Homework
Weekly homework submissions:
Milestone 1 - CL Requirements Attend Class 1 Submit HW1 Submit node preference form Milestone 2 - CL Requirements Watch the Weekly Classes & Recitations Attend at least 33% of all BioClub Node Meetings Submit the HW3 Robot Form 3 Final Project Ideas on the Shared Slide Deck & in your repo Make a post or send a chat message on the Forum Sign the Committed Listener MoU by committing it to your repo
Obsidian Setup for local webpage edits
Why Obsidian? Obsidian does live rendering of markdown, so it is easier to do formatting while doing the homework Obsidian brings the webpages to the local machine, so it is easier to save files, paste screenshots and move things around between folders. What is Obsidian again? Obsidian is a powerful, local-first note-taking and knowledge management app that uses interconnected Markdown files.
Week 1 HW: Principles and Practices
1 Describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. Bio engineering Tool/application: Autonomous Space Biomanufacturing Platform for Active Pharmaceutical Ingredients (APIs) My biological engineering application is an autonomous, space-based biomanufacturing and analysis system capable of producing active pharmaceutical ingredients (APIs) and nutritionally relevant biomolecules during long-duration space missions. The system integrates:
Week 2 HW: DNA read, write, and edit
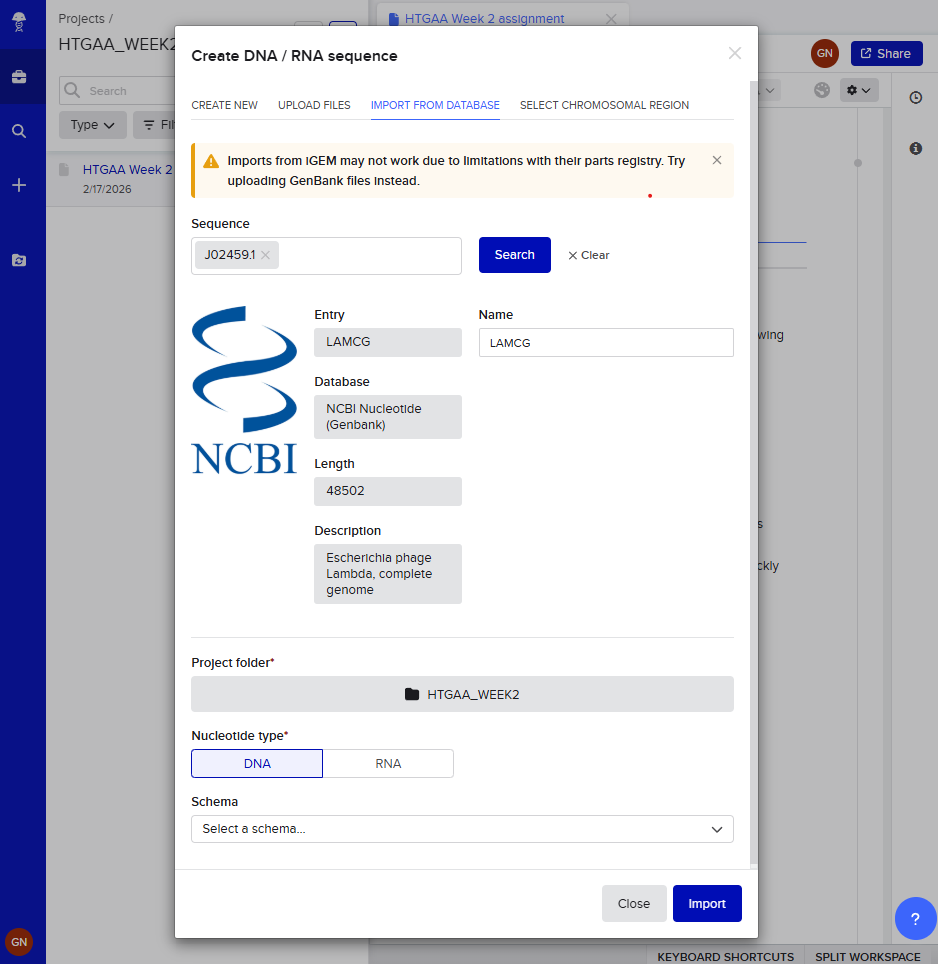
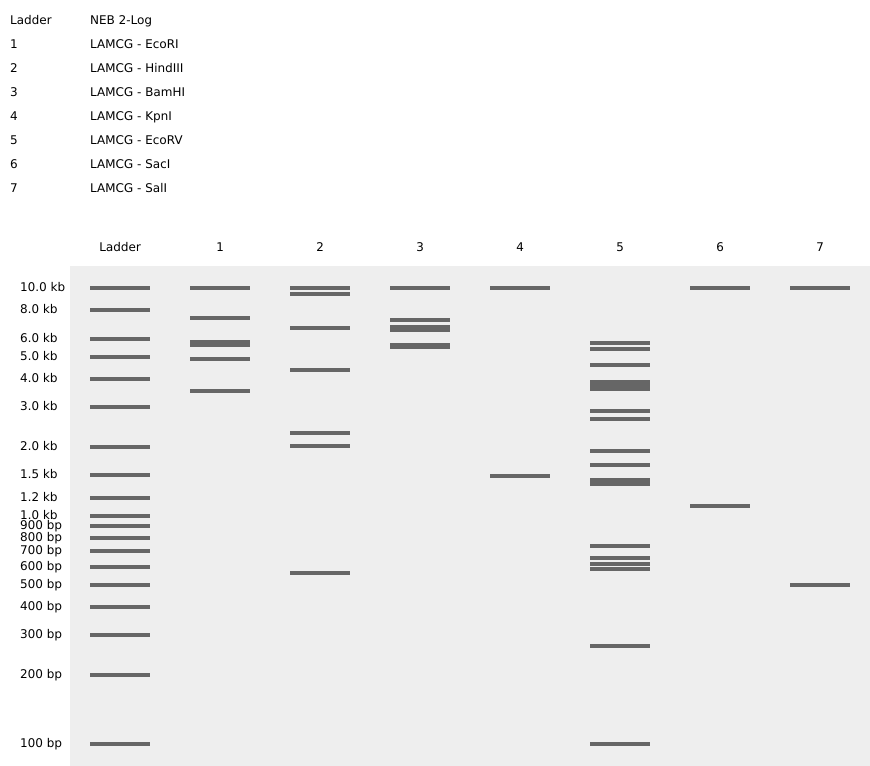
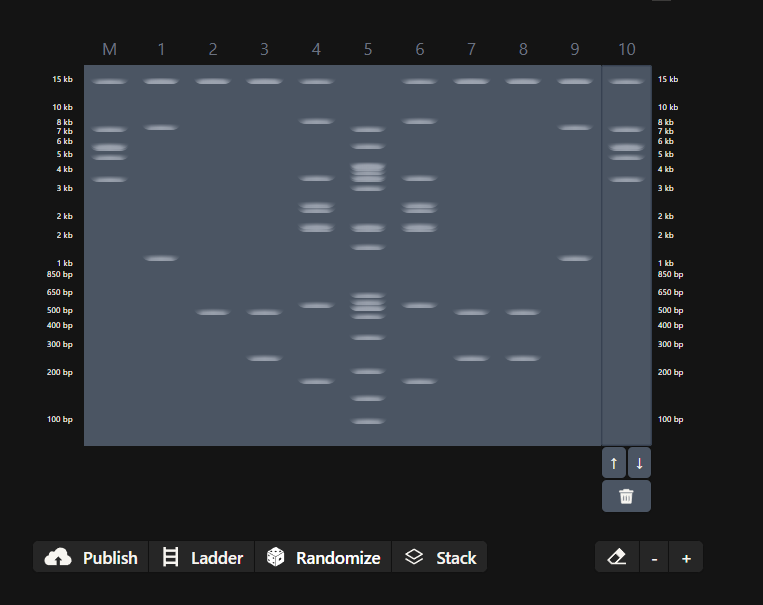
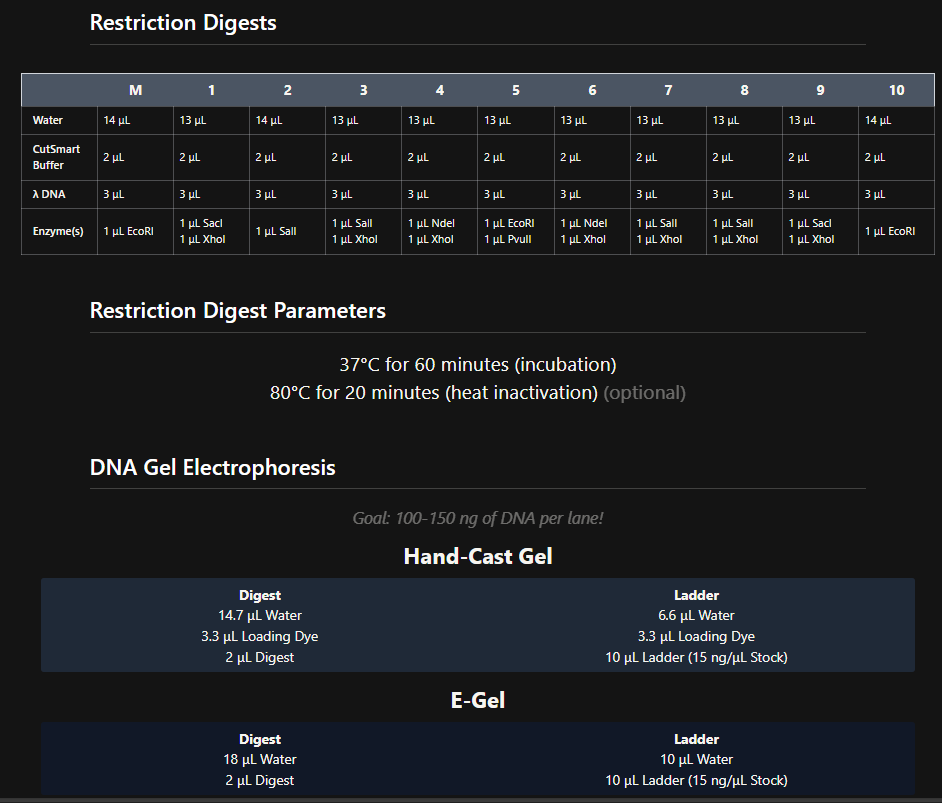
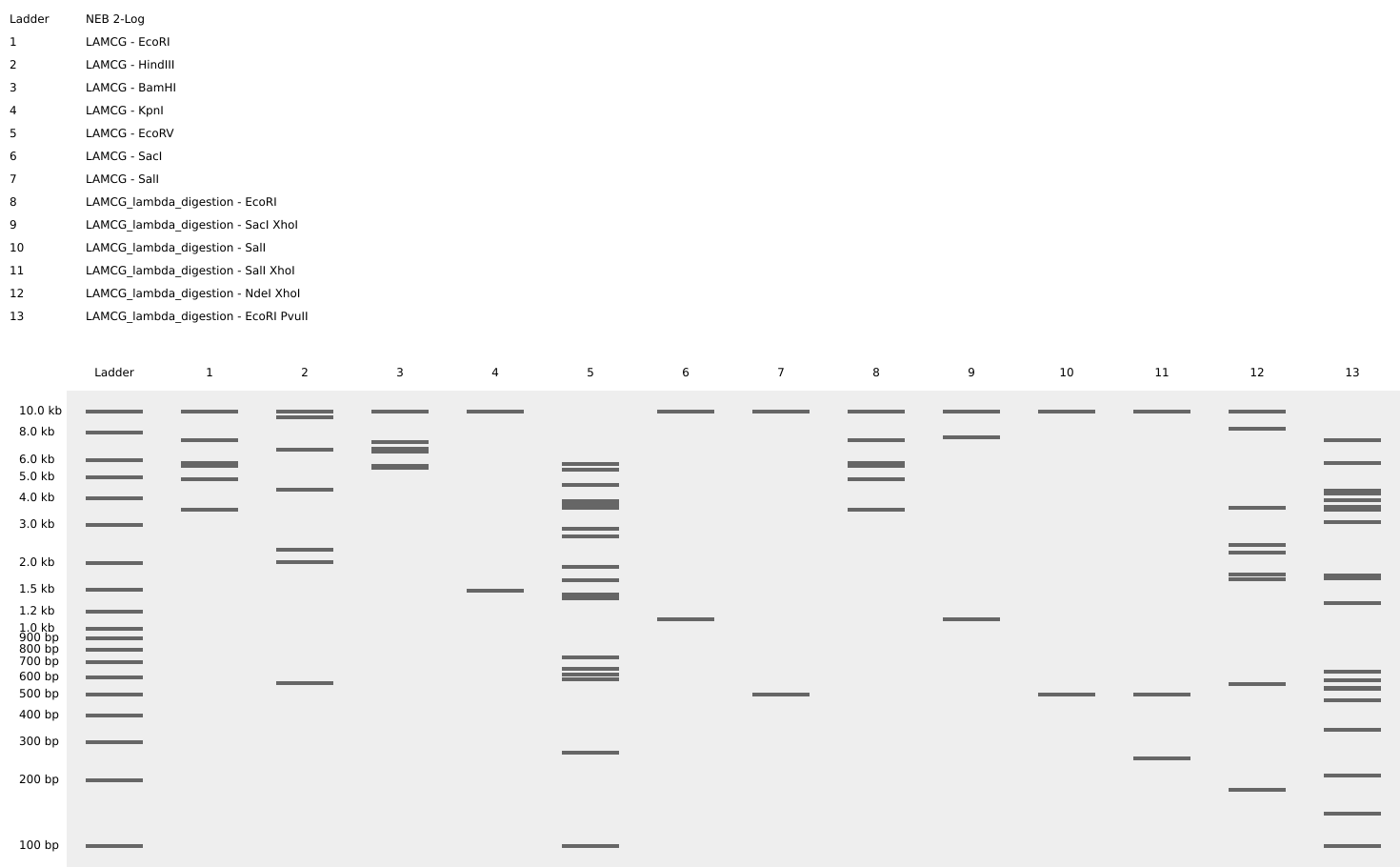
Week 2 - DNA read, write and edit We’re in the deep now, time to get the hands dirty. Part 0 - Basics of Gel electrophoresis - What did the small DNA fragment say to the large one? “Catch me if you can!”
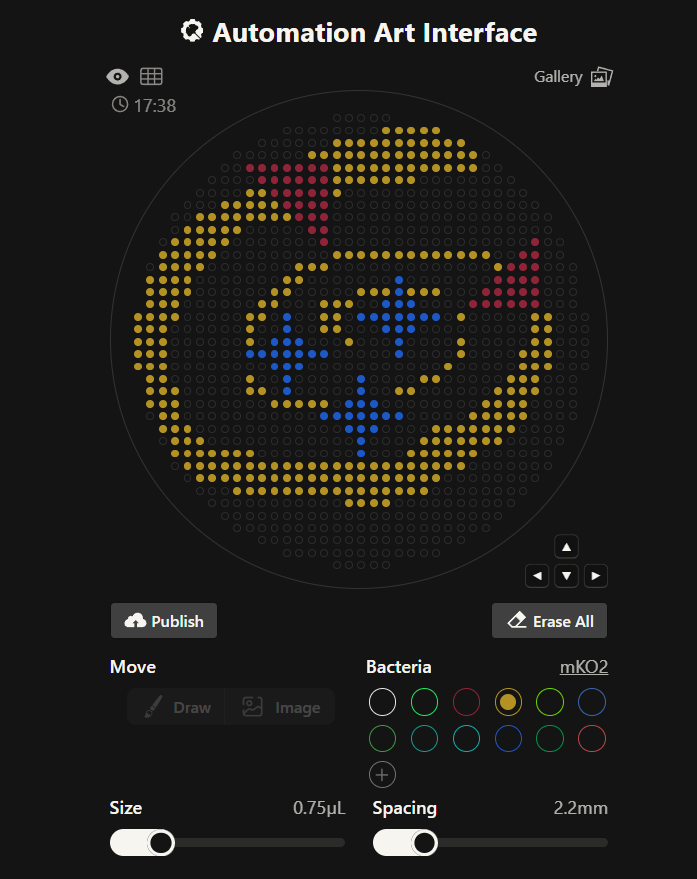
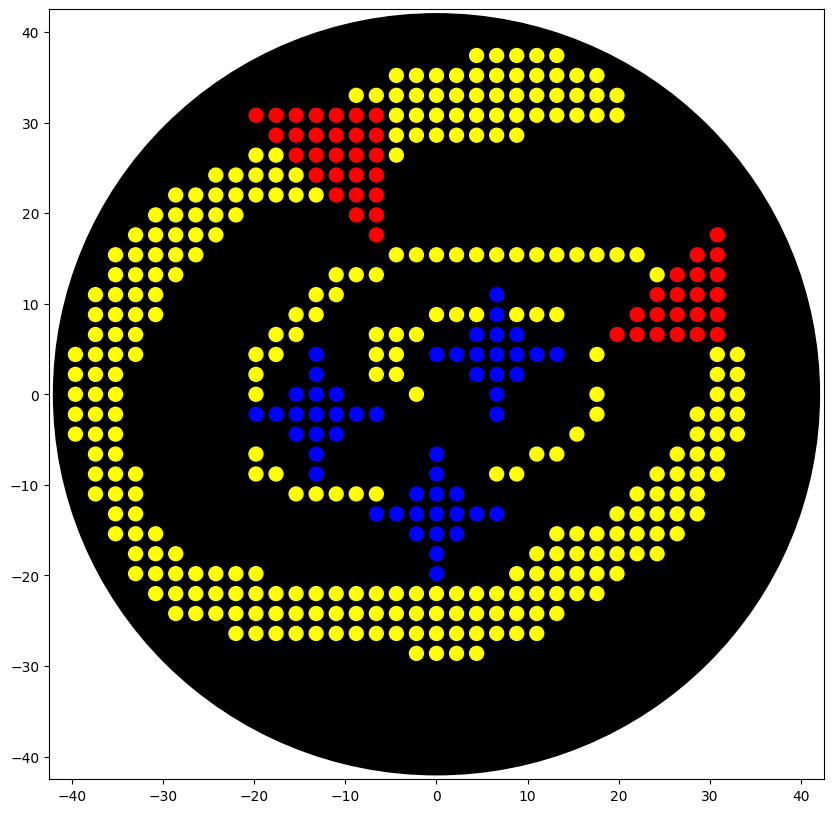
Week 3 lab automation Assignment: Python Script for Opentrons Artwork Review Recitation Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons. Submit form Using AI to generate an image. Image prompt: I am making agar art. I need an image reference. In the image add 3 star flares in the color blue. Lets have an “orbit” around the 3 stars in yellow color, which is an ellipse/spiral that starts thin and gets thicker. In red, lets have a NASA style triangle in the bottom right and top left that is pointing upwards and is small. Give discrete points, not a continous image, similar to bitmap. Use only the colors specified. Give top view.
Week 4 HW: Protein design part 1
Answer conceptual questions Learn basic concept of protein design Brainstorm how to apply these together in the group project Part A - Conceptual questions Amino Acid & Protein Structure Q&A How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) To find the number of molecules, we first determine the mass of protein and then convert that to moles and molecules.
Week 5 HW: Protein design part 2
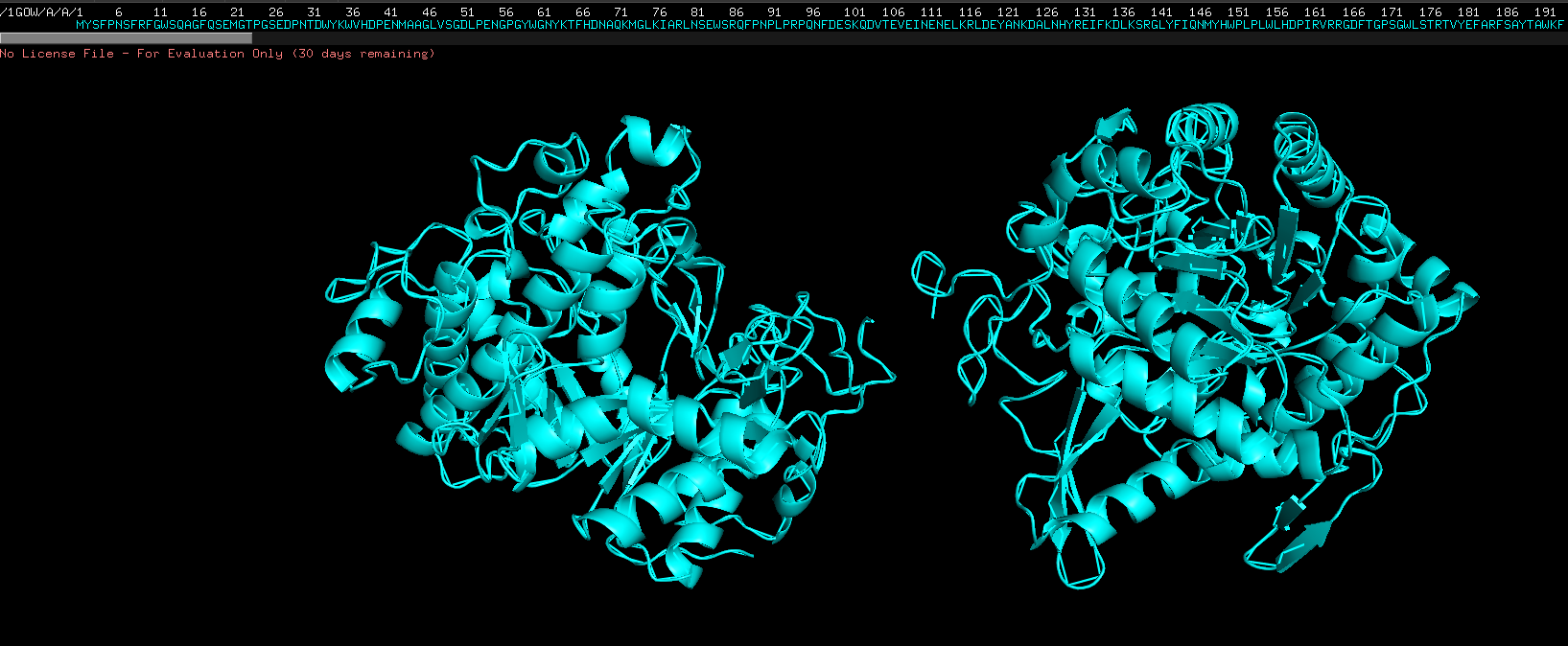
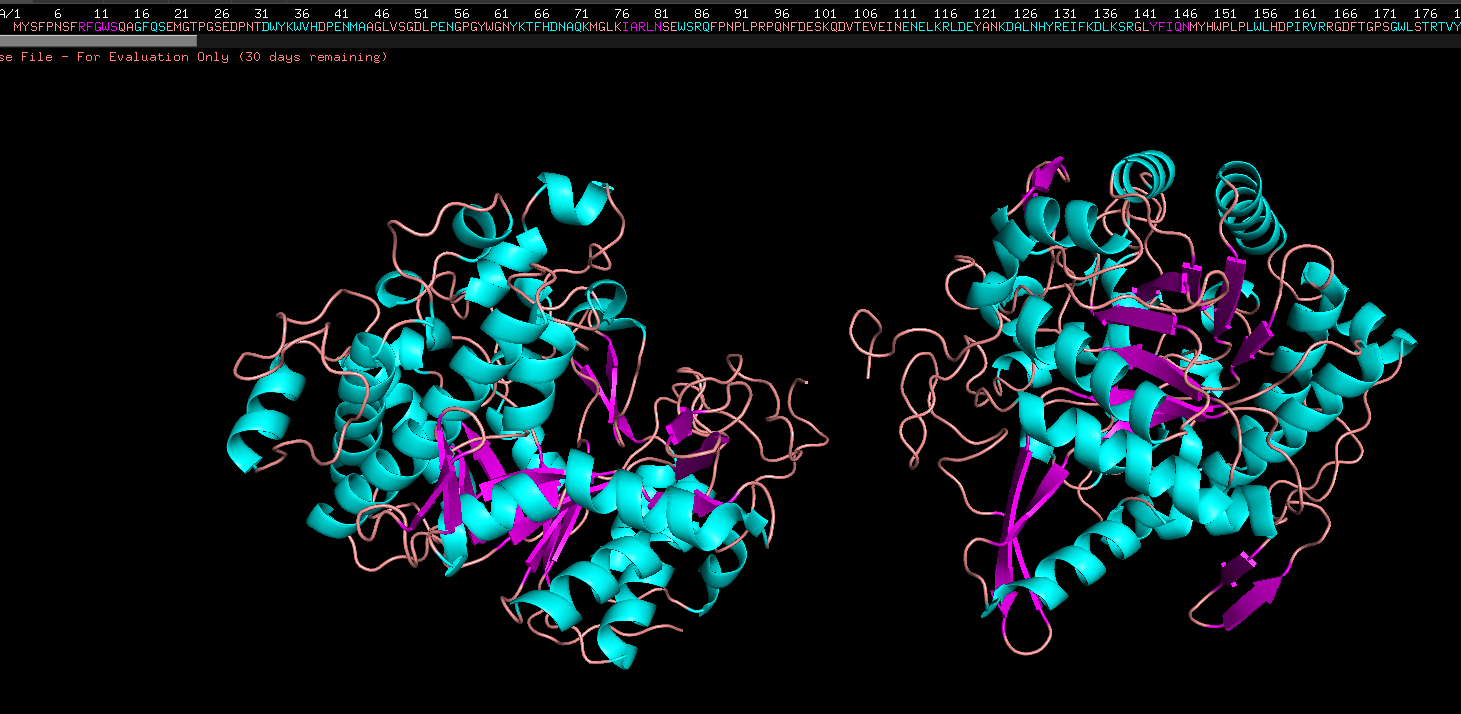
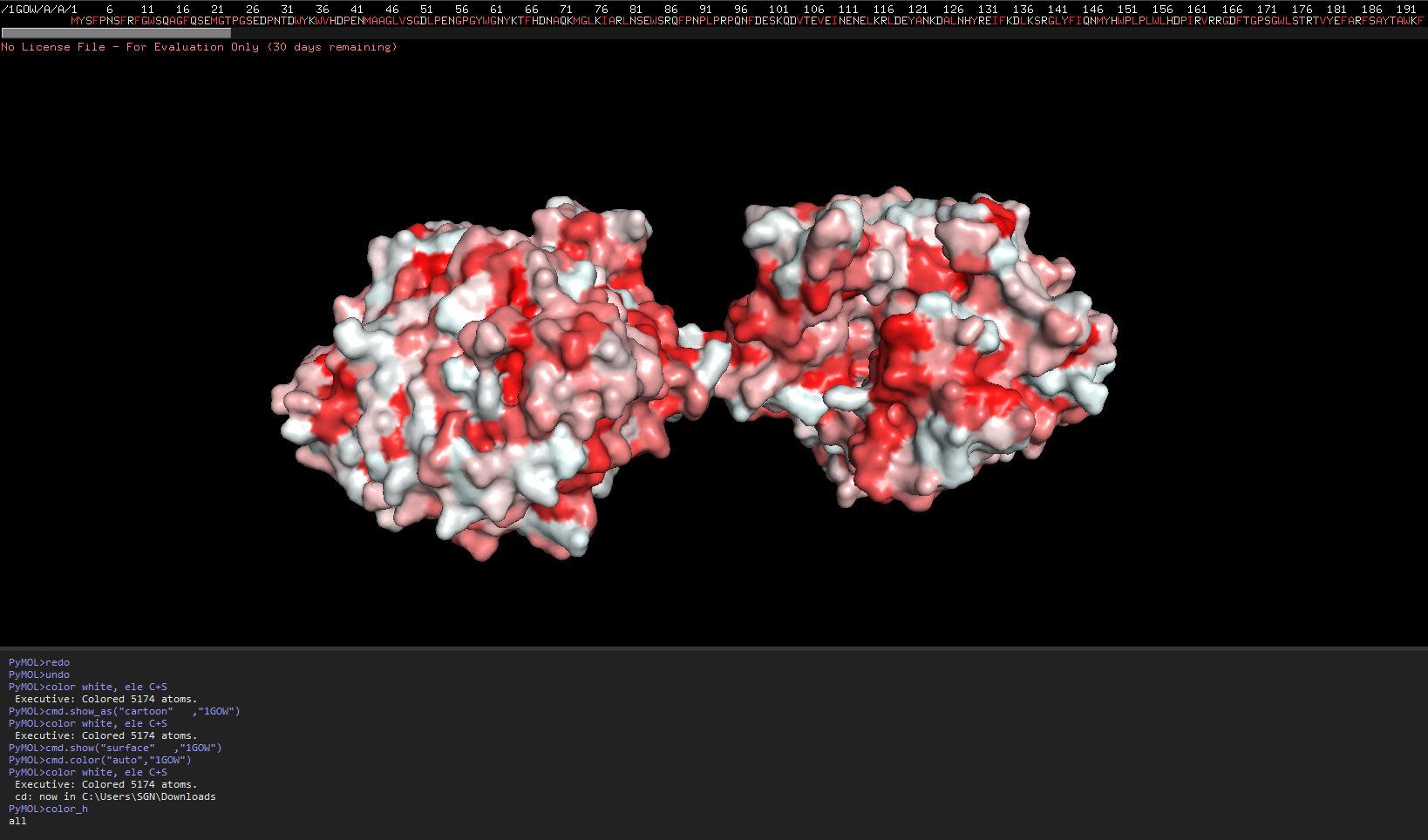
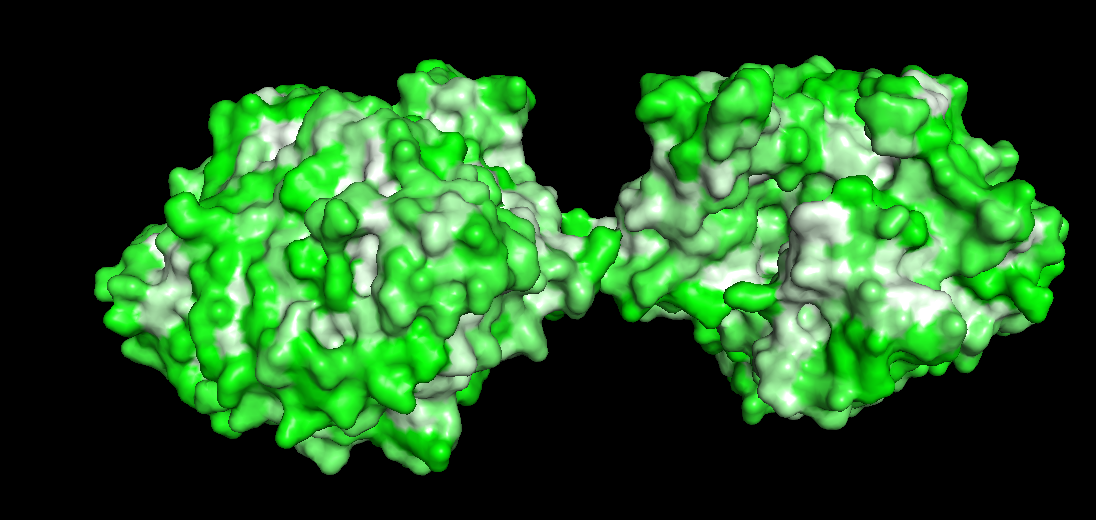
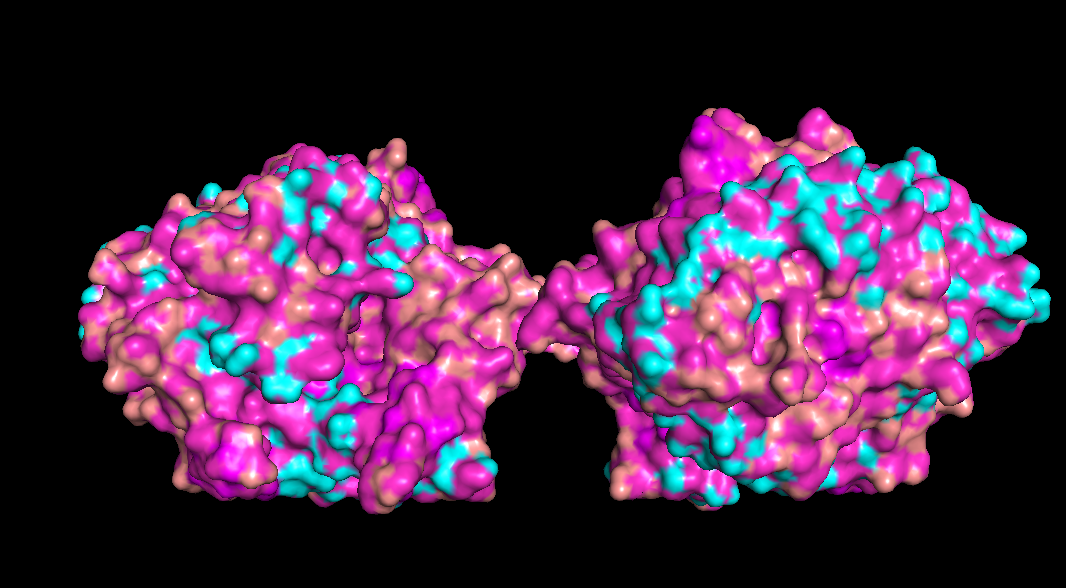
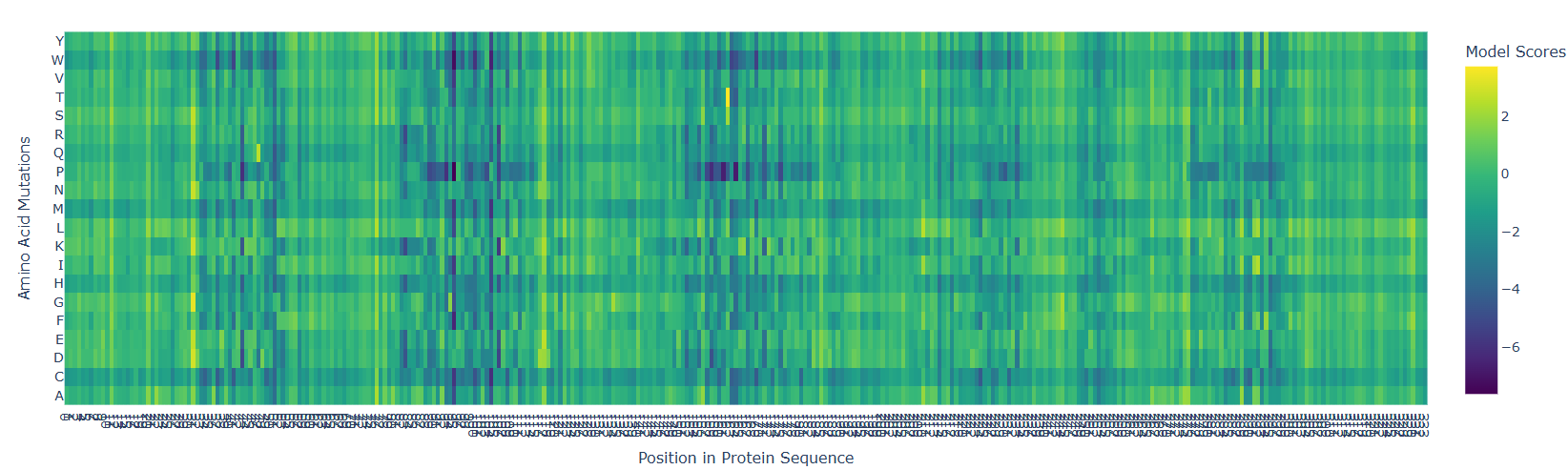
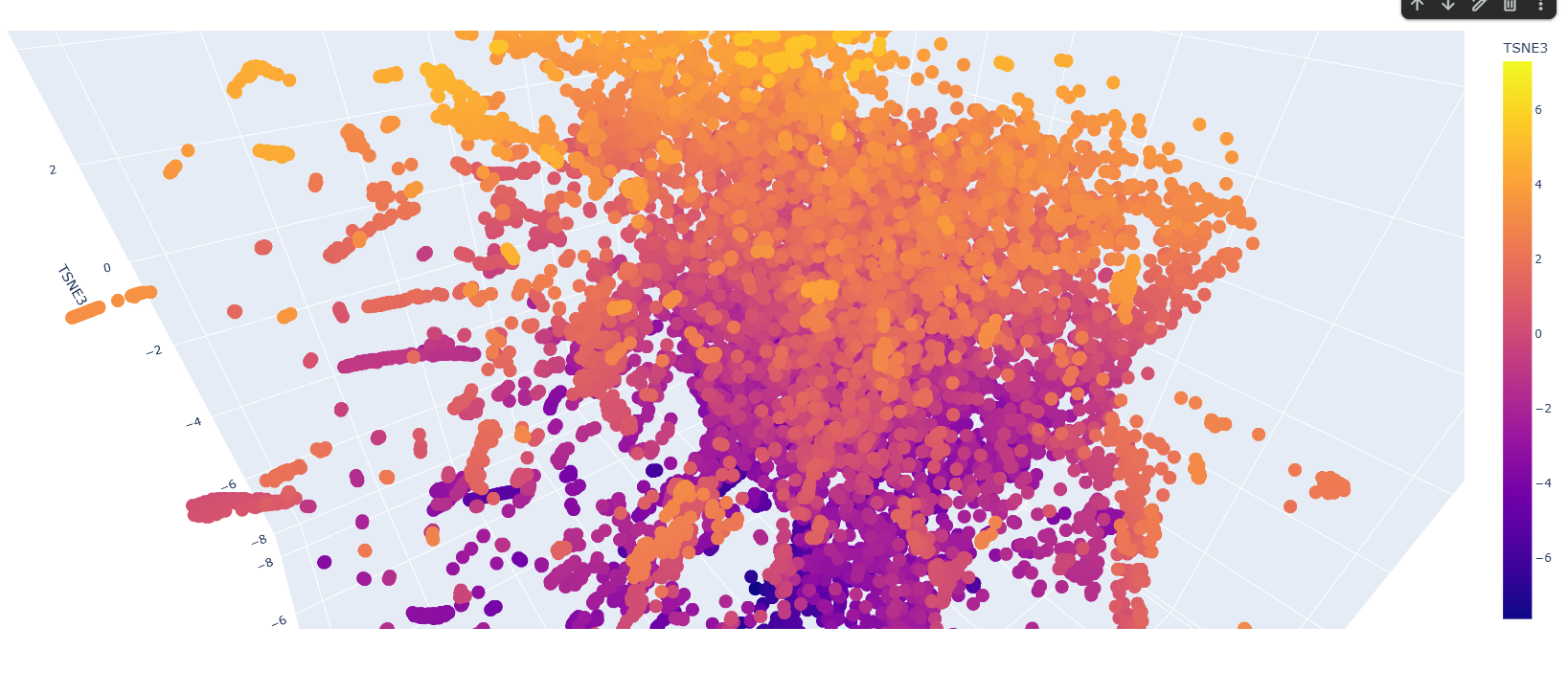
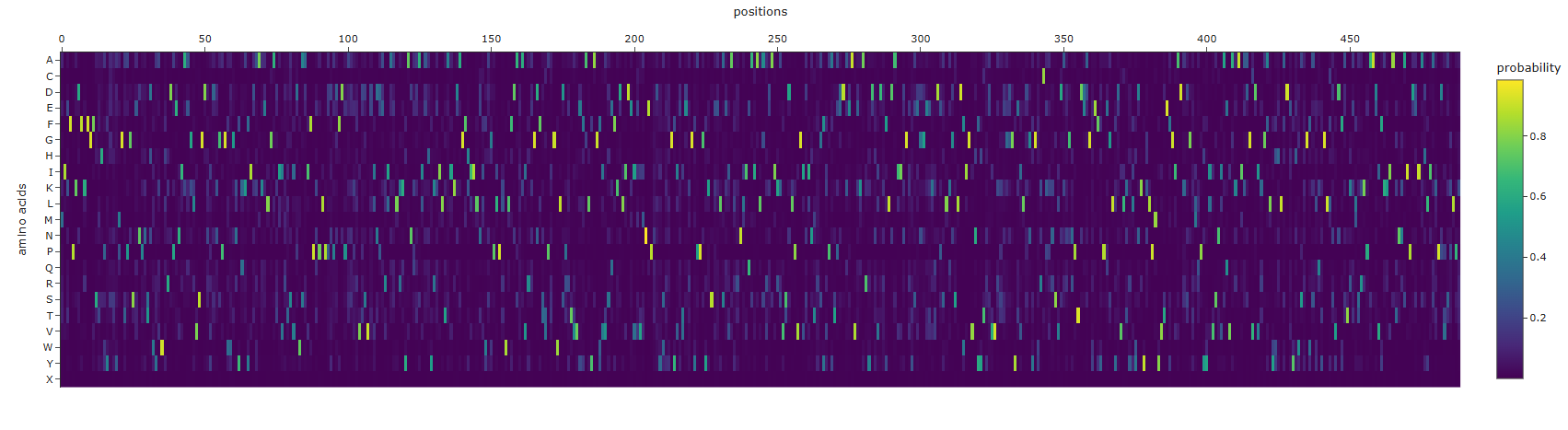
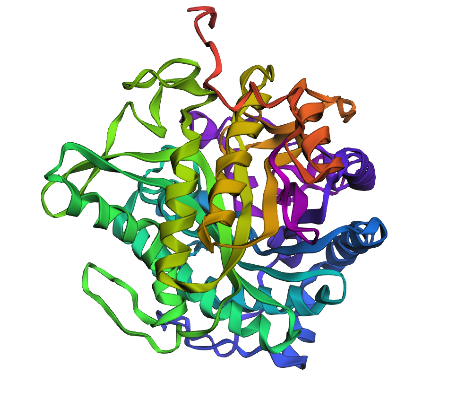
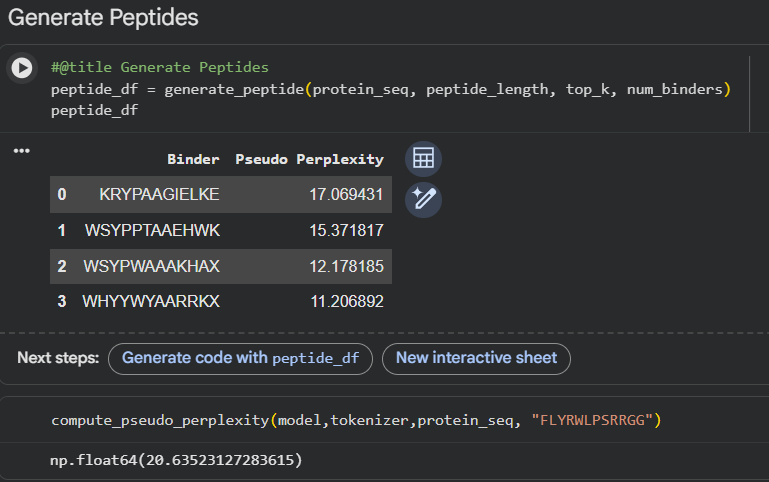
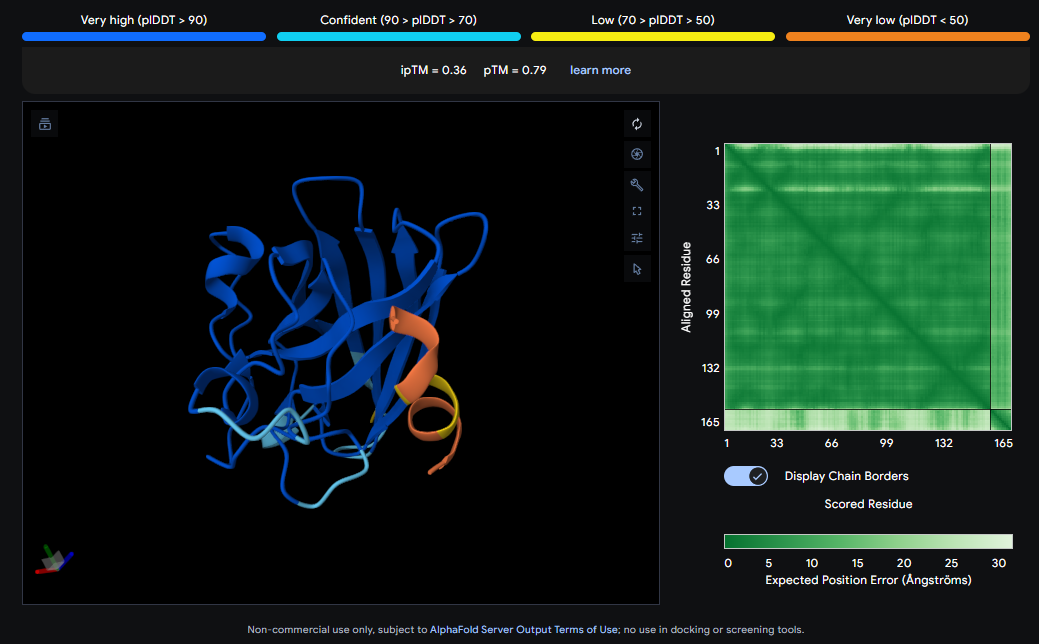
Design short peptides that bind mutant SOD1. Then decide which ones are worth advancing toward therapy. Uniprot SOD1 protein sequence: sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Apply A4V mutation: (ignoring Methionine) The Bolded V used to be A MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Pepmlm Generated peptides and their perplexity score: