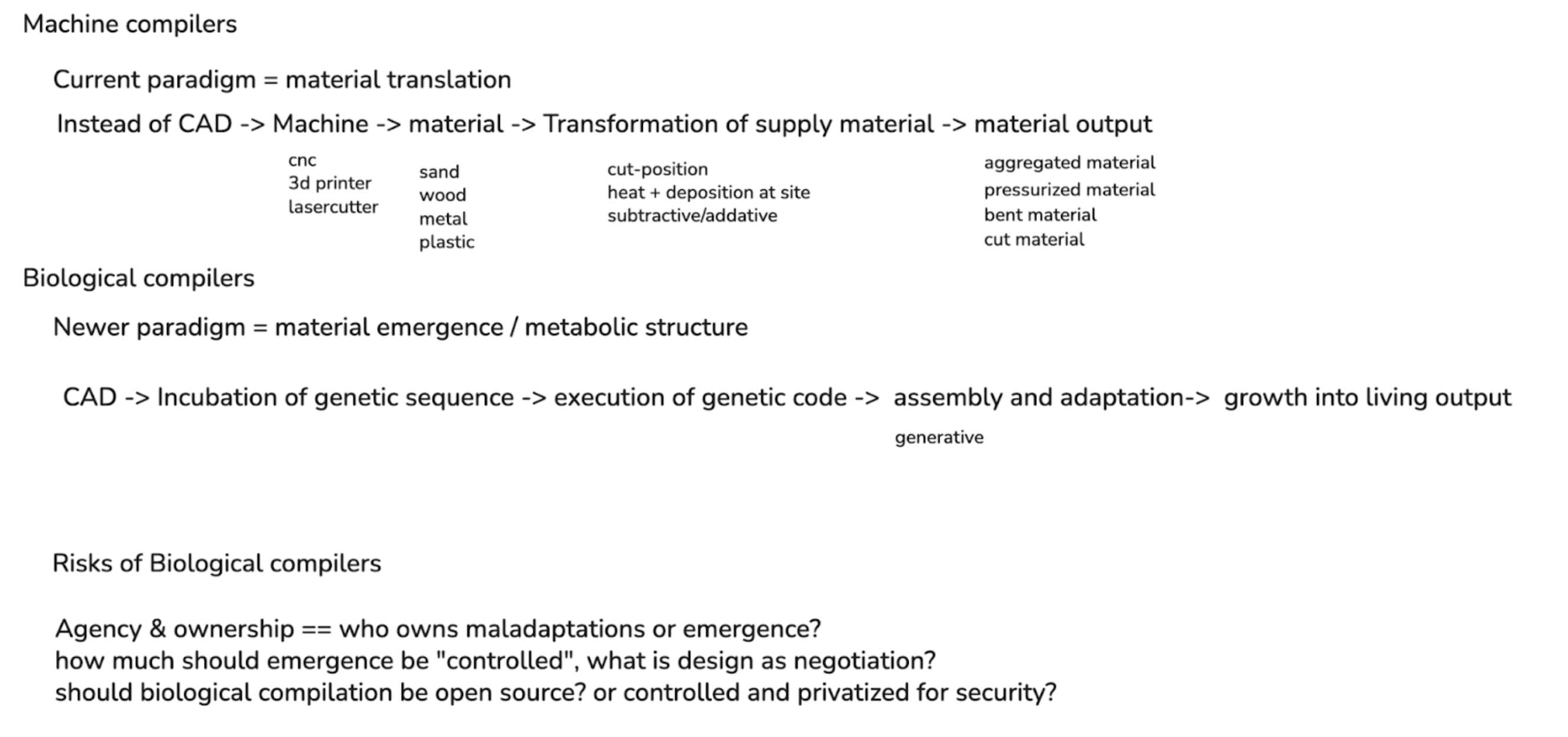
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. Currently, I develop software that creates new paradigms of computer-aided design (CAD) for systems that don’t fit conventional models of making. For example, most current mainstream CAD applications rely on drawing and volumetric representation as the main mechanism of formal shape creation. However, with newer fabrication systems such as robotic printing, zero-gravity printing, pigment printing, and other digital manufacturing advances, the tooling for operating this hardware lags behind the creation potential of these machines.
Homework Questions from Professor Jacobson: [Lecture 2 slides] Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? 1 error per 106 additions, with a throughput of 10 mS per Base Addition. If a human genome is 3.1 gigabase pairs haploid, then 3.1 109 / 10 6 = 3100 errors (3.1 * 1000) It fixes these errors during biological synthesis, where the nucleotides in an error physically dont fit together so it pushes that error out to unjam the system and continue on with further synthesis How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? 1036 base pairs in an average human protein, 3 bp (codon) per protein = 1036 / 3 = ~345 3345 That’s a huge number, but these dont all work physically because of codon bias, mRNA structure, regulatory interference, and translation/folding constraints. Note: Had to ChatGPT this one “Why don’t all codon possibilities work at a protein site?, couldn’t find it in the slides. Homework Questions from Dr. LeProust: [Lecture 2 slides] What’s the most commonly used method for oligo synthesis currently? 1965 Solid phase synthesis of oligos Had to search google separately Why is it difficult to make oligos longer than 200nt via direct synthesis? Because errors accumulate over time, even though oligo synthesis has a 99% success rate, the 1% error rate over time breaks it down with the step-by-step addition. So at 200 nucleotide length, you get a 37% full length molecules, which you can purify away from the rest. Beyond this, the exponential rate of the error makes it not wort the amount of effort. Why can’t you make a 2000bp gene via direct oligo synthesis? A looped run with error rate 99.5% 2000 times == 0.000045 chance of success, because oligo synthesis is all or nothing once a failure occurs. Homework Question from George Church: [Lecture 2 slides] Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
Part 1 Benchling Part 2 Lab Notes: Restriction Digest and Gel Electrophoresis Overview We’re cutting Lambda DNA with different restriction enzymes to create patterns in an agarose gel (inspired by the Latent Figure Protocol). The ladder helps us visualize the DNA fragment sizes based on base pair length.
Part 1 Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Vespers III
The paper presents a fabrication platform for making 3D-printed objects whose surfaces host living bacteria that respond to chemical signals embedded in the print material. The central idea is that a multimaterial inkjet printer can be used not just to control mechanical properties, but to spatially distribute chemical inducers throughout an object — and that bacteria coated onto the surface will “read” those signals and express proteins accordingly. The printer they used (Stratasys Objet Connex500) normally blends a rigid build resin with a sacrificial support resin to handle overhangs. The authors noticed that the support resin (SUP705) is hygroscopic — it absorbs water — which makes it useful for soaking up and slowly releasing chemical solutions like IPTG. By controlling how much support resin appears in each voxel, they could tune how much inducer gets released at any given spot on the surface. Later in the paper they go further and dissolve inducers directly into custom resin formulations loaded into the print cartridges, which lets them place two different chemical signals (IPTG and AHL) independently during a single print job. Bacteria are delivered by spraying a warm hydrogel-cell mixture onto the surface, which gels on contact. The hydrogel keeps cells alive, feeds them, and lets the inducers diffuse through from the print material below. Depending on what genetic circuits the cells carry, they produce visible outputs — blue or magenta pigment via β-galactosidase activity, or fluorescent proteins. The team also tested cells with AND and NAND logic gates, so expression only occurs where both signals are present, or where neither is. They built a computational model to predict how signals diffuse across 3D surfaces over time and how bacteria respond, which they validated against experimental results. The match was reasonable in most regions but broke down close to high-concentration signal sources, partly because some material compositions turned out to lower local pH and suppress expression — something the model didn’t account for.
Part A Why humans eat beef but do not become a cow, eat fish but do not become fish?
Part B: Protein Analysis and Visualization In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
Part A: SOD1 Binder Peptide Design Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. The A4V mutation (Alanine → Valine at residue 4) causes one of the most aggressive forms of familial ALS, subtly destabilizing the N-terminus, perturbing folding energetics, and promoting toxic aggregation.
Subsections of Homework
Week 1 HW: Principles and Practices
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Currently, I develop software that creates new paradigms of computer-aided design (CAD) for systems that don’t fit conventional models of making. For example, most current mainstream CAD applications rely on drawing and volumetric representation as the main mechanism of formal shape creation. However, with newer fabrication systems such as robotic printing, zero-gravity printing, pigment printing, and other digital manufacturing advances, the tooling for operating this hardware lags behind the creation potential of these machines.
While I am a beginner to the synthetic biology space, I see a similar ceiling imposed on the field based on tooling efficacy, with newer tooling like AlphaFold helping to exponentially open the potential of the space via computation. While there are existing “CAD” systems around protein design, there seems to remain a challenge in scaling these biological systems into a formalized design process.
I’m specifically interested in how humans can become part of the emergence of a living system, and how design as a field can change from the mode of human “control” into one of human “negotiation”. When designing with and alongside a living system, the criteria for design change. While gene editing offers significant potential in designing behavior, it also requires considering design over time. The design process extends beyond initial creation into growth: how the cells grow, adapt, and assemble into larger aggregations of structure.
This approach becomes more similar to designing cellular automata at scale, working alongside a living network that can push and pull with your own design goals. This change in theory orients towards design-in-emergence, in which the results of a design-driven process are left open for adaptation of the living system to accommodate.
At the same time, there should still be some intentional design process on the part of the researcher. Rather than relying on chance, computation enables an exploration of design possibilities that translate human intention into instructions that are legible to natural systems through genome editing. This act of “compilation” creates a bridge between high-level, human-authored structures such as a structural lattice into genetic sequences that guide the formation of a system through biological growth processes.
This project idea would look at the potential of biological compilation, i.e. a biocompiler, as an instrument for translating code into living systems, and examining the potential of a metabolic and emergent design process through morphogenesis. Similar to how 3D printing required new file formats and GCODE to translate digital models into machine instructions, biological compilers need new intermediate representations between parametric design and organic genome sequences. I will have to dive more deeply into the technical application of this further in the course, as to what a concrete initial prototype could be for this system of a metabolic and living design process.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
The governance issues relating to the ethical future of biological computation that interest me most are centered on agency and ownership. As fabricated living systems become more adaptive, and as an increasing number of individuals “code” life into being, responsibility becomes increasingly blurred.
1. Accountability and agency Who owns, or is responsible for, maladaptations that arise through emergent biological processes? If a system’s behavior is not fully specified in advance, how should responsibility be assigned when outcomes diverge from intent?
Goal: Create an accountability framework for emergent biocompiled systems.
Sub-goal A: Define liability when organisms deviate from designed behavior through mutation or adaptation.
Sub-goal B: Create certification or validation standards for genetic-to-material compilation accuracy and traceability.
2. Oversight and constraint To what degree is oversight necessary for generative biological systems? While strict guardrails or regulatory constraints may be required to prevent harm or misuse, imposing such limits may also reduce the adaptive and generative potential that makes these systems valuable in the first place.
Goal: Develop oversight mechanisms that balance safety with generative capacity
Sub-goal A: Define operational thresholds at which intervention, shutdown, or containment is required.
Sub-goal B: Define in which safety conditions experimentation would be freely allowed.
3. Ownership and access Should biological compilers be open source and publicly accessible, or privatized and restricted to limit potential harm? While privatization may reduce risk, it also concentrates power and may limit broader participation and positive impact. On the other hand, open access would increase the democratic possibility of the technology but also may amplify its misuse.
Goal: Balance access and control of biological compiler technologies
Sub-goal A: Ensure equitable access to foundational tools and prevent monopolization of biological design infrastructure.
Sub-goal B: Implement access controls or licensing models that preserve research and creative use
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
I will focus on what is done now in the computation space around robotics, because I have more knowledge of it compared to synthetic biology and it has many parallels to a living system.
[Training] While behavior is trained through data, the process of “digital twin” systems is a testing ground for robotic code to learn and adapt in a simulated environment, rather than making costly mistakes in a physical environment.
[Validation] Rather than assume good intent, there is a large need for “red teaming” to try to initiate unwanted behavior from the machine, so as to prevent that behavior from happening in a real environment.
[Deployment] Deployment is restricted to specific zones with clear partnership with local governance areas, [ie Waymo in SF], and has systems for overriding control.
[Access] Access is usually locked down so low-level systems are unable to be modified upon, and only high-level instructions can be inputted so as to not override training behavior and rule logistics.
Based on these precedents, I propose that biological compilation systems:
adopt a governance framework that requires simulated pre-deployment of any experimentation and testing,
perform adversarial tests against maladaptations and unwanted emergent behaviors,
require clearly defined deployment areas with actor-approved governance, and
restrict user-behavior to clear modes of approved interaction mechanisms, and have clear lock-out mechanisms.
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
To make this work, it has to be required for operation to occur.
Money & Publishing: Funding sources (government, nonprofits, companies) and publishers (journals, conferences) must require proper compliance across above conditions for the work to go through.
Regulatory Bodies: International law and local laws must be upheld with biocompilation, and such governance bodies must be created and maintained so as to not fall behind the pace of the emergent technology.
Labs and Developers: Must implement the proper testing, validation, and deployment practices rather than try to fasttrack the work. Licensing must be held to, and implemented correctly.
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
This might already be a solved problem
There is significant enough differences between robotics and synthetic biology that their safety-deployment pipelines should actually not be similar
Organizations might keep things “in-house” rather than going public with information to limit competition and not require proper regulation.
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
The safety pipeline does not consider system goals. For example, if the intended behavior of the original designer is to create a fully autonomous living being, the biocompiler system does not effectively safeguard against intent of goal with the designer. This is a similar risk in robotics and coding development (to what purpose is AI technology developed?).
The unintentional success of a biocompilation system could result in an ignorance of the required supplies needed for the procurement of such a system. For example in software, the abstraction of the “cloud” in compute services fully hides the environmental cost of digital systems through datacenters. A similar result could happen through a biocompiler system, where there no longer is a need to worry about the technical costs of acquiring certain cells and their modification.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Simulation-Based Training Requirements
Adversarial testing
Constrained Deployment, with Override
Access Control
Accountability & Agency
1
2
2
1
• Liability for maladaptation
2
1
2
3
• Traceability / certification
1
2
2
3
2. Oversight & Constraint
2
2
1
3
• Intervention / shutdown thresholds
3
2
1
2
• Conditions for free experimentation
2
2
3
1
Ownership & Access
3
2
2
1
• Prevent monopolization
3
2
2
1
• Preserve research & creative use
3
2
2
1
Other considerations
• Protecting the environment
1
2
3
n/a
• Protecting living systems
1
2
3
n/a
• Feasibility
2
2
3
1
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
My governance actions I designed as part of a process/pipeline, so I think they are all equally necessary to ensure proper deployment of biocompiled code. My main tradeoffs I considered were from what I’ve seen in the software space over the past decade, of the tradeoff between moving quickly to get new technology out (often seen in big tech and startups), compared to the more rigorous and contextual approach of academia with computer science. Citizen scientists and open source developers often sit in this in-between of the two worlds, and so I have them primarily in mind when designing these governance actions and constraints. I imagined it as something similar to `git` for biotech, which allows for control, revision, deployment, testing, and access baked into its functionality (ie CI/CD pipelines).
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
As a committed listener, my main ethical concern I’m faced with after week one is “who am I to learn such skills”. Synthetic biology from the perspective of a newcomer feels akin to taking on a god-like power, and as such I don’t feel nearly qualified to take on these tools. Considering the ethical and moral potential of taking on such power feels nearly insurmountable. Thus, my governance action to respond to this feeling is if there should be something similar to licensure for this technology. Similar to how lawyers need to pass the bar to perform proper legal work, should biotechnologists have to pass a similar certification program? Does one already exist?
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
1 error per 10^6 additions, with a throughput of 10 mS per Base Addition.
If a human genome is 3.1 gigabase pairs haploid, then
3.1 109 / 10 6 = 3100 errors (3.1 * 1000)
It fixes these errors during biological synthesis, where the nucleotides in an error physically dont fit together so it pushes that error out to unjam the system and continue on with further synthesis
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
1036 base pairs in an average human protein, 3 bp (codon) per protein = 1036 / 3 = ~345
3^345
That’s a huge number, but these dont all work physically because of codon bias, mRNA structure, regulatory interference, and translation/folding constraints.
Note: Had to ChatGPT this one “Why don’t all codon possibilities work at a protein site?, couldn’t find it in the slides.
What’s the most commonly used method for oligo synthesis currently?
1965 Solid phase synthesis of oligos
Had to search google separately
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because errors accumulate over time, even though oligo synthesis has a 99% success rate, the 1% error rate over time breaks it down with the step-by-step addition. So at 200 nucleotide length, you get a 37% full length molecules, which you can purify away from the rest. Beyond this, the exponential rate of the error makes it not wort the amount of effort.
Why can’t you make a 2000bp gene via direct oligo synthesis?
A looped run with error rate 99.5% 2000 times == 0.000045 chance of success, because oligo synthesis is all or nothing once a failure occurs.
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Lysine contingency – jurassic park failsafe of not letting the park animals themselves generate lysine, which they need to be provided for. But an adaptation at the park allowed that adaptation of lysine generation to occur.
Ten amino acids animals can’t produce on their own so need to supply are: Arginine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, and Valine
Because Lysine is already an essential amino acid not supplied for most all animals, we all have a lysine contingency and they didn’t really do any gene editing to get this in Jurassic park.
[Given slides #2 & 4 (AA:NA and NA:NA codes)] What code would you suggest for AA:AA interactions?
If NA:NA = A:U, NA:AA = AUG:Met, but for AA:AA this doesnt make sense because their connections are not clear (theyre not “building blocks” as simple as NA -> NA -> AA is compiled), theyre discrete rather than modular units because of their chemical composition
[(Advanced students)] Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
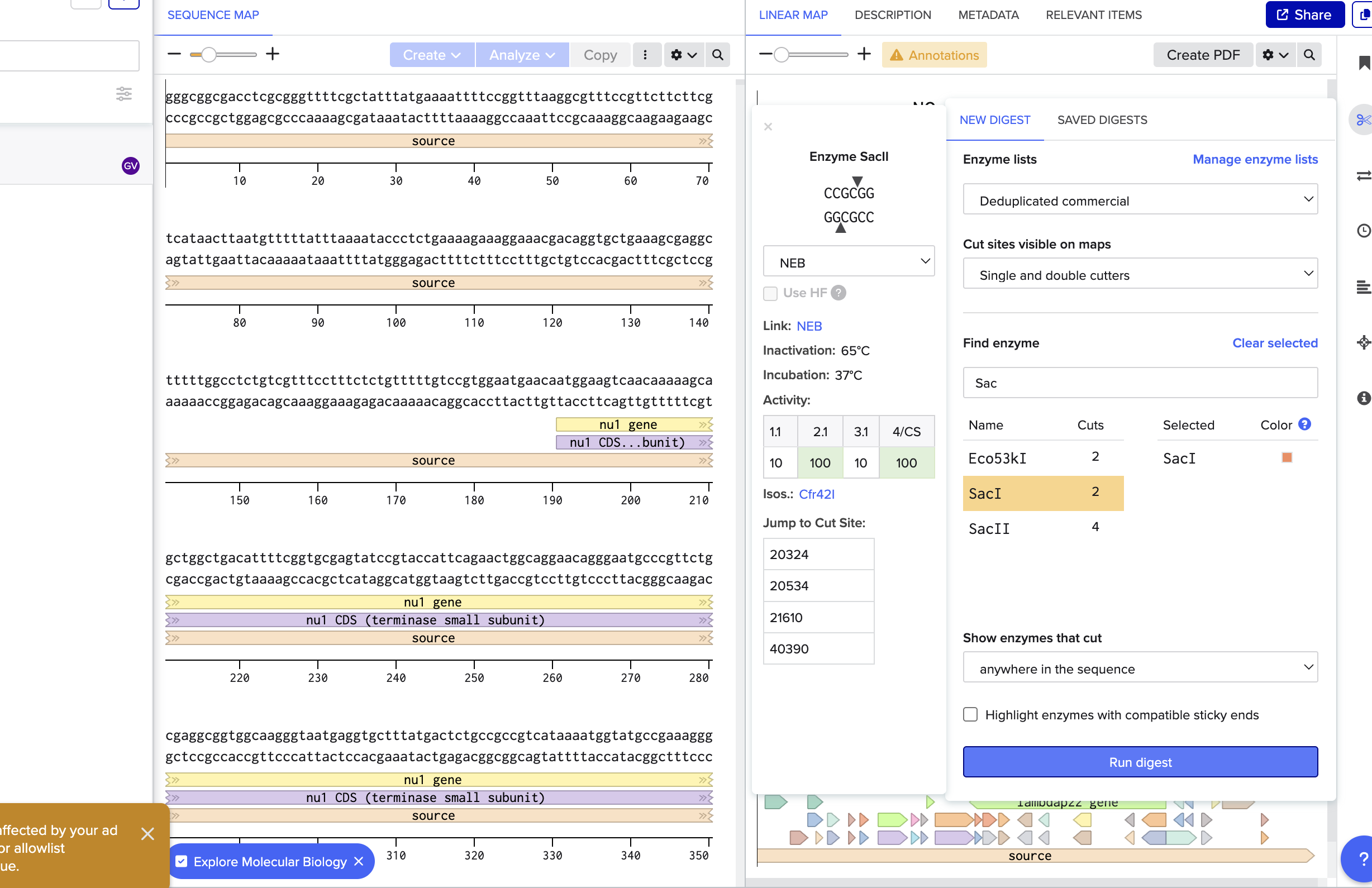
Lab Notes: Restriction Digest and Gel Electrophoresis
Overview
We’re cutting Lambda DNA with different restriction enzymes to create patterns in an agarose gel (inspired by the Latent Figure Protocol). The ladder helps us visualize the DNA fragment sizes based on base pair length.
Enzymes Used
SacI
Fnu4HI
EcoRI
SphI
DpnI
NdeI
AflIII
Digest Setup (per reaction)
Total volume: 50 μl
24 μl nuclease-free water
20 μl Lambda DNA (1 μg)
5 μl NEBuffer (pH buffer to keep the reaction stable)
1 μl restriction enzyme
Procedure
Step 1: Set up the digest
First pipette the water into the tube (used the smaller red tips for DNA)
Add the Lambda DNA, NEBuffer, and restriction enzyme
The restriction enzyme is what actually cuts the DNA at specific sites
NEBuffer maintains the right pH for the enzyme to work
Step 2: Incubation
Put tubes in the heating chamber at 37°C for 1 hour
The temperature is critical for enzyme activity
Step 3: Prepare the gel
Weigh out 0.5g agarose
Put it in a cylinder and add TAE buffer (this makes the gel conductive for electrophoresis)
Step 4: Running the gel
Cover the gel with TAE buffer
Add loading dye to each DNA sample tube
Load the ladder and DNA samples into the gel wells using a pipette
Run electrophoresis to separate fragments by size
Part 3
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above. Example: Get to the original sequence of phage MS2 L-protein from its genome — phage MS2 genome - Nucleotide - NCBI
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why? Example: Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI.
Since organisms have codon usage bias (preference for which codons they use to produce a particular amino acid), codon optimization allows a translation from the original source to the preferred communication method of that organism.
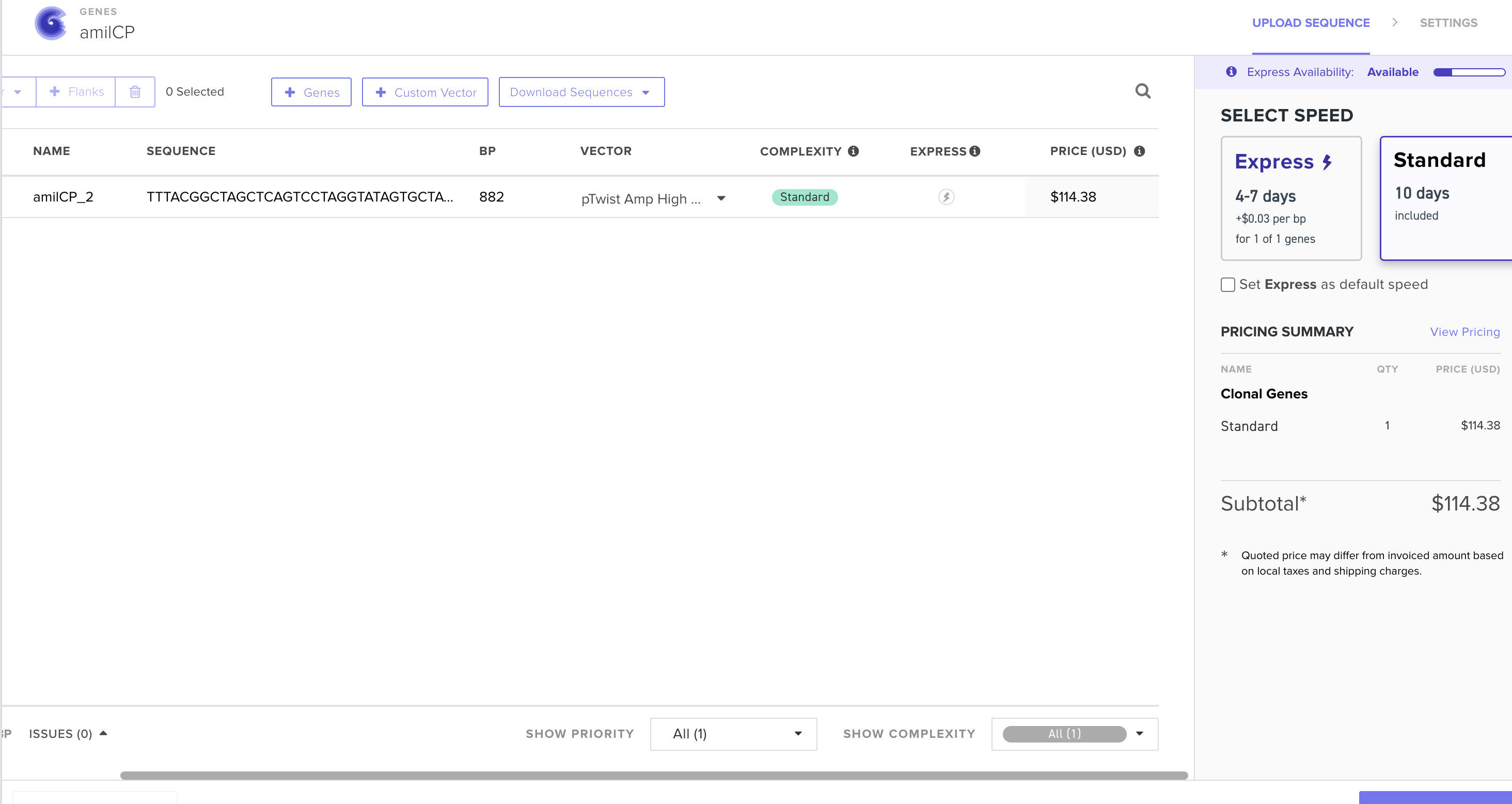
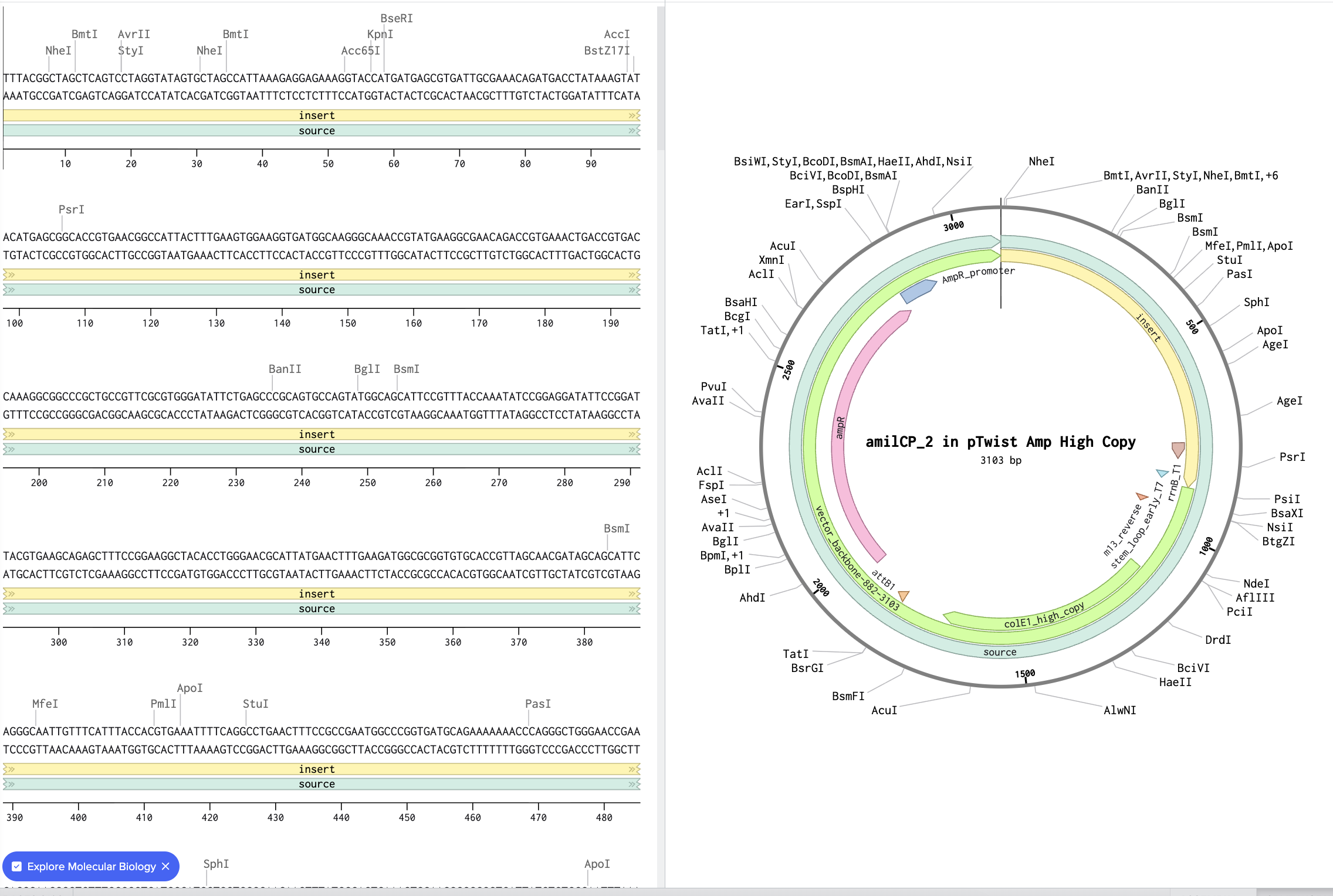
I’m translating the original sequence for E. coli.
What technologies could be used to produce this protein from your DNA? Describe in your words how the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
I could either produce amilCP via in vitro by adding the E. coli lysate to a test tube and mixing the DNA in, but probably easier would be adding this DNA directly to a living E. coli sample in a petri dish, by adding it directly into the host. The cells would then transcribe the DNA into mRNA, which would then create the amino acid sequences which would create amilCP.
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
Right now I’m interested in environmental health data and monitoring, both in water and air quality. I’m primarily interested in pollutants in these regions, but not necessarily biological ones. I’m more interested in metals, plastics, VOCs, temperature, humidity, etc.
From a more theoretical point of view, I would be interested in reading DNA as a data store, but I’m not sure how that really exists today. For the sake of this assignment, I’ll assume there’s a recorded DNA database holding metadata around an object’s provenance and creation.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Oxford Nanopore Technologies. Live-stream sequencing allows real-time processing, so stopping the stream and controlling data input more similar to machinic systems would be beneficial. The portability of it as well allows more diversification if reading in the field is required.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so? Third generation, because it’s loaded in as a long string, rather than needing to post-compute concatenate shorter strings together like 2nd generation technologies.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. The input is synthetic DNA data blocks, which are probably encased in some protective chamber. For preparation, they must be removed from their protection, where you would then do end repair and adapter ligation, and then clean up any non-essential enzymes.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? The mixture is dropped onto a chip with nanopores; the nanopores have an electrical current, and as the DNA is pulled through by a motor protein, the DNA itself disrupts the consistent electrical current. Because each nucleotide has a different size, it causes a variant change in the electrical signal, and that signal is then converted back into the nucleotide sequence digitally.
What is the output of your chosen sequencing technology? There’s the raw output of the electrical signal, and the decoded signal sequence file (FASTQ).
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origami). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs!
Fast-PETase in a cell-free expression
A TphR logic/gate
A chromoprotein such as amilCP to express when the TphR is present
From a theory perspective, I’m interested in a “live-environmental coding” of an object based on the conditions of that space, an “imprint” of it. As a prototype, I think an active signal-remediation loop with microplastic degradation would be interesting. I’m currently thinking of creating a genetic circuit for displaying this protein breakdown in a cell-free system, and dosing that into a porous structure such as 3D-printed ceramic. This would be a plastic-eating structure that shows the color of how it’s changed its environment over time, and when a new unit could be brought in. Could also be interesting in an underwater environment with calcium carbonate, but not sure how quickly denaturing would happen here compared to an air environment. I would like to pair this with software for designing the circuits, from simulating color change over time, and pairing that with different sensor inputs.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Technology choice: I would use Twist Bioscience for several key reasons:
Ready availability – Twist’s commercial platform is accessible and well-established
Parallel synthesis capability – The ability to synthesize many genes in parallel on a silicon chip would allow multiple iterations of the genetic circuit design, enabling me to order multiple variants in a single batch
Iteration efficiency – This parallel approach supports rapid design-build-test cycles for optimizing the circuit
Main limitations:
Cell-free compatibility – I will need to optimize this synthesis method specifically for cell-free expression systems, which may require additional iterations
Cost considerations – Pricing over time could become a factor, especially for multiple design iterations
Accuracy – I shouldn’t encounter issues with synthesis accuracy since all my constructs should be under 1 kb, though I may need to introduce redundancy if needed
Also answer the following questions:
What are the essential steps of your chosen synthesis method? [To be completed]
What are the limitations of your synthesis method (if any) in terms of speed, accuracy, scalability? See main limitations above.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I’d want to edit heat shock protein genes in corals, such as HSP70, to make them more resistant to warming ocean temperatures. This is important for coral health because bleaching occurs when water temperatures rise 1–2°C above normal and corals expel their symbiotic algae, essentially starving them, and this is more likely to rise over the next few decades because of climate change warming ocean surfaces. If improved, heat shock proteins could help corals respond better to temperature spikes, which would improve overall ecosystem health because coral reefs support about 25% of all marine species and provide critical habitat, coastal protection, and fisheries.
To do this, I’d use CRISPR because it allows precise targeting of specific genes.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps? CRISPR uses a guide RNA that matches the DNA sequence you want to change. This guide brings the Cas9 enzyme to the right spot, and Cas9 cuts the DNA. After the cut, the cell tries to repair the break. If you provide a template with the sequence you want, the cell will copy that template and insert your new DNA.
What preparation do you need to do and what is the input for the editing? You need to design a guide RNA that matches the gene being targeted. The main inputs are the Cas9 enzyme, the guide RNA, and a DNA template. You also need to get these into the cells.
What are the limitations of your editing methods in terms of efficiency or precision? CRISPR doesn’t work every time. Either the cells don’t get edited successfully, or only some cells get the edit. Also, Cas9 can cut in the wrong place if there’s a similar sequence in the DNA.
Week 3 Lab & HW
Part 1
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Vespers III
The paper presents a fabrication platform for making 3D-printed objects whose surfaces host living bacteria that respond to chemical signals embedded in the print material. The central idea is that a multimaterial inkjet printer can be used not just to control mechanical properties, but to spatially distribute chemical inducers throughout an object — and that bacteria coated onto the surface will “read” those signals and express proteins accordingly.
The printer they used (Stratasys Objet Connex500) normally blends a rigid build resin with a sacrificial support resin to handle overhangs. The authors noticed that the support resin (SUP705) is hygroscopic — it absorbs water — which makes it useful for soaking up and slowly releasing chemical solutions like IPTG. By controlling how much support resin appears in each voxel, they could tune how much inducer gets released at any given spot on the surface. Later in the paper they go further and dissolve inducers directly into custom resin formulations loaded into the print cartridges, which lets them place two different chemical signals (IPTG and AHL) independently during a single print job.
Bacteria are delivered by spraying a warm hydrogel-cell mixture onto the surface, which gels on contact. The hydrogel keeps cells alive, feeds them, and lets the inducers diffuse through from the print material below. Depending on what genetic circuits the cells carry, they produce visible outputs — blue or magenta pigment via β-galactosidase activity, or fluorescent proteins. The team also tested cells with AND and NAND logic gates, so expression only occurs where both signals are present, or where neither is.
They built a computational model to predict how signals diffuse across 3D surfaces over time and how bacteria respond, which they validated against experimental results. The match was reasonable in most regions but broke down close to high-concentration signal sources, partly because some material compositions turned out to lower local pH and suppress expression — something the model didn’t account for.
Part 2
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
I don’t know which direction I want to go yet for my final project.
If I go the route of the emergent subtractive fabrication, I’d use the Opentrons to characterize my genetic circuit by running arabinose concentration gradients across a 96-well plate to find the induction threshold where the tphr gate reliably drives amIICP expression. I’d design and order the circuit parts through Ginkgo Nebula, and build a simple 3D printed holder to keep PET samples consistently positioned during the degradation assay.
However, if I instead go with the community DNA archive, I would focus more on the software side first — writing an encoder that converts community submissions into .fasta sequences with error correction built in — and then use the Opentrons to optimize the silica encapsulation step, running a matrix of silica-to-DNA ratios to find conditions that actually hold up for long-term storage before casting the final resin tiles.
Example 1: You are creating a custom fabric, and want to deposit art onto specific parts that need to be intertwined in odd ways. You can design a 3D printed holder to attach this fabric to it, and be able to deposit bio art on top. Check out the Opentrons 3D Printing Directory.
Example 2: You are using the cloud laboratory to screen an array of biosensor constructs that you design, synthesize, and express using cell-free protein synthesis.
Echo transfer biosensor constructs and any required cofactors into specified wells.
Bravo stamp in CPFS reagent master mix into all wells of a 96-well / 384-well plate.
Multiflo dispense the CFPS lysate to all wells to start protein expression.
PlateLoc seal the plate.
Inheco incubate the plate at 37°C while the biosensor proteins are synthesized.
XPeel remove the seal.
PHERAstar measure fluorescence to compare biosensor responses.
Why humans eat beef but do not become a cow, eat fish but do not become fish?
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins.
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
fast-petase
Briefly describe the protein you selected and why you selected it.
I selected fast-petase because im considering using it in my final project. I am curious to learn more about the enzymatic breakdown of PET plastics.
How many protein sequence homologs are there for your protein? Hint: Use the pBLAST tool to search for homologs and ClustalOmega to align and visualize them. Tutorial Here
2,930 PETase homologs
Does your protein belong to any protein family?
Cutinase / Dienelactone hydrolase-like
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Method: X-RAY DIFFRACTION
Resolution: 1.57 Å
R-Value Free:
0.140 (Depositor), 0.141 (DCC)
R-Value Work:
0.106 (Depositor), 0.107 (DCC)
R-Value Observed:
0.108 (Depositor)
Are there any other molecules in the solved structure apart from protein?
The solved crystal structure of FAST-PETase (PDB: 7SH6) contains one Sulfate ion (SO4) molecule besides the protein chain
Does your protein belong to any structure classification family?
Hydrolase
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
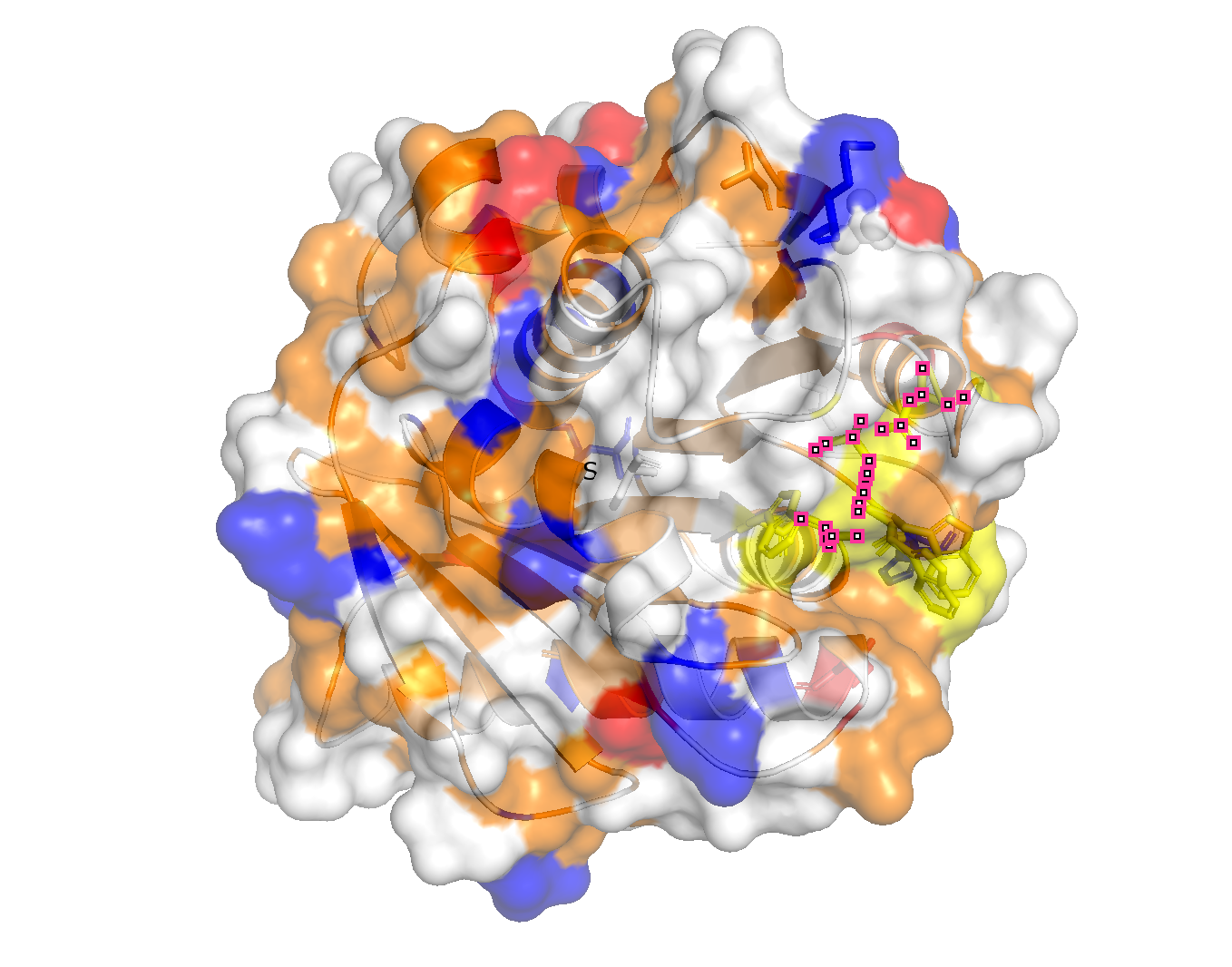
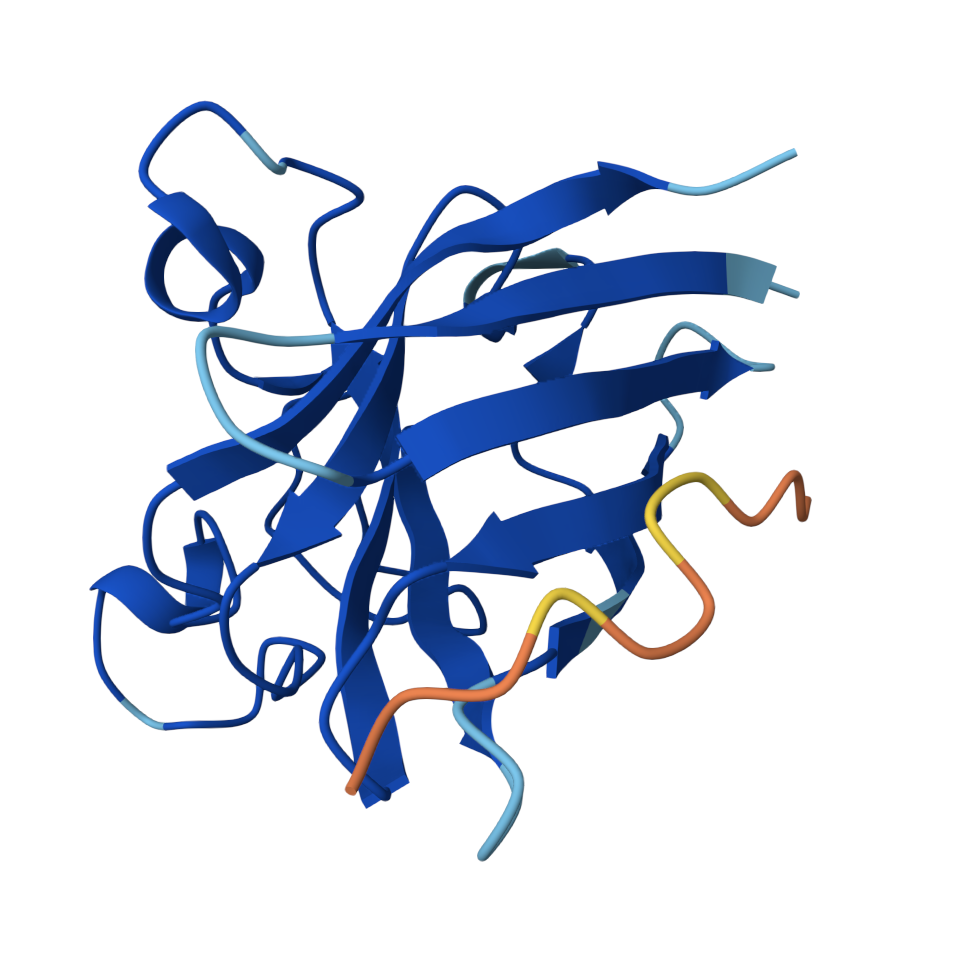
Color the protein by secondary structure. Does it have more helices or sheets?
is organized into 9 β-strands (yellow) and 7 α-helices, so more B-strands.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The hydrophobic tend towards the center (orange) and the hydrophilic towards the outer surface.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
There is one valley in the outer surface as the PET-binding cleft
Part C. Using ML-Based Protein Design Tools
HTGAA ProteinDesign2025
Fold your protein with AlphaFold or ESMFold or Boltz and compare it to the real structure.
Comment on:
Any predicted vs. experimental differences.
It has a 90%+ predicted accuracy for nearly everything
Low-confidence regions and why do you think they are low confidence?
The main low-confidence areas on the loop regions and the His-tag tail.
Inverse-fold your structure with ProteinMPNN
What sequence do you get?
Is it the same as the original sequence you folded?
No, only 52% of the sequence is the same
Why yes or no?
This is possible because many different amino acid combinations can produce the same protein structure, its the structure that matters more than the combinations themselves.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Review the Bacteriophage Final Project Goals:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why you think those tools might help solve your chosen sub-problem.
One or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline
This resource may be useful: HTGAA Protein Engineering Tools
Individually put your plan on your website page
Each group’s short plan for engineering a bacteriophage
Schedule time ( HTGAA Protein Engineering Feedback) to get feedback/discuss your ideas, and put the feedback on your website
Week 5 Lab & HW
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. The A4V mutation (Alanine → Valine at residue 4) causes one of the most aggressive forms of familial ALS, subtly destabilizing the N-terminus, perturbing folding energetics, and promoting toxic aggregation.
Using PepMLM-650M conditioned on the A4V mutant SOD1 sequence, I generated four 12-amino-acid peptides and compared them against the known binder FLYRWLPSRRGG (substituted here as FLYWRLPSRRGG for the run):
Binder
Pseudo Perplexity
WRVPATAVAWKE
8.347
WRYGAAAAEHKE
9.620
WSSYWVGIRLGX
13.782
WRVGVAAVAWKE
11.812
FLYWRLPSRRGG (known)
20.190
Lower perplexity indicates higher model confidence. All four generated peptides achieved lower perplexity than the known binder FLYWRLPSRRGG (20.19), suggesting PepMLM is reasonably confident in these sequences as plausible binders. WRVPATAVAWKE scored best at 8.35.
Part 2: Evaluate Binders with AlphaFold3
Each peptide was submitted alongside the A4V mutant SOD1 sequence as separate chains to AlphaFold Server.
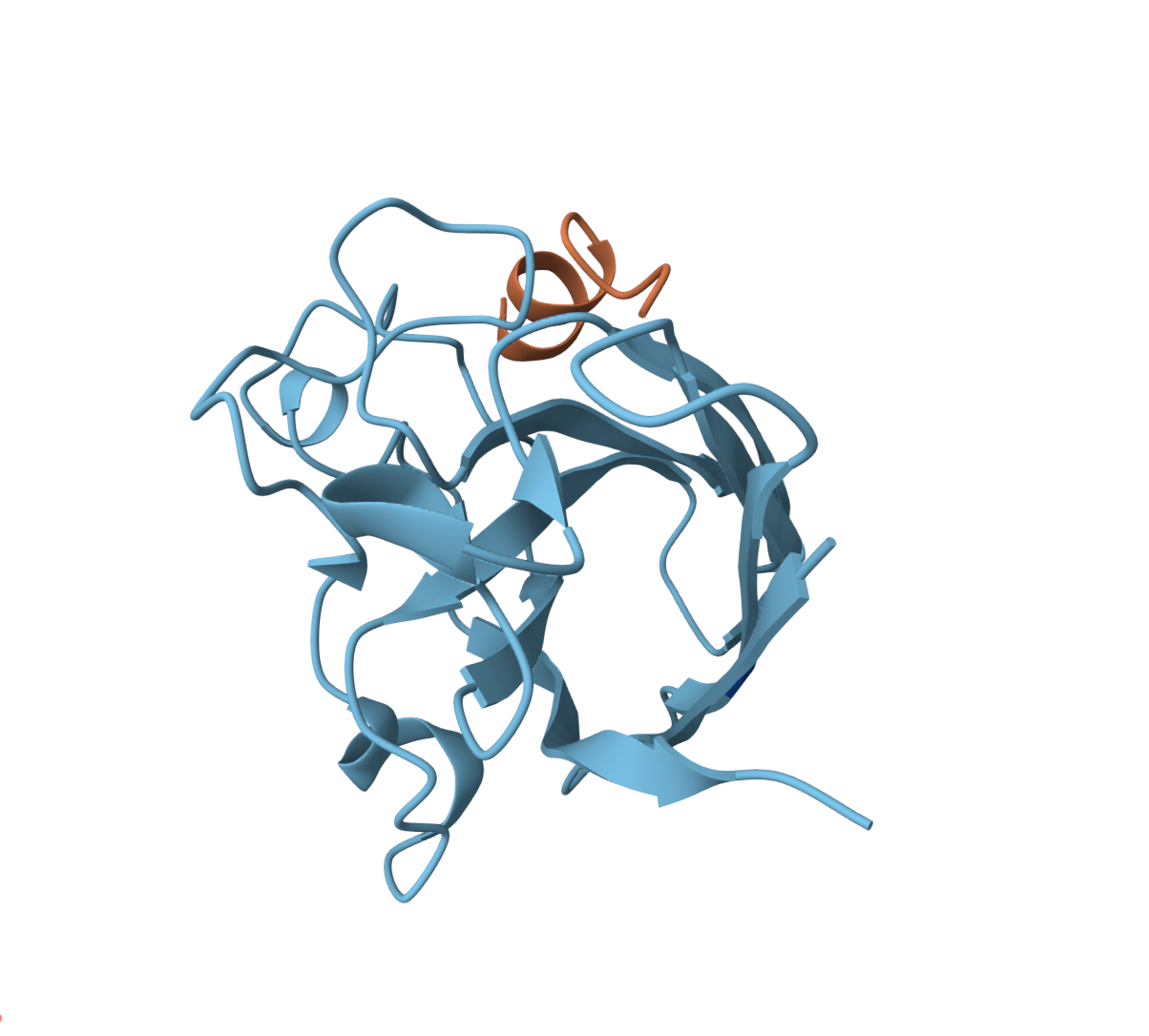
WRVPATAVAWKE — ipTM = 0.34, pTM = 0.83
The peptide docks onto a loop region of the β-barrel at the periphery of the barrel surface. It does not appear to be buried and does not obviously localize near the N-terminus/A4V site.
WRYGAAAAEHKE — ipTM = 0.29, pTM = 0.74
A clear helix forms for this peptide, but it is not well engaged with the protein itself. Overall weak confidence with the lowest ipTM of the set.
WRVGVAAVAWKE — ipTM = 0.26, pTM = 0.85
High pTM confidence but a weak interface, positioned at the edge of the β-barrel. This peptide sits closest to the dimer interface of all candidates.
The ipTM values across all three evaluated peptides (0.26–0.34) are relatively low, indicating weak predicted interface quality. None of the PepMLM-generated peptides clearly localizes to the N-terminus/A4V site. The known binder was not re-evaluated here since its ipTM from the literature would serve as a reference point; none of the generated peptides appear to decisively outperform it structurally.
Part 3: Evaluate Properties with PeptiVerse
Each peptide was evaluated in PeptiVerse with the A4V mutant SOD1 as target.
WRVPATAVAWKE (ipTM = 0.34)
Property
Value
Solubility
Soluble (1.000)
Permeability
Permeable (0.458)
Hemolysis
Non-hemolytic (0.032)
Non-Fouling
Fouling (0.390)
Binding Affinity
Weak (5.902 pKd/pKi)
Length
12 aa
Molecular Weight
1413.6 Da
Net Charge (pH 7)
+0.77
Isoelectric Point
8.75
Hydrophobicity (GRAVY)
-0.18
WRYGAAAAEHKE (ipTM = 0.29)
Property
Value
Solubility
Soluble (1.000)
Permeability
Non-permeable (0.210)
Hemolysis
Non-hemolytic (0.026)
Non-Fouling
Non-fouling (0.677)
Binding Affinity
Weak (5.588 pKd/pKi)
Length
12 aa
Molecular Weight
1388.5 Da
Net Charge (pH 7)
-0.14
Isoelectric Point
6.77
Hydrophobicity (GRAVY)
-1.17
WRVGVAAVAWKE (ipTM = 0.26)
Property
Value
Solubility
Soluble (1.000)
Permeability
Non-permeable (0.229)
Hemolysis
Non-hemolytic (0.062)
Non-Fouling
Fouling (0.152)
Binding Affinity
Medium (7.870 pKd/pKi)
Length
12 aa
Molecular Weight
1371.6 Da
Net Charge (pH 7)
+0.77
Isoelectric Point
8.75
Hydrophobicity (GRAVY)
+0.33
The structural confidence (ipTM) and predicted binding affinity do not correlate cleanly: WRVPATAVAWKE has the highest ipTM (0.34) but only weak predicted affinity (5.9), while WRVGVAAVAWKE has the lowest ipTM (0.26) yet shows medium predicted affinity (7.87). All peptides are predicted to be soluble and non-hemolytic, which is encouraging. None are predicted to be strong binders outright, reflecting that all three fall short of locating the A4V site.
Selected peptide: WRVGVAAVAWKE. Despite its low ipTM, it shows the strongest predicted binding affinity (medium, 7.87 pKd/pKi), is non-hemolytic, and has positive net charge favorable for cytosolic targets. It positions closest to the dimer interface, which is a functionally relevant region for stabilizing the SOD1 homodimer. The main liability is fouling potential (0.152), which would need to be addressed through further optimization before therapeutic advancement.
Part 4: Generate Optimized Peptides with moPPIt
moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward user-specified residue indices while simultaneously optimizing binding and therapeutic properties. Unlike PepMLM, which samples plausible binders from the full target sequence, moPPIt allows precise targeting of a specific region.
For this run, I targeted residues near position 4 (the A4V mutation site) and the dimer interface region, setting peptide length to 12 amino acids with affinity and solubility guidance enabled.
The moPPIt-generated peptides differ from PepMLM peptides in two key ways. First, they are steered toward a specific binding site — the N-terminal A4V region and dimer interface — rather than sampling freely across the entire protein surface. Second, because MOG-DFM optimizes multiple objectives simultaneously (affinity, solubility, hemolysis), the output sequences are less likely to score well on one property while failing on another.
Before advancing any moPPIt peptide toward clinical studies, I would:
Re-evaluate all candidates in AlphaFold3 to confirm localization to the A4V site
Run PeptiVerse analysis to verify therapeutic property predictions
Check for sequence novelty (no overlap with endogenous peptides that could cause off-target effects)
Perform MD simulations to assess binding stability over time
Synthesize the top 1–2 candidates for in vitro binding assays (SPR or ITC) against recombinant A4V SOD1
Test in ALS cell models for reduction of aggregation or rescue of SOD1 function