Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
All of the questions are so interesting, so c :D
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To answer this question, we first need to know how much of meat is made of proteins (therefore, aminoacids). A quick search on Google returned that in 100g of meat we have ~26g of protein (this, of course, changes based on what type of meat we’re talking about, but let’s use this number). Then we have 130g of protein in 500g of meat. If on average an aminioacid is ~100 Daltons (equivalent to 100g/mol) then we have 1.3 mol in 500 grams of meat. Therefore 1.3 x 6 x 10^23 molecules.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Well, we are not literally what we eat. When we eat - a cow or a fish - we are not reusing their nutrients (proteins, carbohydrates) as our own, but we metabolize (break down) these nutrients: proteins are broken down into amino acids, carbohydrates into simple sugars, and fats into fatty acids. Then our cells use these building blocks to grow and survive in a unique way, defined by what is written in our DNA, that therefore, determines who we are as humans.
3. Why are there only 20 natural amino acids?
This is likely the result of an evolutionary optimization where this number covers a wide range of chemical diversity while still not being too costly for the cells to use and synthesise. If the number was lower, it would be harder to cover the diversity needed to manufacture all kinds of proteins; if the number was higher, there would be a need to have more and maybe less efficient cell machinery.
4. Can you make other non-natural amino acids? Design some new amino acids.
Amino acids, by definition, are organic compounds that contain both amino and carboxylic acid functional groups. It’s also a group of chemicals way larger then the only 20 natural amino acids from the genetic code. Therefore, we can design some new aminoacids, so I chose to design
✨magnetic✨ aminoacids (not paramagnetic per se but can make iron bind). These residues whose side chains contain a metal-chelating group capable of binding paramagnetic ions such as Fe2+. Here are my designs (I used Molview to create the molecules):
- Ferrophenylalanine: it’s basically a phenylalanine but the side chain contains a catechol group (two adjacent hydroxyl groups on an aromatic ring), which is known to strongly chelate Fe(III) through bidentate coordination.
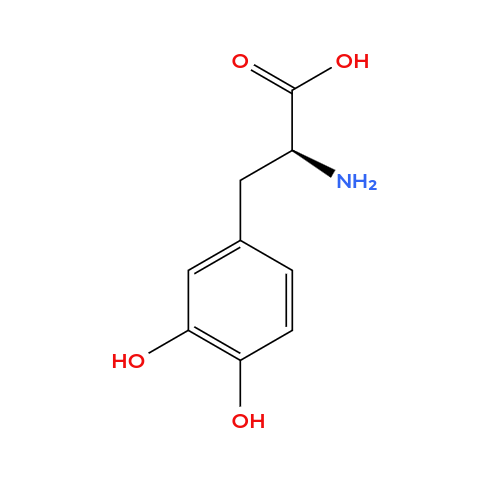
- Bipyridylalanine: it’s an alanine but the side chain contanis a bipyridine ring (two linked pyridines, two nitrogens pointing inward), this forms stable complexes with paramagnetic metal ions.
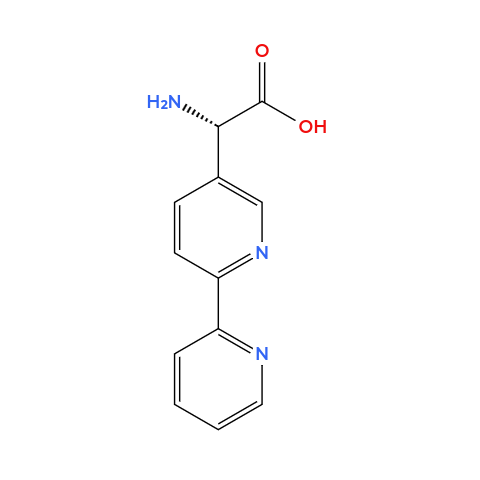
Note: I used ChatGPT to help me understand how to design these magnetic aminoacids, that is, how to design a side chain able to be metal binding (prompt [translated from portuguese]: I want to make other non-natural amino acids and I need help thinking of side chains that are metal-binding, an aminoacid able to bind metals like iron)
5. Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes and life, amino acids could come from pre-biotic chemistry: an atmosphere with water, methane, ammonia and hydrogen gas. The Milley-Urey experiment showed that when you hit such atmosphere with an electrical discharge (simulating lightning), amino acids can form. Therefore, the experiment shows that the formation of amino acids does not need complex organic molecules to start.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect the handedness to be left, and the opposite is true: if you make an α-helix using L-amino acids, the handednesse will be right.
7. Can you discover additional helices in proteins?
Because helix formation depends on specific backbone geometry and hydrogen bond distances, only a finite number of stable helices are possible. While some theoretical helices (such as the γ-helix) have been proposed, most are energetically unfavorable. However, through protein design, it may be possible to stabilize rare or artificial helical conformations.
8. Why are most molecular helices right-handed?
Most molecular helices in biology are right-handed because biological polymers are constructed from homochiral building blocks. In proteins, L-amino acids favor right-handed α-helices due to optimal backbone dihedral angles and minimal steric clashes. In DNA, the D-sugar stereochemistry biases the backbone geometry and base stacking interactions toward a right-handed double helix. Thus, chirality combined with energy minimization determines the predominant handedness of biological helices.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because β-strands have exposed backbone hydrogen bond donors and acceptors that can form intermolecular hydrogen bonds. When multiple strands align, they form extended sheet structures that are energetically stabilized. The primary driving force for aggregation, therefore is hydrogen bonding and hydrophobic effect.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases involve proteins that misfold into β-sheet–rich conformations. β-strands can align and form highly ordered cross-β structures stabilized by extensive intermolecular hydrogen bonding and hydrophobic interactions. Because these β-sheets can extend indefinitely, they form elongated fibrils that accumulate in tissues. Because amyloid β-sheets are highly ordered, stable, and versatile, it is possible to use it as materials, such as nanofibrils.
Part B. Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I chose human hemoglobin (specifically one of the alpha subunit - PDB: 7VDE), the protein responsable for carrying oxygen in the blood and also the reason why blood is red. I chose it because of its historical importance for structural biology and also because I want to work with it in one of my HTGAA projects.
2. Identify the amino acid sequence of your protein.MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The alpha unit is 142 residues long; the most frequent amino acid is alanine (21 times) - How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Hemoglobin (alpha subunit) is paralog to the beta subunit and also has some orthologs: BLAST identified 245 homologs, all Hemoglobin subunit alpha from different organisms. - Does your protein belong to any protein family?
Yes, to the globin family.
3. Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The PDB I’m using (human hemoglobin - 7VDE) was solved in 2021, the quality is not that good (3.60 Å) but is Cryo-EM so it’s pretty solid. - Are there any other molecules in the solved structure apart from protein?
Yes, the heme group. - Does your protein belong to any structure classification family?
Yes, Globin or Globin-like (Scoop)
4. Open the structure of your protein in any 3D molecule visualization software:
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
- Color the protein by secondary structure. Does it have more helices or sheets?
It has more helices. It has no sheets. - Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
When I use the surface mode, I can se the hydrophic residues are a bit less exposed than the hydrophilic ones. - Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes! Where oxygen binds!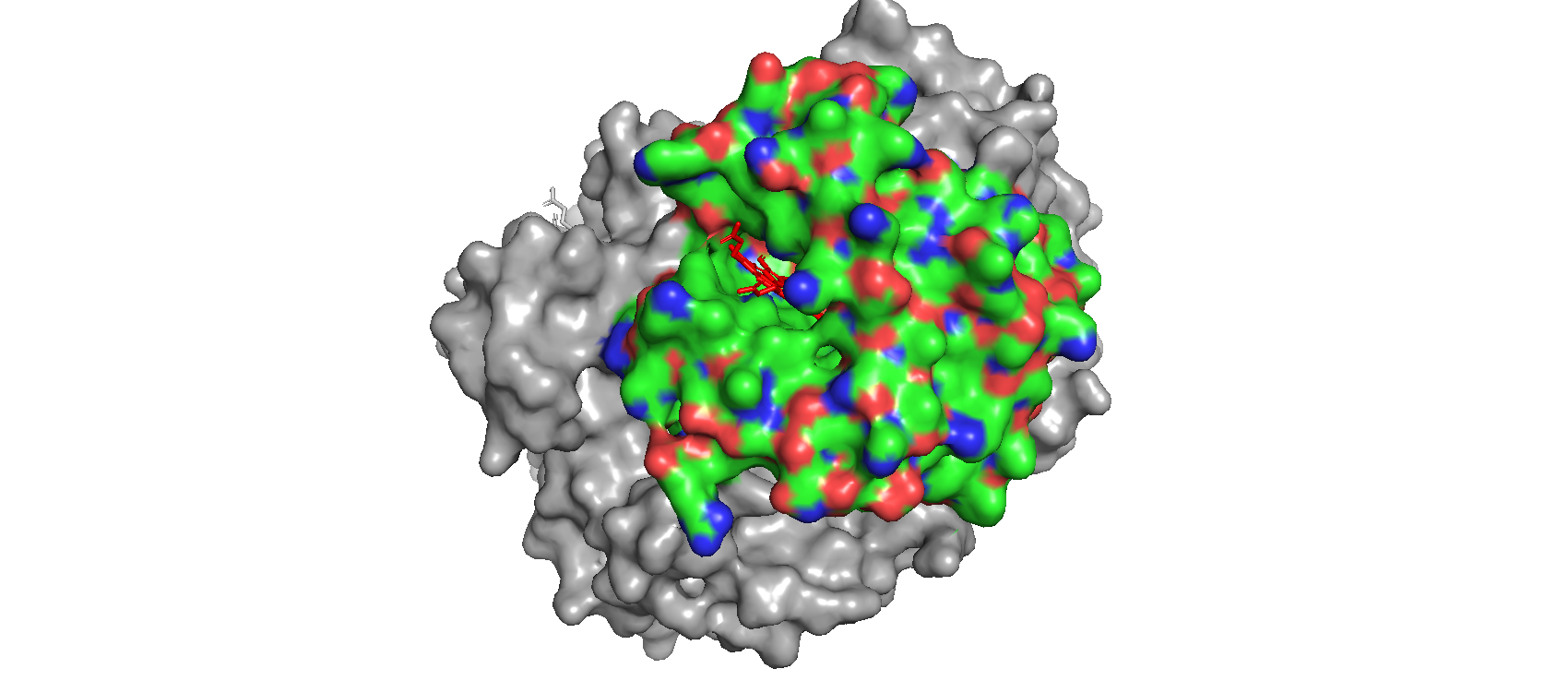 I’m using (and I’ll be using for the whole homework) the subunit alpha of human hemoglobin. Something interesting that I noticed with the mutation heatmap is that changing residues to cystein usually causes a loss of stability. For exampleA124C (Alanine at position 124 mutated to Cysteine) exhibited the lowest LLR of -5.4675 mutation scan heatmap
I’m using (and I’ll be using for the whole homework) the subunit alpha of human hemoglobin. Something interesting that I noticed with the mutation heatmap is that changing residues to cystein usually causes a loss of stability. For exampleA124C (Alanine at position 124 mutated to Cysteine) exhibited the lowest LLR of -5.4675 mutation scan heatmap
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
1. Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
I’m using (and I’ll be using for the whole homework) the subunit alpha of human hemoglobin. Something interesting that I noticed with the mutation heatmap is that changing residues to cystein usually causes a loss of stability. For example A124C (Alanine at position 124 mutated to Cysteine) exhibited the lowest LLR of -5.4675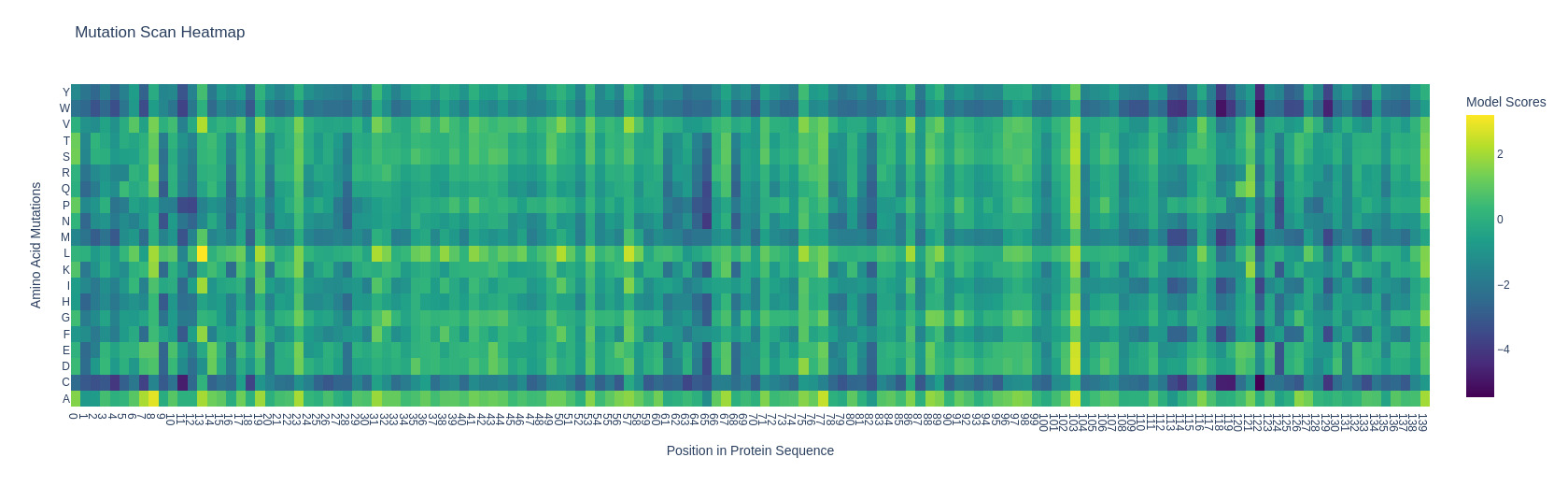
2. Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality.
- Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, they do. It’s possible to see some clusters forming from similar proteins: when the sequence of aminoacids is transformed into a vector (embedding), similar proteins stick together. One example is given on the next question. - Place your protein in the resulting map and explain its position and similarity to its neighbors.
I used the gemini chat to help me highlight my protein. Interestingly, the proteins sitting next to mine (alpha subunit of human hemoglobin) were almost always hemoglobins or other types of globins, like myoglobin. Below it is shown an example. (some proteins showed “automated matches” instead of an actual name, I’m not sure why)
C2. Protein Folding
Folding a Protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
- Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Our ideas can be found here;