Week 2 Lab & HW
Part 1
Benchling
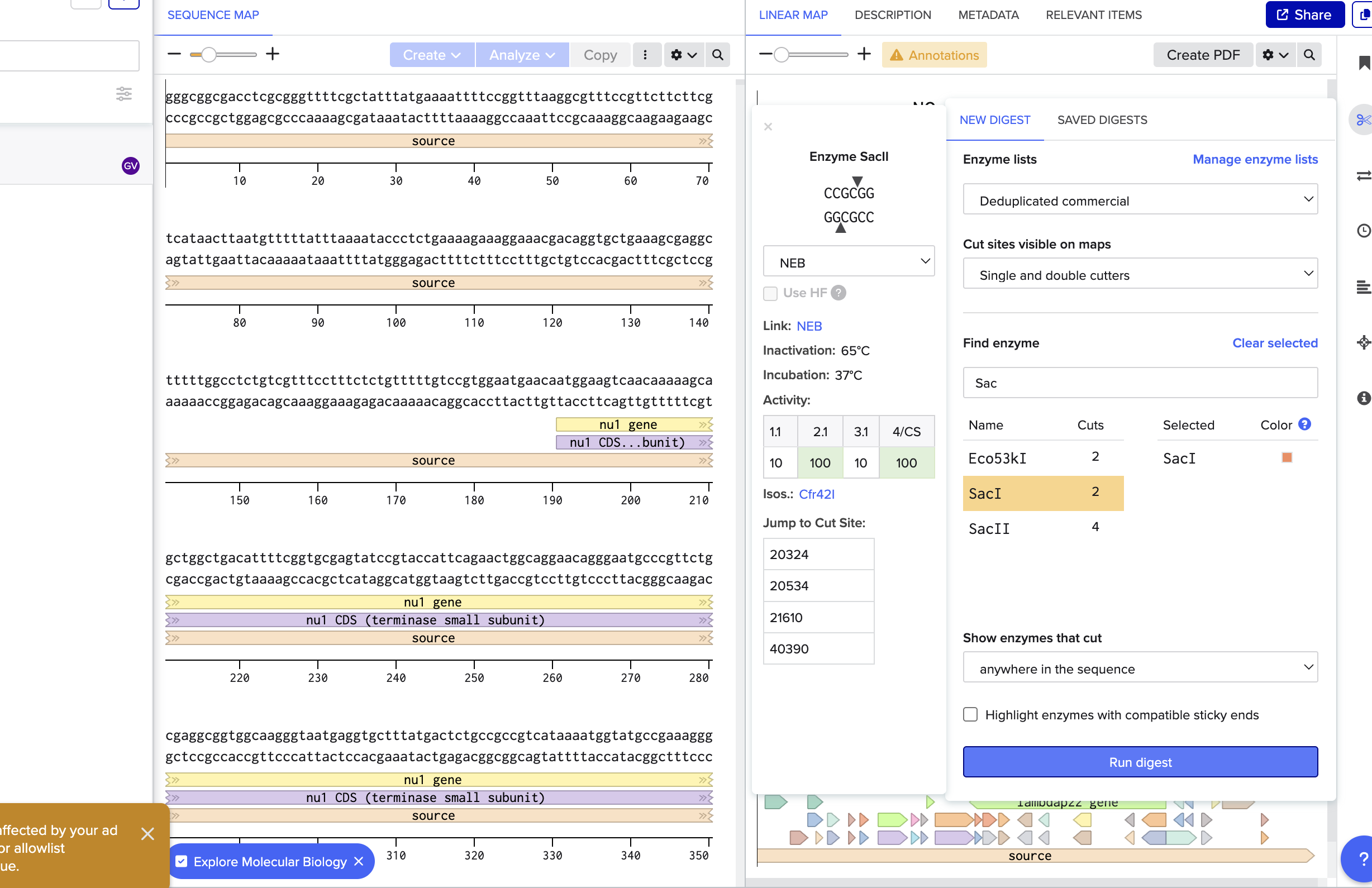

Part 2
Lab Notes: Restriction Digest and Gel Electrophoresis
Overview
We’re cutting Lambda DNA with different restriction enzymes to create patterns in an agarose gel (inspired by the Latent Figure Protocol). The ladder helps us visualize the DNA fragment sizes based on base pair length.
Enzymes Used
- SacI
- Fnu4HI
- EcoRI
- SphI
- DpnI
- NdeI
- AflIII
Digest Setup (per reaction)
Total volume: 50 μl
- 24 μl nuclease-free water
- 20 μl Lambda DNA (1 μg)
- 5 μl NEBuffer (pH buffer to keep the reaction stable)
- 1 μl restriction enzyme
Procedure
Step 1: Set up the digest
- First pipette the water into the tube (used the smaller red tips for DNA)
- Add the Lambda DNA, NEBuffer, and restriction enzyme
- The restriction enzyme is what actually cuts the DNA at specific sites
- NEBuffer maintains the right pH for the enzyme to work
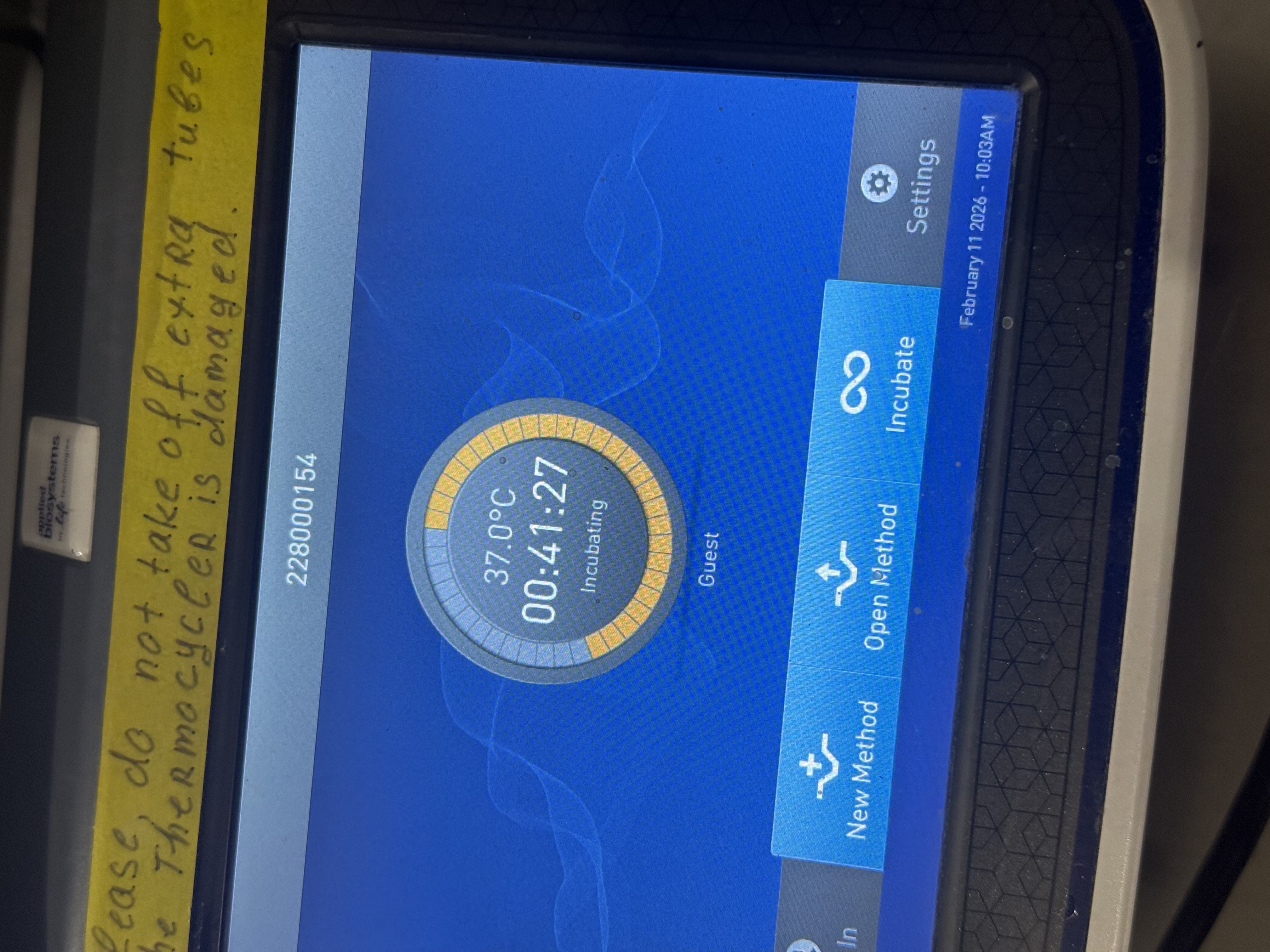
Step 2: Incubation
- Put tubes in the heating chamber at 37°C for 1 hour
- The temperature is critical for enzyme activity
Step 3: Prepare the gel
- Weigh out 0.5g agarose
- Put it in a cylinder and add TAE buffer (this makes the gel conductive for electrophoresis)
Step 4: Running the gel
- Cover the gel with TAE buffer
- Add loading dye to each DNA sample tube
- Load the ladder and DNA samples into the gel wells using a pipette
- Run electrophoresis to separate fragments by size






Part 3
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
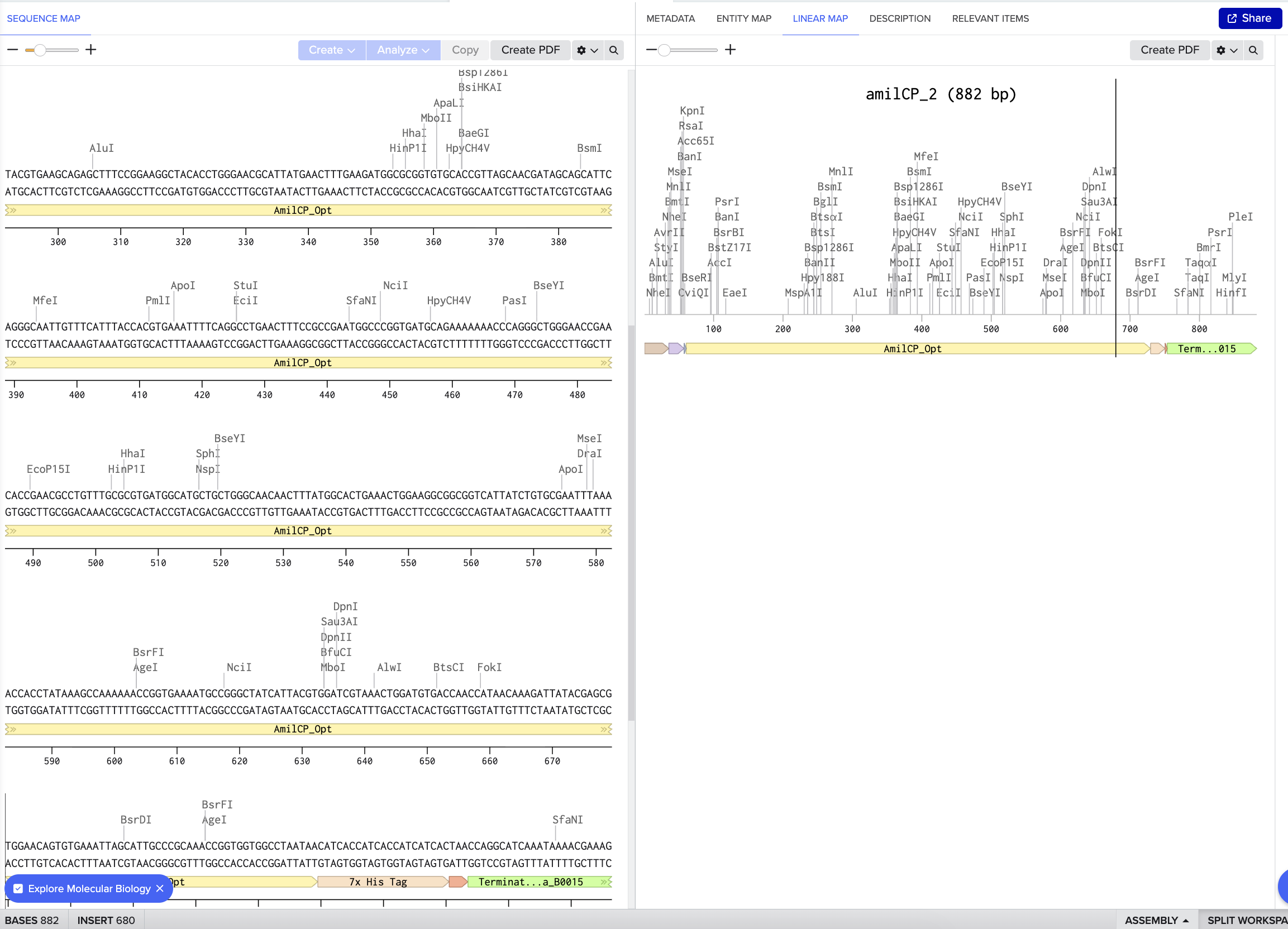
AmilCP — reef-building coral Acropora millepora
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Example: Get to the original sequence of phage MS2 L-protein from its genome — phage MS2 genome - Nucleotide - NCBI
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Example: Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI.
Since organisms have codon usage bias (preference for which codons they use to produce a particular amino acid), codon optimization allows a translation from the original source to the preferred communication method of that organism.
I’m translating the original sequence for E. coli.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words how the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
I could either produce amilCP via in vitro by adding the E. coli lysate to a test tube and mixing the DNA in, but probably easier would be adding this DNA directly to a living E. coli sample in a petri dish, by adding it directly into the host. The cells would then transcribe the DNA into mRNA, which would then create the amino acid sequences which would create amilCP.
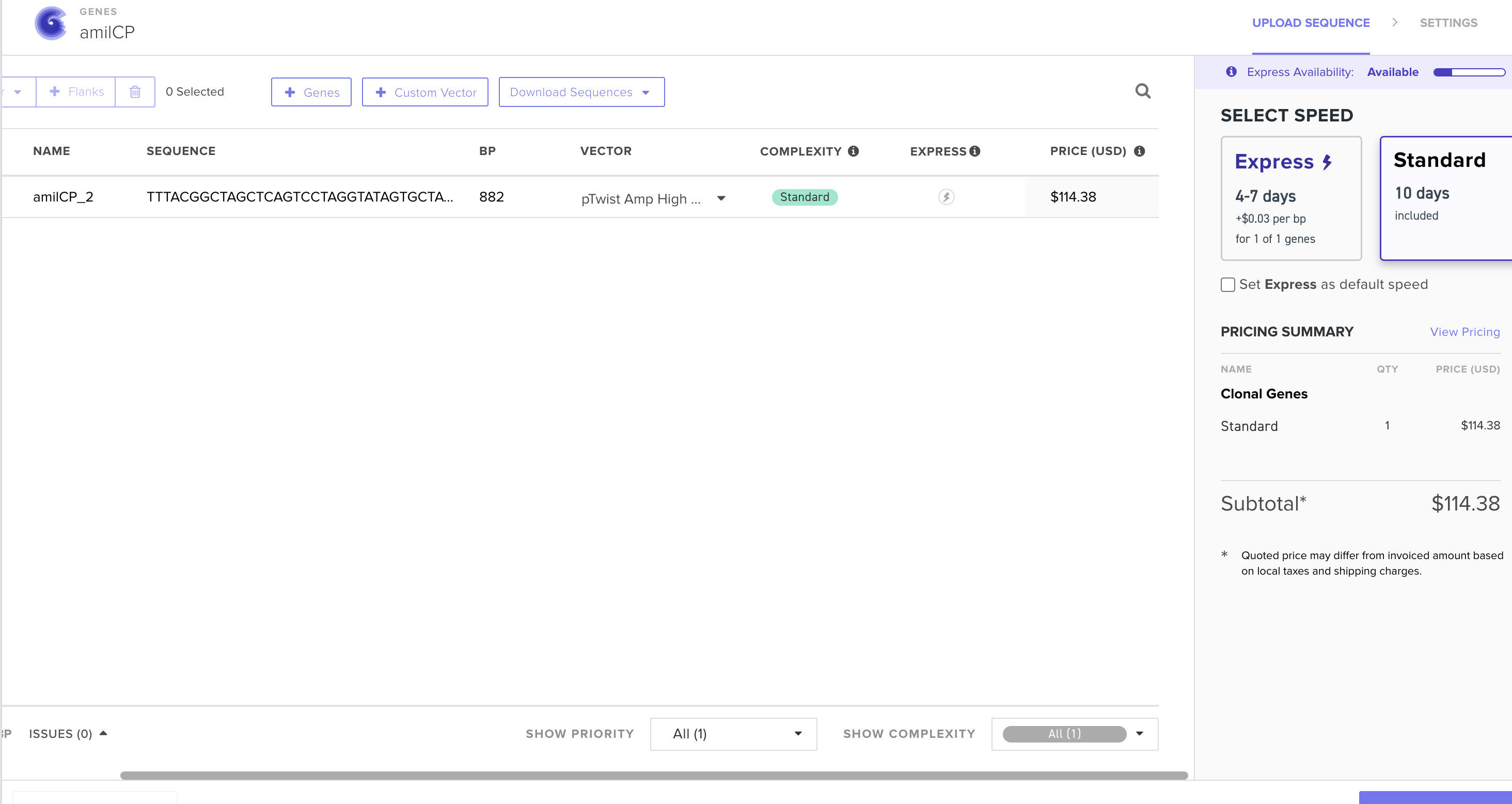
Benchling link: seq-Y9OCshTKaHht34vERJXL
Part 4

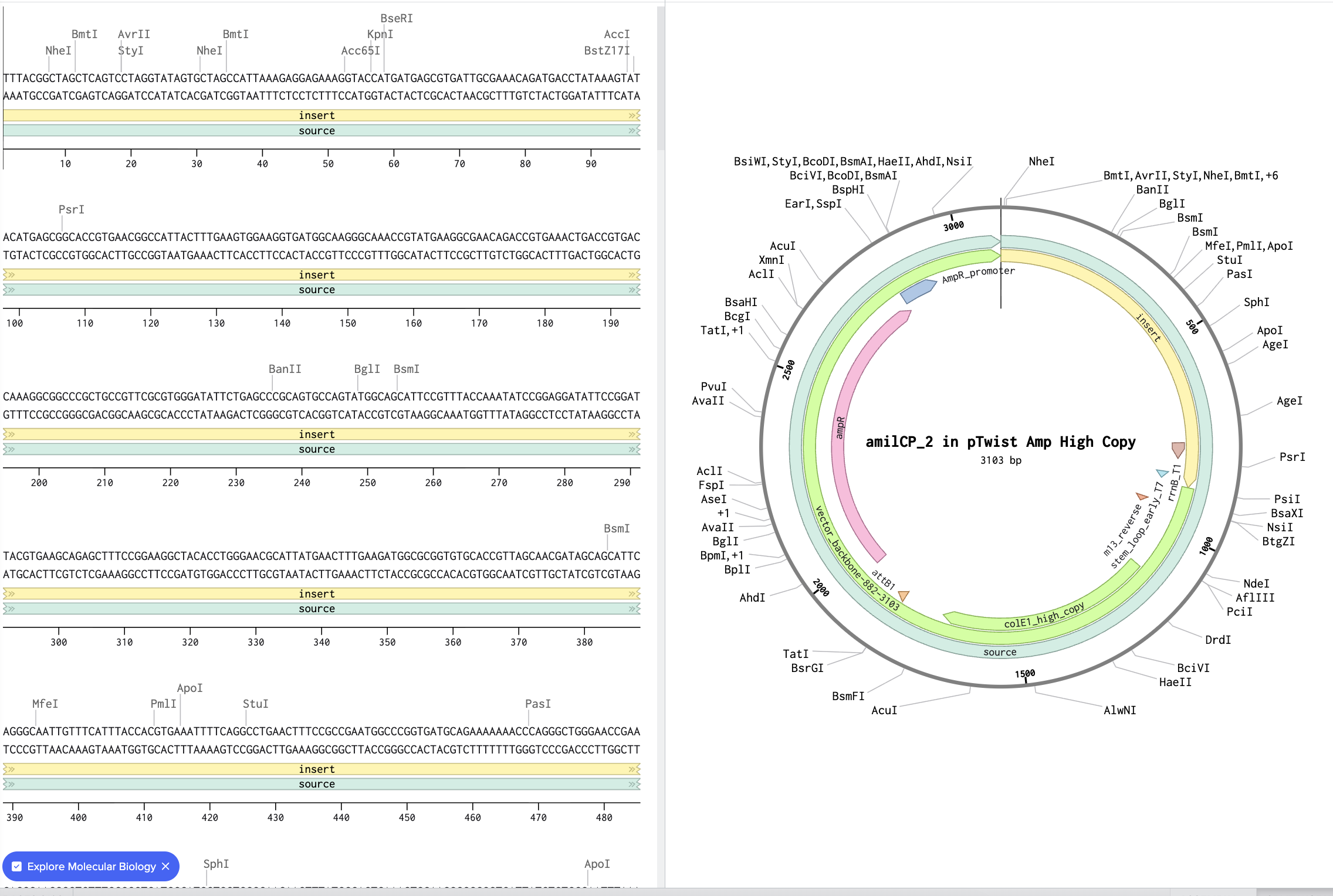
Part 5
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
Right now I’m interested in environmental health data and monitoring, both in water and air quality. I’m primarily interested in pollutants in these regions, but not necessarily biological ones. I’m more interested in metals, plastics, VOCs, temperature, humidity, etc.
From a more theoretical point of view, I would be interested in reading DNA as a data store, but I’m not sure how that really exists today. For the sake of this assignment, I’ll assume there’s a recorded DNA database holding metadata around an object’s provenance and creation.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Oxford Nanopore Technologies. Live-stream sequencing allows real-time processing, so stopping the stream and controlling data input more similar to machinic systems would be beneficial. The portability of it as well allows more diversification if reading in the field is required.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
Third generation, because it’s loaded in as a long string, rather than needing to post-compute concatenate shorter strings together like 2nd generation technologies.What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input is synthetic DNA data blocks, which are probably encased in some protective chamber. For preparation, they must be removed from their protection, where you would then do end repair and adapter ligation, and then clean up any non-essential enzymes.What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
The mixture is dropped onto a chip with nanopores; the nanopores have an electrical current, and as the DNA is pulled through by a motor protein, the DNA itself disrupts the consistent electrical current. Because each nucleotide has a different size, it causes a variant change in the electrical signal, and that signal is then converted back into the nucleotide sequence digitally.What is the output of your chosen sequencing technology?
There’s the raw output of the electrical signal, and the decoded signal sequence file (FASTQ).
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origami). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs!
- Fast-PETase in a cell-free expression
- A TphR logic/gate
- A chromoprotein such as amilCP to express when the TphR is present
From a theory perspective, I’m interested in a “live-environmental coding” of an object based on the conditions of that space, an “imprint” of it. As a prototype, I think an active signal-remediation loop with microplastic degradation would be interesting. I’m currently thinking of creating a genetic circuit for displaying this protein breakdown in a cell-free system, and dosing that into a porous structure such as 3D-printed ceramic. This would be a plastic-eating structure that shows the color of how it’s changed its environment over time, and when a new unit could be brought in. Could also be interesting in an underwater environment with calcium carbonate, but not sure how quickly denaturing would happen here compared to an air environment. I would like to pair this with software for designing the circuits, from simulating color change over time, and pairing that with different sensor inputs.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Technology choice: I would use Twist Bioscience for several key reasons:
- Ready availability – Twist’s commercial platform is accessible and well-established
- Parallel synthesis capability – The ability to synthesize many genes in parallel on a silicon chip would allow multiple iterations of the genetic circuit design, enabling me to order multiple variants in a single batch
- Iteration efficiency – This parallel approach supports rapid design-build-test cycles for optimizing the circuit
Main limitations:
- Cell-free compatibility – I will need to optimize this synthesis method specifically for cell-free expression systems, which may require additional iterations
- Cost considerations – Pricing over time could become a factor, especially for multiple design iterations
- Accuracy – I shouldn’t encounter issues with synthesis accuracy since all my constructs should be under 1 kb, though I may need to introduce redundancy if needed
Also answer the following questions:
- What are the essential steps of your chosen synthesis method? [To be completed]
- What are the limitations of your synthesis method (if any) in terms of speed, accuracy, scalability? See main limitations above.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I’d want to edit heat shock protein genes in corals, such as HSP70, to make them more resistant to warming ocean temperatures. This is important for coral health because bleaching occurs when water temperatures rise 1–2°C above normal and corals expel their symbiotic algae, essentially starving them, and this is more likely to rise over the next few decades because of climate change warming ocean surfaces. If improved, heat shock proteins could help corals respond better to temperature spikes, which would improve overall ecosystem health because coral reefs support about 25% of all marine species and provide critical habitat, coastal protection, and fisheries.
To do this, I’d use CRISPR because it allows precise targeting of specific genes.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
CRISPR uses a guide RNA that matches the DNA sequence you want to change. This guide brings the Cas9 enzyme to the right spot, and Cas9 cuts the DNA. After the cut, the cell tries to repair the break. If you provide a template with the sequence you want, the cell will copy that template and insert your new DNA.What preparation do you need to do and what is the input for the editing?
You need to design a guide RNA that matches the gene being targeted. The main inputs are the Cas9 enzyme, the guide RNA, and a DNA template. You also need to get these into the cells.What are the limitations of your editing methods in terms of efficiency or precision?
CRISPR doesn’t work every time. Either the cells don’t get edited successfully, or only some cells get the edit. Also, Cas9 can cut in the wrong place if there’s a similar sequence in the DNA.