to small variations in liquid handling. This paper demonstrates how the Opentrons-2 liquid handling robot can be used to automate protein crystallization experiments, specifically sitting-drop crystallization in 24-well plates.
Question 2 I intend to use the Opentrons robot to automate the fabrication, assembly, and testing of paper-based biosensors that detect environmental signals (such as carbon monoxide). The core idea is to use automation to precisely deposit reagents onto paper substrates in reproducible patterns, enabling scalable production and systematic testing of biosensor designs. I do need to research more to undderstand exactly what is possible or not with OpenTrons.
Part 3: Final Project Ideas
It’s all on the slides!
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
All of the questions are so interesting, so c :D 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) To answer this question, we first need to know how much of meat is made of proteins (therefore, aminoacids). A quick search on Google returned that in 100g of meat we have ~26g of protein (this, of course, changes based on what type of meat we’re talking about, but let’s use this number). Then we have 130g of protein in 500g of meat. If on average an aminioacid is ~100 Daltons (equivalent to 100g/mol) then we have 1.3 mol in 500 grams of meat. Therefore 1.3 x 6 x 10^23 molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish? Well, we are not literally what we eat. When we eat - a cow or a fish - we are not reusing their nutrients (proteins, carbohydrates) as our own, but we metabolize (break down) these nutrients: proteins are broken down into amino acids, carbohydrates into simple sugars, and fats into fatty acids. Then our cells use these building blocks to grow and survive in a unique way, defined by what is written in our DNA, that therefore, determines who we are as humans.
3. Why are there only 20 natural amino acids? This is likely the result of an evolutionary optimization where this number covers a wide range of chemical diversity while still not being too costly for the cells to use and synthesise. If the number was lower, it would be harder to cover the diversity needed to manufacture all kinds of proteins; if the number was higher, there would be a need to have more and maybe less efficient cell machinery.
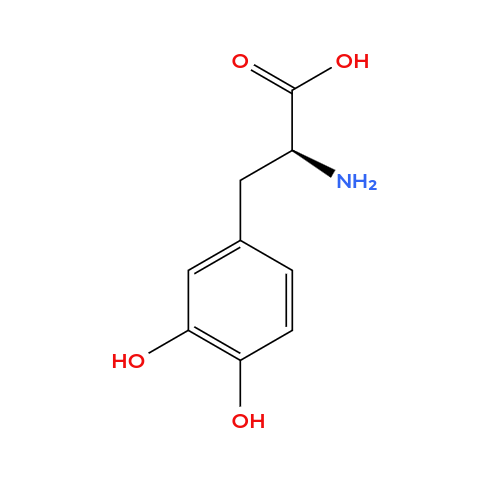
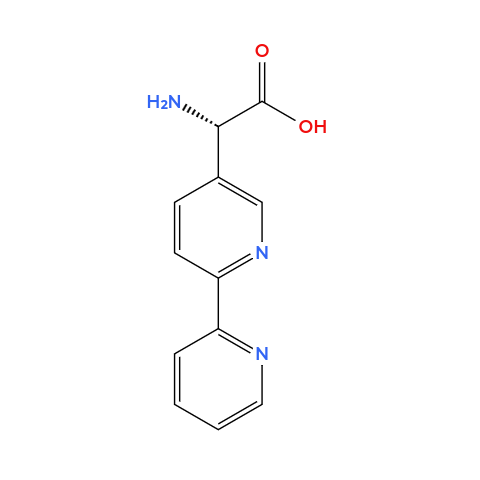
4. Can you make other non-natural amino acids? Design some new amino acids. Amino acids, by definition, are organic compounds that contain both amino and carboxylic acid functional groups. It’s also a group of chemicals way larger then the only 20 natural amino acids from the genetic code. Therefore, we can design some new aminoacids, so I chose to design
✨magnetic✨ aminoacids (not paramagnetic per se but can make iron bind). These residues whose side chains contain a metal-chelating group capable of binding paramagnetic ions such as Fe2+. Here are my designs (I used Molview to create the molecules):
Ferrophenylalanine: it’s basically a phenylalanine but the side chain contains a catechol group (two adjacent hydroxyl groups on an aromatic ring), which is known to strongly chelate Fe(III) through bidentate coordination.
Bipyridylalanine: it’s an alanine but the side chain contanis a bipyridine ring (two linked pyridines, two nitrogens pointing inward), this forms stable complexes with paramagnetic metal ions.
Note: I used ChatGPT to help me understand how to design these magnetic aminoacids, that is, how to design a side chain able to be metal binding (prompt [translated from portuguese]: I want to make other non-natural amino acids and I need help thinking of side chains that are metal-binding, an aminoacid able to bind metals like iron)
5. Where did amino acids come from before enzymes that make them, and before life started? Before enzymes and life, amino acids could come from pre-biotic chemistry: an atmosphere with water, methane, ammonia and hydrogen gas. The Milley-Urey experiment showed that when you hit such atmosphere with an electrical discharge (simulating lightning), amino acids can form. Therefore, the experiment shows that the formation of amino acids does not need complex organic molecules to start.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? I would expect the handedness to be left, and the opposite is true: if you make an α-helix using L-amino acids, the handednesse will be right.
7. Can you discover additional helices in proteins? Because helix formation depends on specific backbone geometry and hydrogen bond distances, only a finite number of stable helices are possible. While some theoretical helices (such as the γ-helix) have been proposed, most are energetically unfavorable. However, through protein design, it may be possible to stabilize rare or artificial helical conformations.
8. Why are most molecular helices right-handed? Most molecular helices in biology are right-handed because biological polymers are constructed from homochiral building blocks. In proteins, L-amino acids favor right-handed α-helices due to optimal backbone dihedral angles and minimal steric clashes. In DNA, the D-sugar stereochemistry biases the backbone geometry and base stacking interactions toward a right-handed double helix. Thus, chirality combined with energy minimization determines the predominant handedness of biological helices.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? β-sheets tend to aggregate because β-strands have exposed backbone hydrogen bond donors and acceptors that can form intermolecular hydrogen bonds. When multiple strands align, they form extended sheet structures that are energetically stabilized. The primary driving force for aggregation, therefore is hydrogen bonding and hydrophobic effect.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Many amyloid diseases involve proteins that misfold into β-sheet–rich conformations. β-strands can align and form highly ordered cross-β structures stabilized by extensive intermolecular hydrogen bonding and hydrophobic interactions. Because these β-sheets can extend indefinitely, they form elongated fibrils that accumulate in tissues. Because amyloid β-sheets are highly ordered, stable, and versatile, it is possible to use it as materials, such as nanofibrils.
Part B. Protein Analysis and Visualization
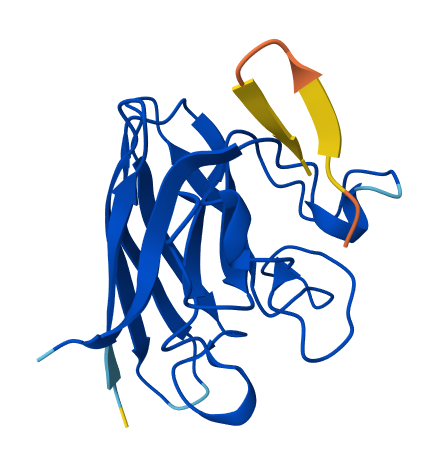
1. Briefly describe the protein you selected and why you selected it. I chose human hemoglobin (specifically one of the alpha subunit - PDB: 7VDE), the protein responsable for carrying oxygen in the blood and also the reason why blood is red. I chose it because of its historical importance for structural biology and also because I want to work with it in one of my HTGAA projects.
2. Identify the amino acid sequence of your protein. MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. The alpha unit is 142 residues long; the most frequent amino acid is alanine (21 times)
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Hemoglobin (alpha subunit) is paralog to the beta subunit and also has some orthologs: BLAST identified 245 homologs, all Hemoglobin subunit alpha from different organisms.
Does your protein belong to any protein family? Yes, to the globin family.
3. Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å) The PDB I’m using (human hemoglobin - 7VDE) was solved in 2021, the quality is not that good (3.60 Å) but is Cryo-EM so it’s pretty solid.
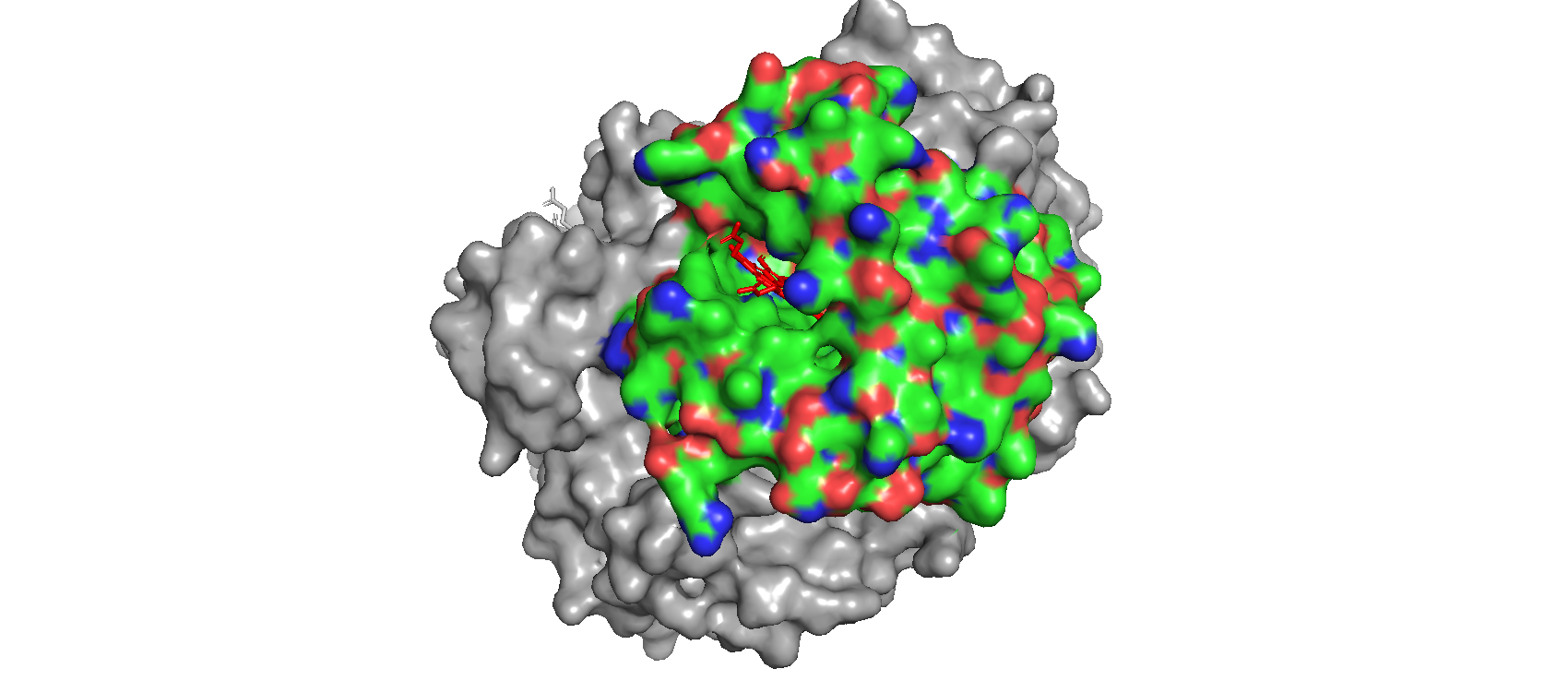
Are there any other molecules in the solved structure apart from protein? Yes, the heme group.
Does your protein belong to any structure classification family? Yes, Globin or Globin-like (Scoop)
4. Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets? It has more helices. It has no sheets.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? When I use the surface mode, I can se the hydrophic residues are a bit less exposed than the hydrophilic ones.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? Yes! Where oxygen binds!
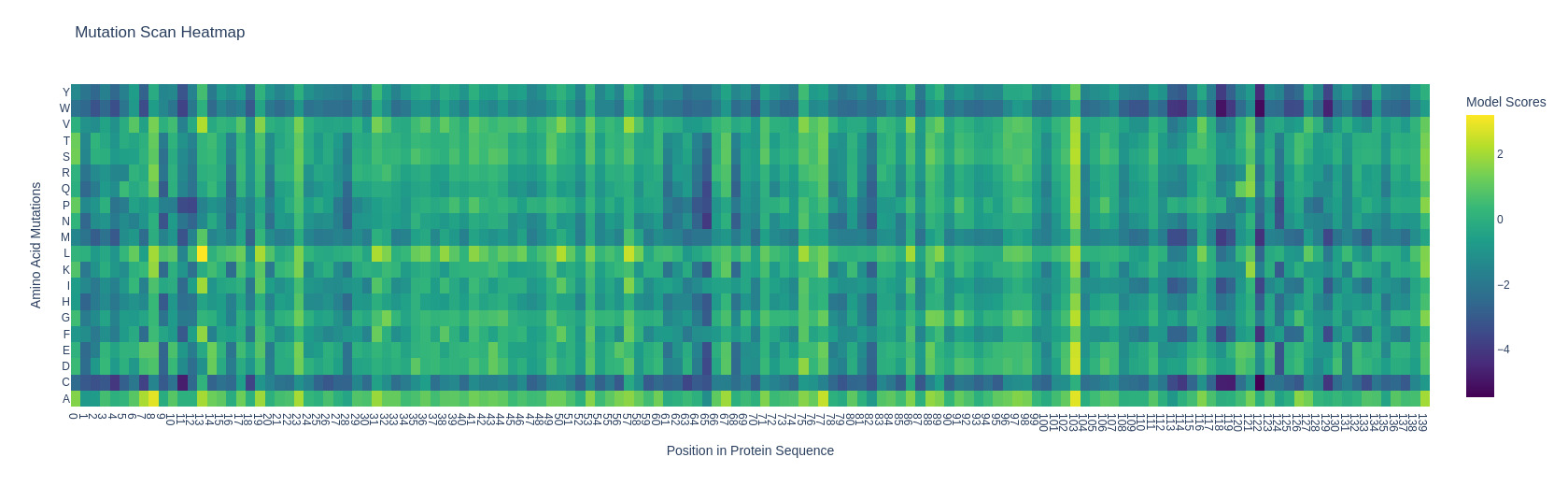
I’m using (and I’ll be using for the whole homework) the subunit alpha of human hemoglobin. Something interesting that I noticed with the mutation heatmap is that changing residues to cystein usually causes a loss of stability. For exampleA124C (Alanine at position 124 mutated to Cysteine) exhibited the lowest LLR of -5.4675 mutation scan heatmap
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
1. Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out) I’m using (and I’ll be using for the whole homework) the subunit alpha of human hemoglobin. Something interesting that I noticed with the mutation heatmap is that changing residues to cystein usually causes a loss of stability. For example A124C (Alanine at position 124 mutated to Cysteine) exhibited the lowest LLR of -5.4675
2. Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins? Yes, they do. It’s possible to see some clusters forming from similar proteins: when the sequence of aminoacids is transformed into a vector (embedding), similar proteins stick together. One example is given on the next question.
Place your protein in the resulting map and explain its position and similarity to its neighbors. I used the gemini chat to help me highlight my protein. Interestingly, the proteins sitting next to mine (alpha subunit of human hemoglobin) were almost always hemoglobins or other types of globins, like myoglobin. Below it is shown an example. (some proteins showed “automated matches” instead of an actual name, I’m not sure why)
C2. Protein Folding
Folding a Protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
I first generated the mutated sequence: SOD1 A4V MATK**V**VCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I then used the PepMLM-650M notebook to generate peptide binders conditioned on the mutant SOD1 sequence. I set the peptide length to 12 amino acids and generated candidate binders from the model output. The resulting peptides and their pseudo-perplexity values are shown below.
Binder
Pseudo Perplexity
WRYYAVALALKE
11.254131
WLVYVAAARLKE
11.480680
KRYYAAGAAWWE
14.88393
WHYPAVVARHWE
12.81336
FLYRWLPSRRGG
As the article mentions it, pseudo perplexity (PPL) is used to score binders.
Part 2: Evaluate Binders with AlphaFold3
I evaluated the predicted complexes between mutant SOD1 (A4V) and each generated peptide using AlphaFold3. Here is the table with the results:
Peptide
ipTM
Binding location
WRYYAVALALKE
0.25
outer surface of the β-barrel
WLVYVAAARLKE
0.24
outer surface of the β-barrel
KRYYAAGAAWWE
0.45
outer surface of the β-barrel, but a bit more interesting because it has a higher ipTM shows secondary structure
WHYPAVVARHWE
0.29
outer surface of the β-barrel, also shows a bit of secondary structure
FLYRWLPSRRGG
0.34
outer surface of the β-barrel
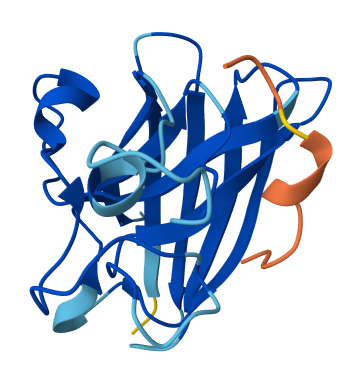
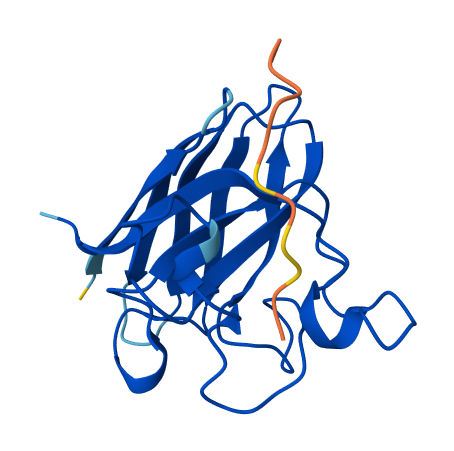
All of the peptides appear to contact basically only the outer surface of the β-barrel, which I guess is not ideal. Here are some pictures:
WRYYAVALALKE
WLVYVAAARLKE
KRYYAAGAAWWE
WHYPAVVARHWE
FLYRWLPSRRGG
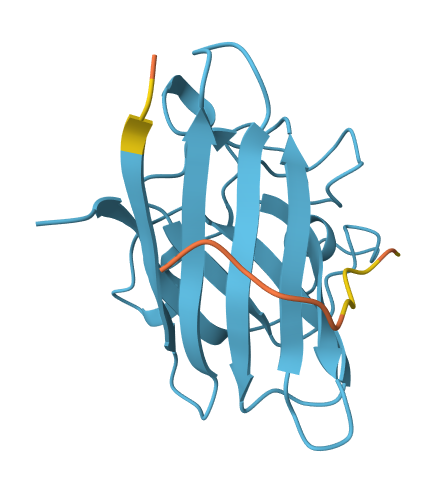
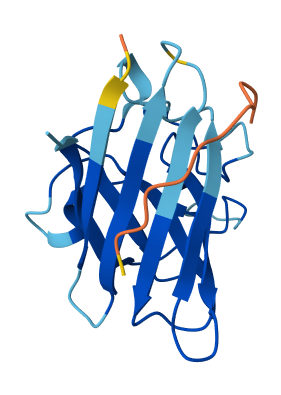
Across all candidates, the peptides consistently localized to the outer surface of the SOD1 β-barrel, without clear engagement of the N-terminal region where the A4V mutation is located, nor obvious interaction with the dimer interface. In all cases, the peptides appeared largely surface-bound rather than deeply buried, suggesting weak or nonspecific interactions.
The ipTM values for all complexes were relatively low (0.24–0.45), indicating limited confidence in the predicted protein–peptide interfaces. Among the candidates, KRYYAAGAAWWE showed the highest ipTM (0.45) and displayed some degree of secondary structure, which may suggest a slightly more organized interaction compared to the other peptides. However, even this value remains below the range typically associated with strong or reliable binding predictions.
Overall, the PepMLM-generated peptides did not show strong or well-localized binding to functionally relevant regions of SOD1. Compared to the known binder, none of the generated peptides clearly exceeded its predicted interaction quality. In fact, both the generated peptides and the known binder appeared to produce relatively weak or diffuse binding modes in AlphaFold, suggesting that this system may be challenging to model accurately using this approach. Also, all binders were generally colored yellow to orange, indicating lower confidence on the prediction of the peptide’s structure itself.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide
Binding Affinity (pKd/pKi)
Solubility
Hemolysis Prob.
Net Charge (pH 7)
Molecular Weight (Da)
GRAVY
WRYYAVALALKE
6.958
Soluble
0.082
0.77
1482.7
0.15
WLVYVAAARLKE
6.231
Soluble
0.065
0.77
1418.7
0.61
KRYYAAGAAWWE
6.706
Soluble
0.052
0.77
1471.6
-0.79
WHYPAVVARHWE
5.873
Soluble
0.036
-0.06
1550.7
-0.59
FLYRWLPSRRGG
5.968
Soluble
0.047
2.76
1507.7
-0.71
Across all candidates, PeptiVerse predicted high solubility (probability = 1.000) and low hemolysis probability (~0.03–0.05), suggesting that none of the peptides present immediate toxicity or formulation concerns. However, all peptides were predicted to have weak binding affinity (pKd/pKi ≈ 5.8–6.7), which is at least (😟) consistent with the AlphaFold3 results, where all complexes showed relatively low ipTM values (0.24–0.45) and surface-bound interactions on the β-barrel.
From a developability perspective, KRYYAAGAAWWE and WHYPAVVARHWE appear particularly favorable due to their moderate net charge (~0), high solubility, and low hemolysis probability. In contrast, FLYRWLPSRRGG has a significantly higher positive charge (+2.76), which may increase nonspecific interactions despite acceptable solubility and low hemolysis prediction.
No clear correlation was observed between structural confidence and predicted binding affinity. For example, KRYYAAGAAWWE, which had the highest ipTM in AlphaFold3 (0.45), showed relatively weak predicted affinity (6.706), while WHYPAVVARHWE displayed the strongest predicted affinity (5.873) despite a lower ipTM (0.29). This suggests that structural confidence alone is not sufficient to identify the strongest binders.
Overall, the results indicate that the generated peptides tend to form weak, nonspecific interactions, both structurally and energetically. If selecting one peptide to advance, I would choose KRYYAAGAAWWE, as it provides the best balance between structural confidence (highest ipTM) and favorable therapeutic properties, even though its predicted binding affinity remains modest. This makes it the most reasonable starting point for further optimization.
Week 6 HW: Genetic Circuits Part I
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains Phusion DNA polymerase, a enzyme that synthesizes DNA (this one also has proofreading activity). It also includes HF buffer, which provides the optimal pH and salt concentration (MgCl2) required for enzyme activity, as well as dNTPs, which are the nucleotide building blocks used to synthesize the new DNA strands. Glycerol helps stabilize the enzyme, and DMSO may be added to improve amplification of GC-rich regions by reducing secondary structure formation.
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature is mainly determined by the primer melting temperature (Tm), which depends on primer length, GC content, sequence composition, and buffer salt concentration.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR generates linear DNA fragments by amplifying a specific region through repeated cycles of denaturation, primer annealing, and extension by DNA polymerase. Restriction enzyme digestion generates linear fragments by cutting existing DNA at specific recognition sites. PCR is preferable when amplifying a specific sequence or adding custom overhangs, while restriction digests are preferable when suitable cut sites are already present in the DNA.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson Assembl only works if the sequences you want to clone have homologous overlaps, typically around 20–40 base pairs. So you ensure that it will work by designing PCR primers that include overlap sequences complementary to the neighboring fragment or vector.
5. How does the plasmid DNA enter the E. coli cells during transformation?
There are two famous methods for transformation (the process in which the plasmid enters the cell):
Chemical Transformation (Heat Shock): where cells are treated with cold divalent cations and then briefly exposed to high temperatures (42°C) to create transient pores in the membrane.
Electroporation: electrical pulse are used to create temporary pores in the membrane, allowing DNA entry.
6.Describe another assembly method in detail (such as Golden Gate Assembly)
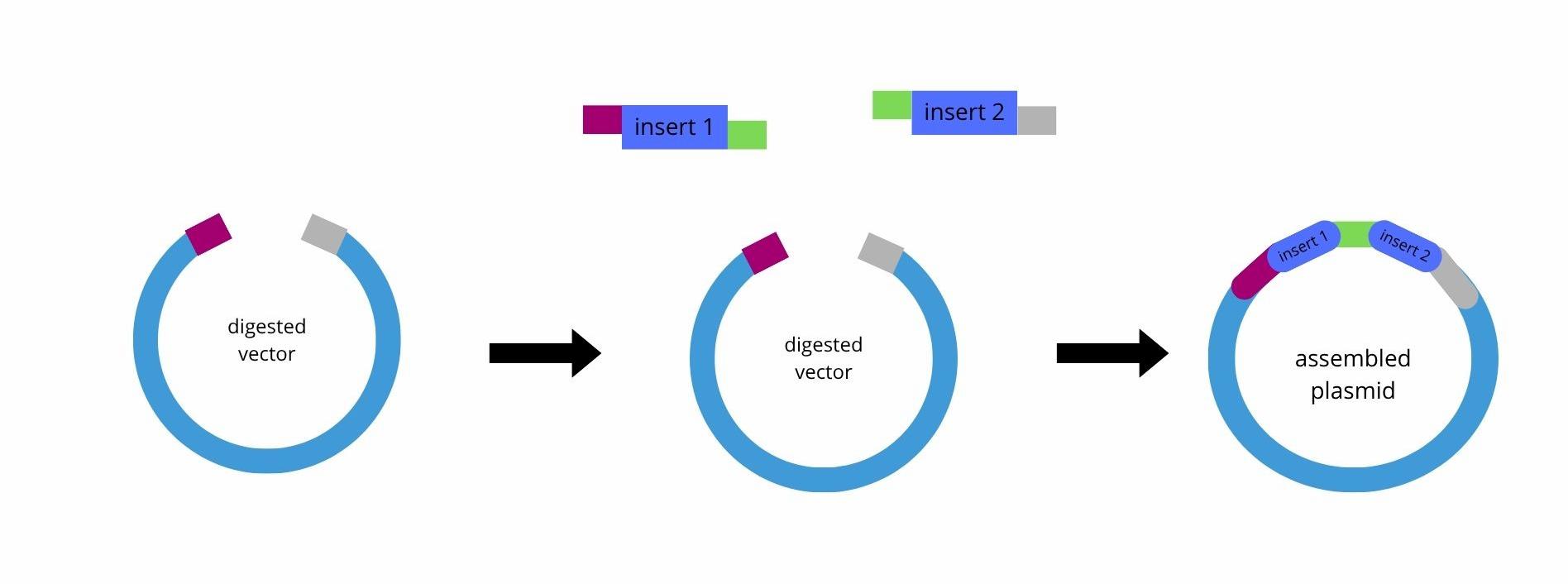
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online). Golden Gate Assembly is a DNA assembly method that uses Type IIS restriction enzymes, such as BsaI or BsmBI, together with DNA ligase to join multiple DNA fragments in a defined order. Unlike standard restriction enzymes, the Type IIS enzymes cut outside of their recognition sequence, which possibilizes the creation of custom single-stranded overhangs. These overhangs can be designed so that each fragment only ligates to the correct neighboring fragment, and this is important if you want an assembly more directional (where the order matters, for example).
During the reaction, the DNA fragments are first digested by the restriction enzyme, producing complementary sticky ends. DNA ligase then joins these compatible ends. Because the recognition sites are removed after correct ligation, the final assembled product is no longer cut in subsequent cycles, which increases assembly efficiency. The reaction is typically performed in thermocycling steps alternating between digestion (for example, 37°C) and ligation (16°C).
Assignment: Asimov Kernel
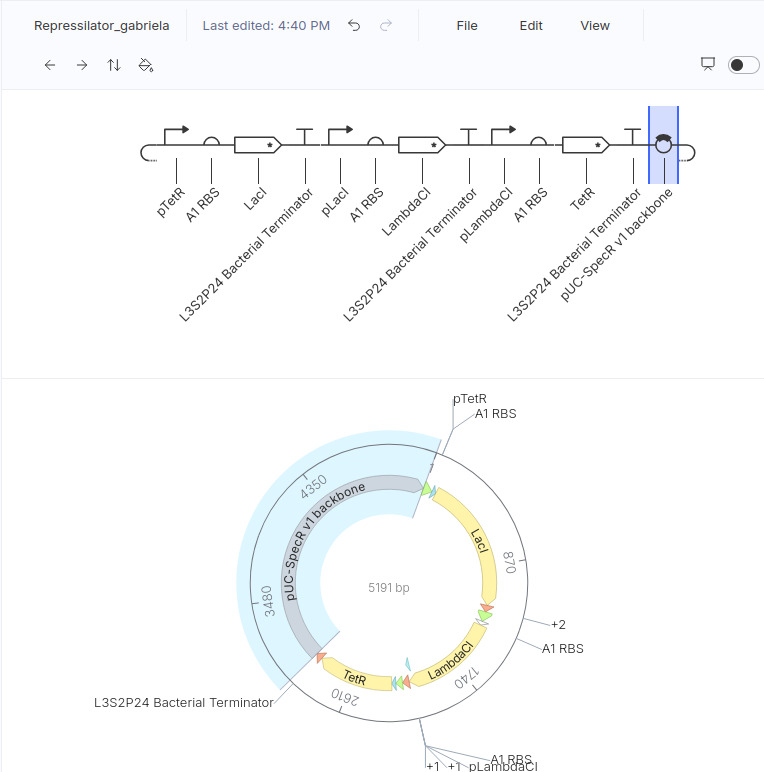
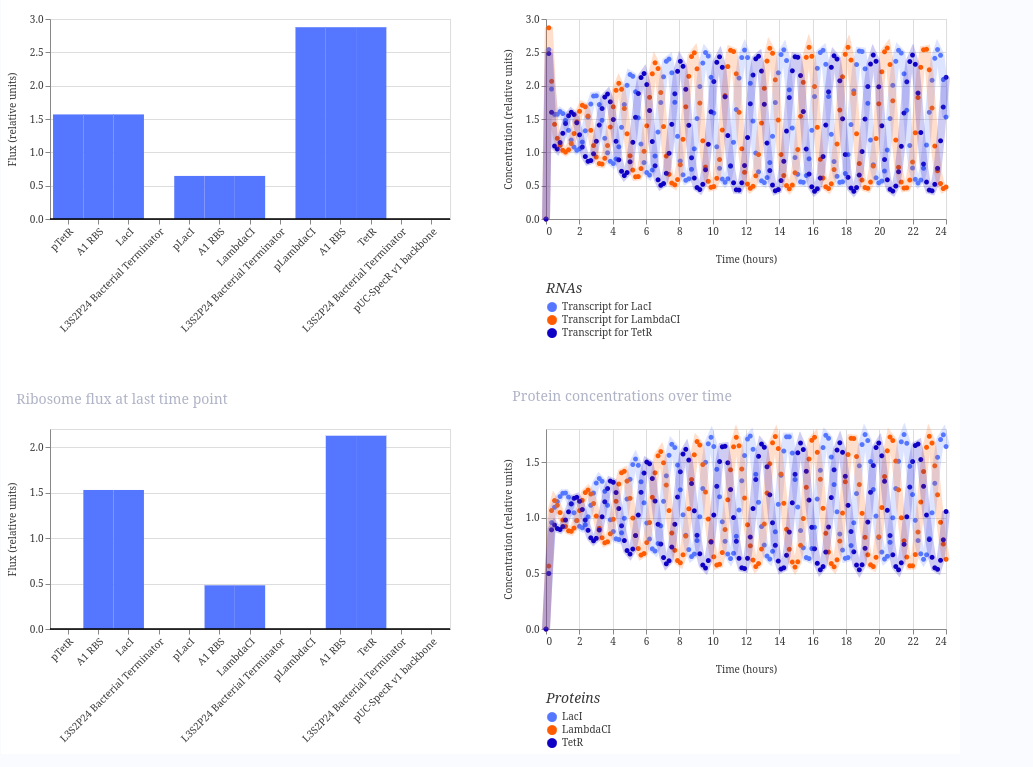
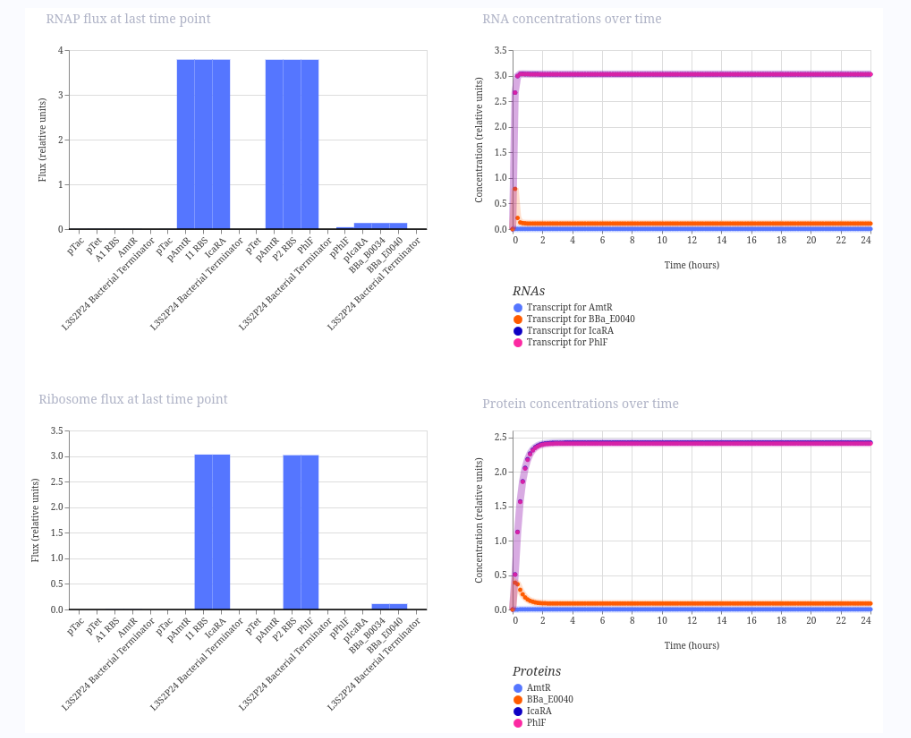
Repressilator
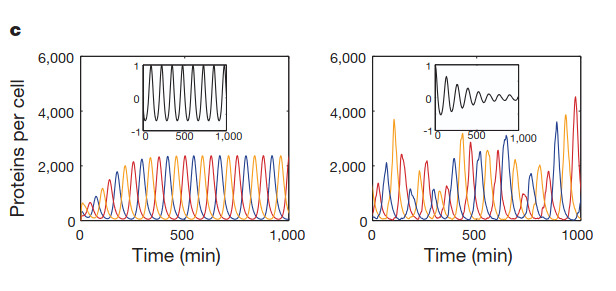
I ran the repressilator using a 24-hour duration because it generated plots such as the one published by Elowitz in the year 2000.
This is my design (copied from the demo):
These are the plots generated:
And this is a figure taken from the Elowitz’s article
My constructs
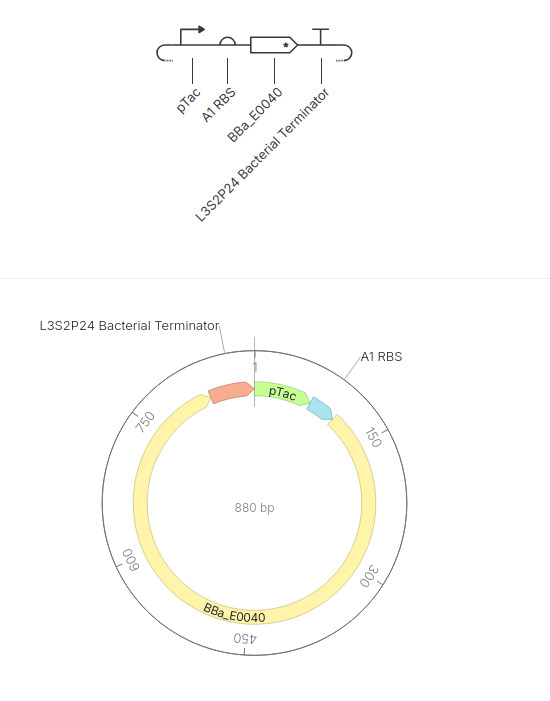
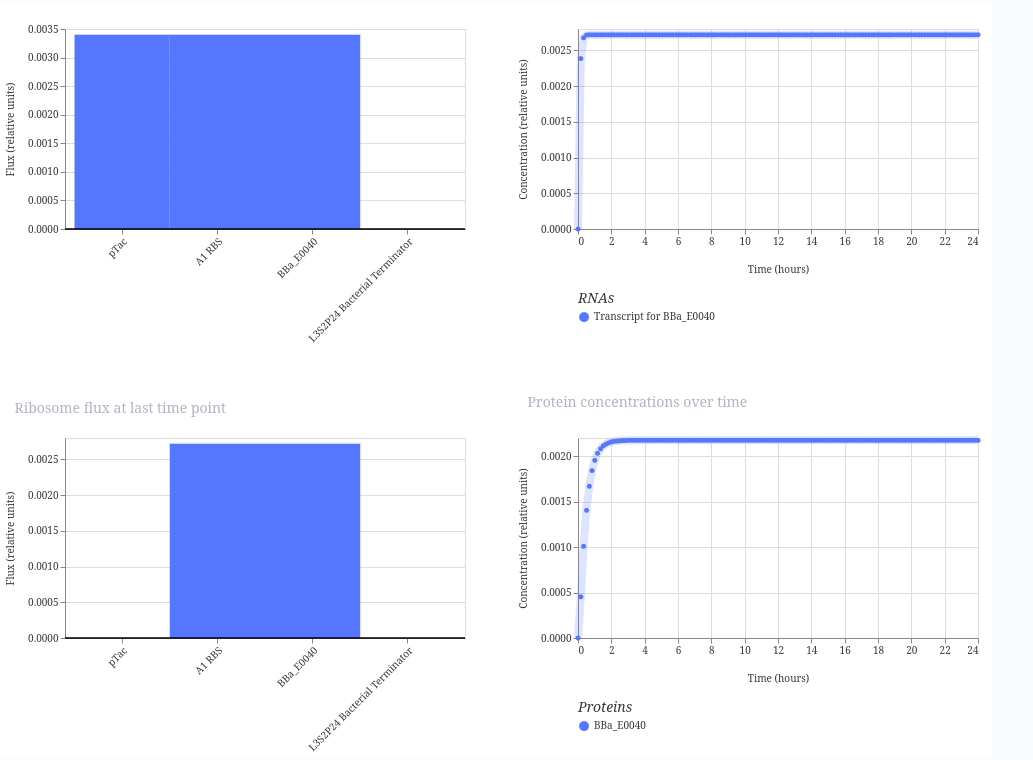
Simple GFP expression construct
This construct is a simple inducible reporter plasmid where the pTac promoter controls expression of GFP (BBa_E0040), allowing fluorescence to increase when the promoter is activated.
The result is fluorescence to increase over time when the promoter is active. Here is the result:
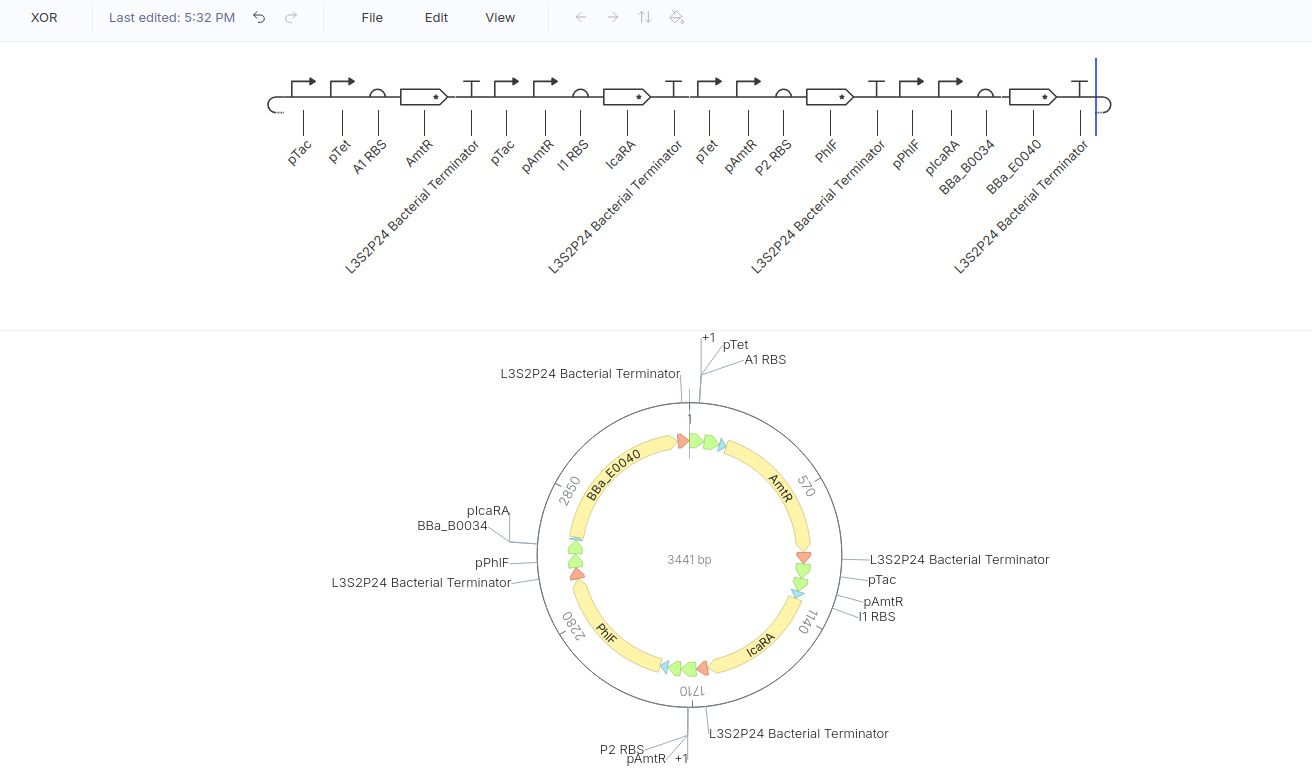
XOR gate
BasicallY: Output = ON only if Input A OR Input B are ON (if both are ON , then the output is OFF )
Therefore, the truth table can be written as
Input A
Input B
Output
0
0
0
0
1
1
1
0
1
1
1
0
Here is my design:
Here is the result:
This is not exactly what I expected as it only shows the 1 1 0 case, but I’m not sure how to modulate it.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? An advantage of IANNs is that gene expression can vary continuously, meaning that—unlike Boolean functions, whose outputs are limited to ON or OFF states—IANNs can weigh inputs and produce graded responses based on their relative strengths, enabling more nuanced, flexible, and biologically realistic decision-making within the cell.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. IANNs can be used to detect cancer by integrating multiple biomarkers as inputs and producing a specific, graded output. For example, biomarkers such as PSA (prostate), CA-125 (ovarian), CEA (colorectal), HER2 and BRCA1/2 (breast), and AFP (liver) could serve as weighted inputs, allowing the circuit to distinguish between cancer types based on their combined expression patterns rather than simple presence or absence. The output could be the expression of a fluorescent reporter for diagnosis or a therapeutic protein, such as one that induces apoptosis, only when the integrated signal crosses a defined threshold. However, limitations include variability and noise in biomarker expression, difficulty in precisely tuning and maintaining stable weights in vivo, potential off-target interactions with native cellular pathways, and challenges in safely delivering and controlling such complex systems in patients.
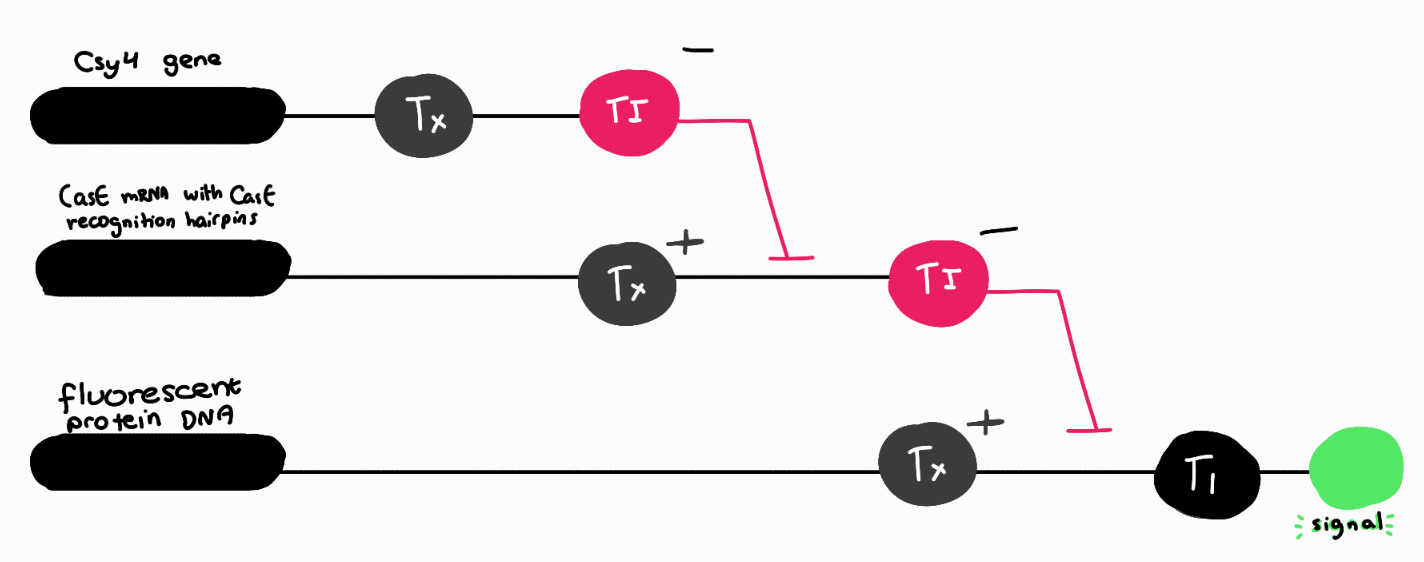
3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts? Existing fungal materials include mycelium-based packaging (used as a sustainable alternative to Styrofoam), building materials like bricks and insulation, leather-like fabrics (e.g., Mylo), and even furniture. I know about an NGO that uses mycelium to make surf-related products like boards, showing how versatile and lightweight the material can be. Their main advantages are that they are biodegradable, renewable, and require low energy to produce, but disadvantages include lower mechanical strength, sensitivity to moisture, and challenges in scaling and standardization compared to plastics or concrete.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria? We might genetically engineer fungi to produce stronger materials, sense environmental signals, or self-heal, making them useful for smart construction or environmental monitoring. Fungi are advantageous over bacteria because they naturally grow into large, structured networks (mycelium), making them ideal for materials, and they can secrete complex proteins and enzymes more efficiently. However, they are slower to grow and harder to genetically manipulate than bacteria.
Week 9 HW: Cell-Free Systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis (CFPS) offers much greater flexibility and experimental control than in vivo expression because protein production occurs outside living cells, typically in a test tube using cellular extracts. This means variables such as DNA concentration, ion composition, temperature, cofactors, chaperones, and energy substrates can be precisely adjusted without worrying about cell viability, metabolism, or toxicity. Another notable example is time, as to perform CFPS takes 1 –2 days, whereas in vivo protein expression may take 1–2 weeks. Here are two examples of when CFPS can be efficiently used:
Toxic proteins: proteins that would kill or stress living cells can still be produced using CFPS
Proteins with non-canonical aminoacids: as the system used allows way more flexibility and just choosing its components, CFPS facilitates translation with non-canonical aminoacids
2. Describe the main components of a cell-free expression system and explain the role of each component. A cell-free expression system usually contains: a cell extract (lysate), which contains all the molecular machinery (polymerases, ribosomes, tRNAS etc.) needed for transcription and translation; a template that provides the coding sequence of the target protein, it could be DNA or mRNA; the aminoacids, which are the building blocks for the target protein; an energy regeneration system (common sources are phosphoenol pyruvate, acetyl phosphate, and creatine phosphate) which is important for maintaining high-level protein production, preventing the accumulation of inhibitory phosphatesnucleotides; (ATP,GTP,CTP,UTP) which are requires for transcription; also salts and ccofactors such as Mg2+, so the enzymes can work properly.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment. Because a cell-free system requires energy (ATP) to make the reactions happen, but it doesn’t contain any metabolism to continue the production of ATP thus mantaining the protein synthesis. Therefore, an energy provision system is needed. A common method to maintain ATP supply is using an ATP regeneration system based on phosphocreatine and creatine kinase.
Phosphocreatine + ADP → ATP + Creatine
This continuously converts ADP back into ATP during the experiment.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems (for example E. coli lysate) are faster, cheaper, and produce high yields, but they are less suitable for proteins requiring complex post-translational modifications (PTMs). A good protein to produce in this system would be GFP because it is small, simple and folds quickly.
Eukaryotic systems (for example from CHO cells, rabbit reticulocyte, or insect extracts) are better for proteins requiring disulfide bonds, glycosylation, or other types of PTMs. A good example would be a histore, as it has many PTMs.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
It’s already been described that the lack of a natural membrane would impede the synthesis of membrane proteins. Therefore, to make this synthesis fesasible, the setup should include adding membrane-mimicking structures such as micelle-forming detergents, nanodiscs, liposomes, or exogenous microsomes
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
I imagine each component mentioned on question 2 can go wrong. One common component that can lower the yield is the conditions of the DNA template, such as low concentration, degradation, or a weak promoter, which can be addressed by checking DNA integrity and increasing the amount or improving the construct design. Another common issue can be reaction conditions, especially incorrect ions such as magnesium and potassium, or temperature settings, so a possible troubleshooting strategy is to systematically optimize these variables in test reactions. A third reason is the lack of energy coming from a disfunctional energy provision system, which causes translation to stop early; in this case, adding or improving an energy regeneration system, such as phosphoenolpyruvate or phosphocreatine with the appropriate enzyme, can help maintain continuous protein synthesis.
Homework question from Kate Adamala
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
A wearable bracelet containing a freeze-dried cell-free biosensor that changes color in the presence of carbon monoxide, serving as a modern “canary in the coal mine” for mine workers.
How will the idea work, in more detail? Write 3-4 sentences or more.
The bracelet would contain a small patch or microfluidic chamber embedded in the band, loaded with a freeze-dried cell-free expression system programmed with a carbon monoxide–responsive sensing circuit. When activated, the system begins functioning and continuously samples air diffusing through a gas-permeable membrane. Upon exposure to dangerous concentrations of carbon monoxide, the biosensor triggers expression of a chromogenic or fluorescent reporter protein, causing the bracelet window to visibly change color from green to red.
What societal challenge or market need will this address?
Carbon monoxide poisoning remains a major public health and occupational safety problem because the gas is odorless, colorless, and often undetectable without specialized equipment. This wearable design could protect miners, firefighters, industrial workers, mechanics, and even families using gas heaters or stoves in poorly ventilated environments.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The main limitations are activation, reaction lifetime, and one-time use. To address activation, the bracelet could include a sealed hydration pouch that the user presses before entering a hazardous environment, initiating the reaction only when needed. For stability, the freeze-dried components would remain shelf-stable for weeks to months inside a sealed moisture-proof compartment. For the one-time use problem, maybe the sensing patch could be designed to be replacable.
Homework question from Ally Huang
Background Information
Inspired by Project Hail Mary by Andy Weir, this proposal explores how biological systems might survive extreme radiation during deep-space travel. In the novel, the microorganism “Astrophage” not only survives near stars, but also feeds on stellar energy itself, meaning it must tolerate enormous amounts of radiation — something even Rocky and his species did not fully understand, which lead to a disaster (not spoiling it!). In real biology, some organisms such as tardigrades possess proteins that protect their DNA from radiation damage. Understanding these mechanisms is important for future long-duration missions, where protecting biological systems from cosmic radiation will be essential for human survival and biotechnology in space.
Molecular or Genetic Target
The tardigrade DNA-protective protein Dsup (Damage suppressor protein) and radiation-damaged plasmid DNA exposed to UV radiation.
Relationship Between the Target and the Space Biology Question
Cosmic radiation is one of the greatest biological dangers in deep-space missions because it damages DNA, proteins, and cells. The tardigrade protein Dsup is known to reduce DNA damage caused by radiation on Earth. By studying whether Dsup can protect DNA in a freeze-dried cell-free system, we can investigate whether protective biomolecules could help stabilize biological tools used during long-term missions. This is scientifically interesting because it combines synthetic biology, astrobiology, and radiation biology while testing whether biological resilience mechanisms can function outside living cells in extreme environments.
Hypothesis or Research Goal
I hypothesize that adding the Dsup protein to a freeze-dried BioBits® cell-free expression reaction will reduce radiation-induced damage to DNA templates and improve protein production after UV exposure. The reasoning is based on studies showing that Dsup binds chromatin and helps shield DNA from hydroxyl radicals generated by radiation. In space, freeze-dried biological systems may be used for diagnostics, medicine production, or environmental monitoring, but radiation could degrade their DNA components during storage and transport. If Dsup improves the stability of cell-free reactions after radiation exposure, it could become part of future protective biotechnology systems for space exploration. More broadly, this experiment explores whether natural evolutionary solutions from extremophiles could inspire biotechnology for surviving harsh extraterrestrial environments.
Experimental Plan
Freeze-dried BioBits® reactions containing a fluorescent reporter plasmid will be prepared in two groups: one expressing Dsup and one without Dsup (control). Samples will be exposed to different levels of UV radiation to simulate DNA damage. After rehydration, fluorescence intensity will be measured using the P51 Molecular Fluorescence Viewer as an indicator of successful protein expression. Additional controls will include non-irradiated samples. The experiment will compare fluorescence levels between groups to determine whether Dsup improves the resistance of the cell-free system to radiation-induced damage.
For my final project I would like to measure a few things:
whether the biosensor can detect carbon monoxide (CO) in a reliable and quantifiable way. The main readout will be the signal generated after CO binding, such as fluorescence, or a visible color change, depending on the final design. By exposing the system to known CO concentrations, I can build a calibration curve and evaluate sensitivity, detection range, and response time.
also if the protein sensor itself is being produced correctly and remains functional. For this, I would use SDS-PAGE to confirm protein size and purity, and UV-visible spectroscopy to verify heme incorporation and observe spectral changes upon CO binding. To test specificity, I would compare the response to CO with other gases or environmental factors such as CO2 and humidity.
Waters Part I — Molecular Weight
Question 1 Using the online calculator, the calculated molecular weight (unmodified) is 28006.60
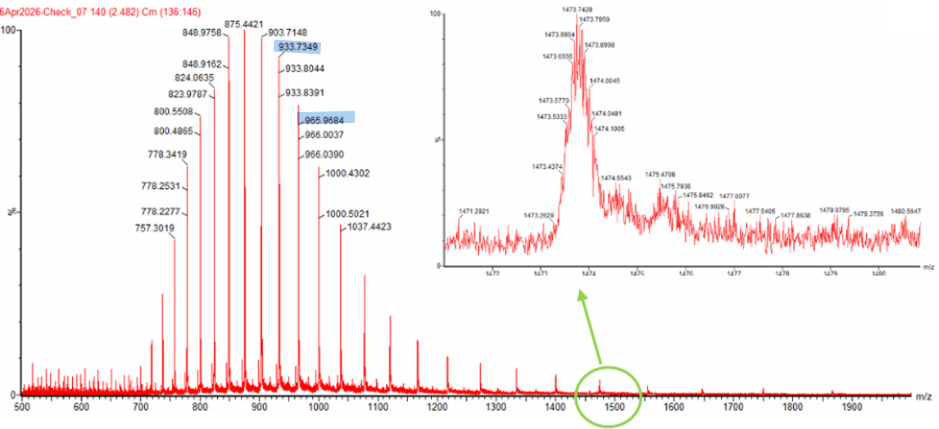
Question 2 The two adjacent peaks I selected from the LC-MS spectrum at m/z 933.7349 and 965.9684.
To determine $z$, we use
[
z = \frac{m/z_{n+1}}{(m/z_n - m/z_{n+1})}
]
Using the two adjacent peaks at $m/z = 933.7349$ and $m/z = 965.9684$, we assign
$m/z_{n+1} = 933.7349$ and $m/z_n = 965.9684$, so
Thus, the charge state of the peak at $m/z = 965.9684$ is $29+$, and the adjacent peak at $m/z = 933.7349$ corresponds to $30+$.
To determine the MW of the protein we can use the relationship between $\frac{m}{z_n}$, $MW$, and $z$:
[
\frac{m}{z} = \frac{MW + z}{z}
]
we rearrange to solve for $MW$:
[
MW = z\left(\frac{m}{z}\right) - z
]
Using $z = 29$ and the peak at $m/z = 965.9684$:
[
MW = 29(965.9684) - 29
]
[
MW = 27984.08\ \text{Da}
]
Therefore, the molecular weight of the protein is 27984 Da
We can calculate the accuracy of the measurement using both this number 27984 Da, the predicted MW of 28006.60 Da and the formula:
$$ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $$
$$ \text{Accuracy} = \frac{|27984 - 28006.6|}{28006.6} = 8 \times 10^{-4}$$
question 2.
After manually counting 26 lysine (K) and arginine (R) residues in the eGFP sequence, and confirming that none were followed by proline (which blocks trypsin cleavage), I expected complete tryptic digestion to produce 27 peptides in total. However, when I analyzed the sequence using the ExPASy PeptideMass tool, the software displayed fewer peptides than expected. I believe this discrepancy occurred because the tool parameters were set to show only peptides with masses greater than 500 Da, causing smaller peptide fragments generated during digestion to be excluded from the output.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Unfortunately, I didn’t have a chance to contribute because I never received the personalized URL. I guess I’ll have to make up for it by becoming a TA this fall :)
That said, I really loved the idea of collaborative bioart. My favorite part was the upper right section with the gene expression, it looks so cool.
For next year, I think it could be even more engaging if there were multiple canvases or themes (for example, one per node or topic), so more people could contribute and explore different ideas within the same project.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Question 1
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) Gives the system the molecular machines (ribosomes, tRNAs, enzymes) needed for transcription and translation. The T7 RNA polymerase is known for a highly efficient, single-subunit enzyme utilized for rapid, high-yield in vitro RNA synthesis
Salts/Buffer
Potassium Glutamate Mimics intracellular ionic conditions and stabilizes ribosomes and enzymes for efficient protein synthesis.
HEPES-KOH pH 7.5 Maintains a stable pH environment, which is critical for optimal enzyme activity during transcription and translation.
Magnesium Glutamate Supplies Mg2+ ions, essential cofactors for ribosome function, tRNA charging, and enzymatic reactions in protein synthesis.
Potassium phosphate monobasic Importanbt for buffering capacity and phosphate balance, essential to support metabolic and enzymatic reactions.
Potassium phosphate dibasic Works with the monobasic form to maintain pH stability and provide phosphate for energy-related processes.
Energy / Nucleotide System
Ribose Serves as a precursor for nucleotide synthesis and supports regeneration of nucleotides during the reaction.
Glucose Fuels energy regeneration pathways, helping sustain ATP production over time.
AMP A nucleotide building block that participates in energy metabolism and nucleotide recycling.
CMP Provides cytidine nucleotides required for RNA synthesis.
GMP Supplies guanine nucleotides for RNA transcription.
UMP Provides uridine nucleotides necessary for RNA synthesis.
Guanine Acts as a precursor for GTP synthesis, which is required for both transcription and translation processes.
Translation Mix (Amino Acids)
17 Amino Acid Mix Supplies most of the amino acids required for protein synthesis.
Tyrosine Added separately due to stability or solubility constraints, ensuring sufficient availability for translation.
Cysteine Provided separately because it is prone to oxidation, ensuring proper incorporation into proteins.
Additives
Nicotinamide Supports redox balance and enzyme function by contributing to NAD⁺/NADH-related metabolic processes.
Backfill
Nuclease Free Water Adjusts the final reaction volume and ensures no degradation of nucleic acids occurs.
Question 2
The 1-hour PEP–NTP system supplies fully formed NTPs and uses PEP as a high-energy donor, enabling fast, high-rate transcription and translation but with rapid resource depletion. In contrast, the 20-hour NMP–ribose–glucose system starts from NMPs and simple carbon sources, relying on metabolic enzymes in the lysate to regenerate NTPs more slowly. This makes it much more sustainable over long periods, though with lower instantaneous expression rates.
Question 3
Transcription can still occur because guanine can be salvaged and converted enzymatically into GMP (and then GTP) within the lysate, supplying the necessary nucleotide for the RNA synthesis.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Fluorescent Protein Properties
sfGFP
sfGFP (superfolder GFP) is engineered for extremely efficient folding, even under stressful conditions, making it highly robust in cell-free systems. It also matures relatively quickly, allowing strong fluorescence signals to appear early during incubation.
mRFP1
mRFP1 has a slower maturation time compared to many green fluorescent proteins, which can delay fluorescence development in long incubations. Its folding efficiency is also lower than newer red fluorescent proteins, potentially reducing total signal output.
mKO2
mKO2 is a bright orange fluorescent protein with fast maturation, which improves early fluorescence detection. However, it is somewhat sensitive to acidic conditions, meaning pH changes during long incubations may decrease fluorescence intensity.
mTurquoise2
mTurquoise2 is known for its very high quantum yield and brightness, making it highly sensitive for fluorescence readout. However, cyan fluorescent proteins can be more sensitive to oxidative conditions and folding stress in cell-free reactions.
mScarlet-I
mScarlet-I is an improved red fluorescent protein with exceptionally high brightness and fast maturation relative to older red proteins. Its efficient folding and photostability make it well suited for long-term fluorescence measurements.
Electra2
Electra2 is designed for strong fluorescence output and improved performance in engineered systems, but like many fluorescent proteins, its chromophore formation depends on oxygen availability. Limited oxygen diffusion in sealed reactions may therefore reduce fluorescence development.
Hypothesis
I hypothesize that increasing the concentration of molecular chaperones and optimizing magnesium ion concentration in the cell-free mastermix will improve folding efficiency and fluorescence intensity for mRFP1 during a 36-hour incubation. Because mRFP1 folds less efficiently and matures more slowly than several other fluorescent proteins, improved protein folding conditions should increase the amount of correctly folded fluorescent protein produced. Additionally, maintaining sufficient magnesium levels may support translation efficiency and overall protein stability, leading to stronger fluorescence signals over time.