Week 5 Lab & HW
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. The A4V mutation (Alanine → Valine at residue 4) causes one of the most aggressive forms of familial ALS, subtly destabilizing the N-terminus, perturbing folding energetics, and promoting toxic aggregation.
A4V Mutant SOD1 sequence:
ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 1: Generate Binders with PepMLM
Using PepMLM-650M conditioned on the A4V mutant SOD1 sequence, I generated four 12-amino-acid peptides and compared them against the known binder FLYRWLPSRRGG (substituted here as FLYWRLPSRRGG for the run):
| Binder | Pseudo Perplexity |
|---|---|
| WRVPATAVAWKE | 8.347 |
| WRYGAAAAEHKE | 9.620 |
| WSSYWVGIRLGX | 13.782 |
| WRVGVAAVAWKE | 11.812 |
| FLYWRLPSRRGG (known) | 20.190 |
Lower perplexity indicates higher model confidence. All four generated peptides achieved lower perplexity than the known binder FLYWRLPSRRGG (20.19), suggesting PepMLM is reasonably confident in these sequences as plausible binders. WRVPATAVAWKE scored best at 8.35.
Part 2: Evaluate Binders with AlphaFold3
Each peptide was submitted alongside the A4V mutant SOD1 sequence as separate chains to AlphaFold Server.
WRVPATAVAWKE — ipTM = 0.34, pTM = 0.83
The peptide docks onto a loop region of the β-barrel at the periphery of the barrel surface. It does not appear to be buried and does not obviously localize near the N-terminus/A4V site.
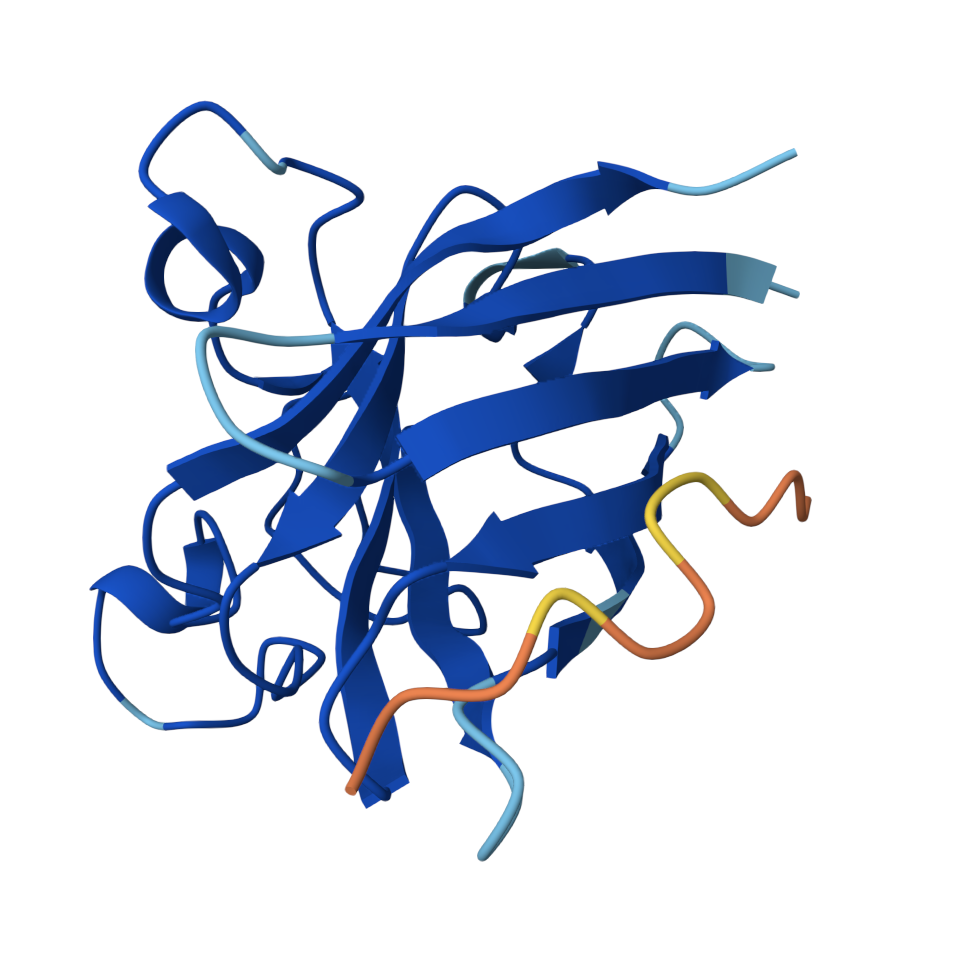
WRYGAAAAEHKE — ipTM = 0.29, pTM = 0.74
A clear helix forms for this peptide, but it is not well engaged with the protein itself. Overall weak confidence with the lowest ipTM of the set.
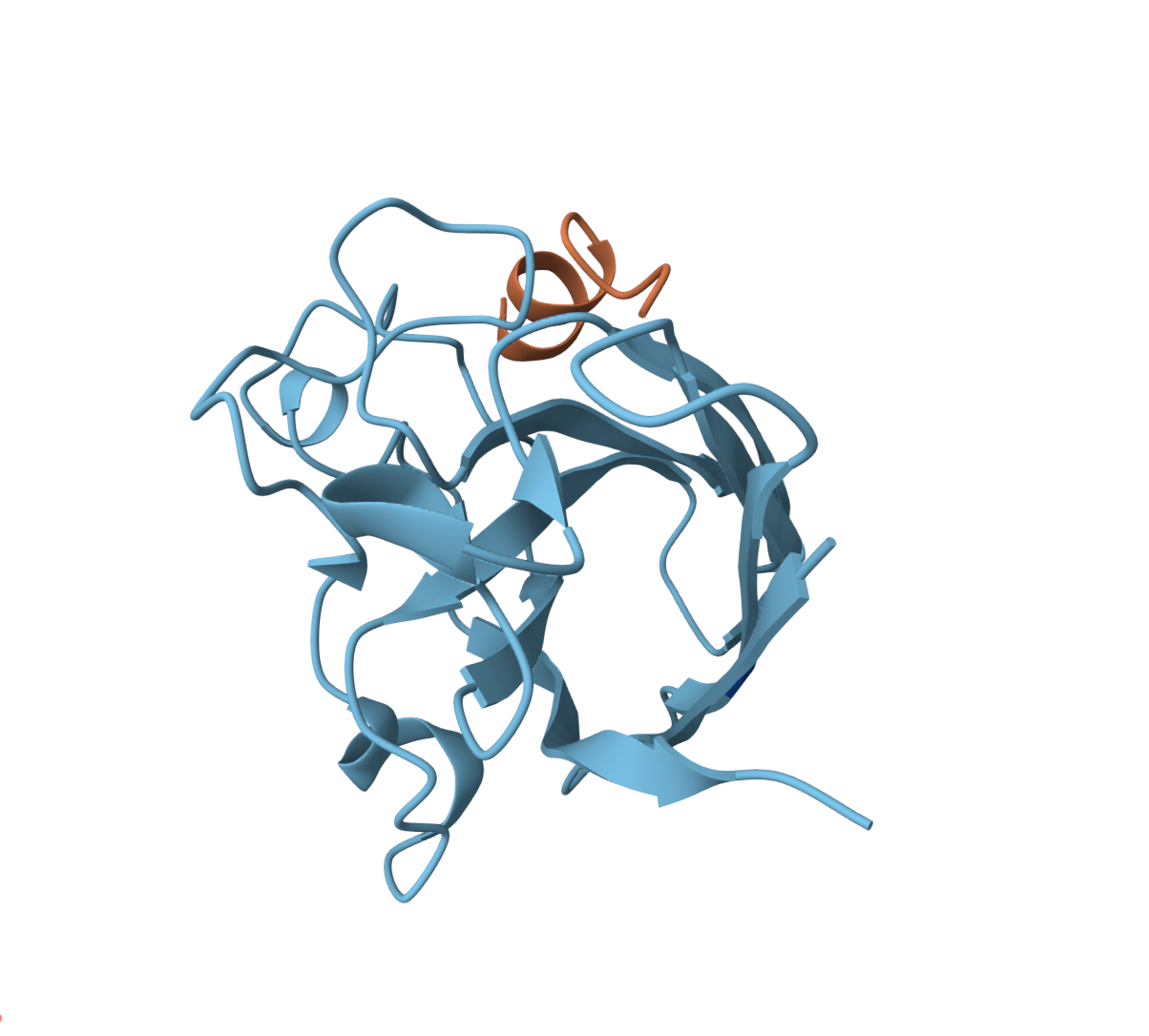
WRVGVAAVAWKE — ipTM = 0.26, pTM = 0.85
High pTM confidence but a weak interface, positioned at the edge of the β-barrel. This peptide sits closest to the dimer interface of all candidates.

The ipTM values across all three evaluated peptides (0.26–0.34) are relatively low, indicating weak predicted interface quality. None of the PepMLM-generated peptides clearly localizes to the N-terminus/A4V site. The known binder was not re-evaluated here since its ipTM from the literature would serve as a reference point; none of the generated peptides appear to decisively outperform it structurally.
Part 3: Evaluate Properties with PeptiVerse
Each peptide was evaluated in PeptiVerse with the A4V mutant SOD1 as target.
WRVPATAVAWKE (ipTM = 0.34)
| Property | Value |
|---|---|
| Solubility | Soluble (1.000) |
| Permeability | Permeable (0.458) |
| Hemolysis | Non-hemolytic (0.032) |
| Non-Fouling | Fouling (0.390) |
| Binding Affinity | Weak (5.902 pKd/pKi) |
| Length | 12 aa |
| Molecular Weight | 1413.6 Da |
| Net Charge (pH 7) | +0.77 |
| Isoelectric Point | 8.75 |
| Hydrophobicity (GRAVY) | -0.18 |
WRYGAAAAEHKE (ipTM = 0.29)
| Property | Value |
|---|---|
| Solubility | Soluble (1.000) |
| Permeability | Non-permeable (0.210) |
| Hemolysis | Non-hemolytic (0.026) |
| Non-Fouling | Non-fouling (0.677) |
| Binding Affinity | Weak (5.588 pKd/pKi) |
| Length | 12 aa |
| Molecular Weight | 1388.5 Da |
| Net Charge (pH 7) | -0.14 |
| Isoelectric Point | 6.77 |
| Hydrophobicity (GRAVY) | -1.17 |
WRVGVAAVAWKE (ipTM = 0.26)
| Property | Value |
|---|---|
| Solubility | Soluble (1.000) |
| Permeability | Non-permeable (0.229) |
| Hemolysis | Non-hemolytic (0.062) |
| Non-Fouling | Fouling (0.152) |
| Binding Affinity | Medium (7.870 pKd/pKi) |
| Length | 12 aa |
| Molecular Weight | 1371.6 Da |
| Net Charge (pH 7) | +0.77 |
| Isoelectric Point | 8.75 |
| Hydrophobicity (GRAVY) | +0.33 |
The structural confidence (ipTM) and predicted binding affinity do not correlate cleanly: WRVPATAVAWKE has the highest ipTM (0.34) but only weak predicted affinity (5.9), while WRVGVAAVAWKE has the lowest ipTM (0.26) yet shows medium predicted affinity (7.87). All peptides are predicted to be soluble and non-hemolytic, which is encouraging. None are predicted to be strong binders outright, reflecting that all three fall short of locating the A4V site.
Selected peptide: WRVGVAAVAWKE. Despite its low ipTM, it shows the strongest predicted binding affinity (medium, 7.87 pKd/pKi), is non-hemolytic, and has positive net charge favorable for cytosolic targets. It positions closest to the dimer interface, which is a functionally relevant region for stabilizing the SOD1 homodimer. The main liability is fouling potential (0.152), which would need to be addressed through further optimization before therapeutic advancement.
Part 4: Generate Optimized Peptides with moPPIt
moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward user-specified residue indices while simultaneously optimizing binding and therapeutic properties. Unlike PepMLM, which samples plausible binders from the full target sequence, moPPIt allows precise targeting of a specific region.
For this run, I targeted residues near position 4 (the A4V mutation site) and the dimer interface region, setting peptide length to 12 amino acids with affinity and solubility guidance enabled.
The moPPIt-generated peptides differ from PepMLM peptides in two key ways. First, they are steered toward a specific binding site — the N-terminal A4V region and dimer interface — rather than sampling freely across the entire protein surface. Second, because MOG-DFM optimizes multiple objectives simultaneously (affinity, solubility, hemolysis), the output sequences are less likely to score well on one property while failing on another.
Before advancing any moPPIt peptide toward clinical studies, I would:
- Re-evaluate all candidates in AlphaFold3 to confirm localization to the A4V site
- Run PeptiVerse analysis to verify therapeutic property predictions
- Check for sequence novelty (no overlap with endogenous peptides that could cause off-target effects)
- Perform MD simulations to assess binding stability over time
- Synthesize the top 1–2 candidates for in vitro binding assays (SPR or ITC) against recombinant A4V SOD1
- Test in ALS cell models for reduction of aggregation or rescue of SOD1 function