Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
SOD1_A4V Sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
| Peptide | Perplexity Score |
|---|---|
| WHYYVAVVRLGE | 36.758428 |
| WLYPPTAVAHKK | 14.060910 |
| WRYYPVALAHKK | 11.940822 |
| HRYPAVVVEHKE | 16.230484 |
| FLYRWLPSRRGG | 20.635231 |
Part 2: Evaluate Binders with AlphaFold3
| Binder | ipTM Score | Binding Site | Evaluation |
|---|---|---|---|
| 1 | 0.71 | 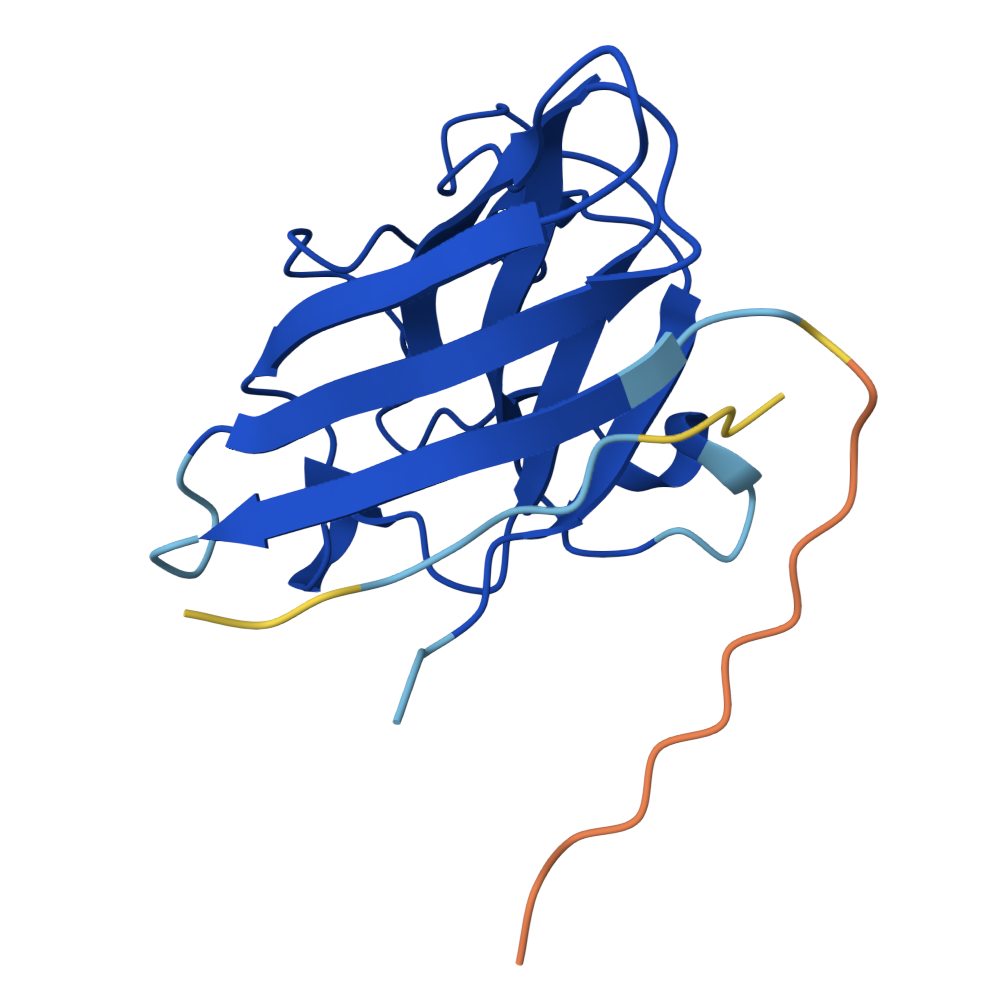
| Binder 1 associates near the β barrel at the surface of the protein. |
| 2 | 0.44 | 
| Like Binder 1, Binder 2 associates near the β barrel at the surface of the protein. However, this binder has a much lower ipTM score, meaning the confidence in this generated structure is much lower. |
| 3 | 0.31 | 
| Binder 3 associates across the β barrel and disordered region at the surface of the SOD1 protein. |
| 4 | 0.30 | 
| Binder 4 associates at the surface of the disordered region. |
| 5 | 0.39 | 
| Binder 5 also associates at the surface of the disordered region. |
Binders 1 and 2 have stronger ipTM values than Binder 5, the known binder provided for this exercise. All binders associate near the surface of the SOD1 protein, not integrating into the protein interior. The ipTM value for Binder 1 is relatively strong (0.71), meaning there is high confidence in that association between SOD1 and the binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
| Binder | Binding Affinity | Solubility | Hemolysis Probability | Net Charge | Molecular Weight |
|---|---|---|---|---|---|
| 1 | Weak binding affinity (6.150 pKd/pKi) | Soluble (1.000) | Non-hemolytic (0.095) | -0.15 | 1491 Da |
| 2 | Weak binding affinity (5.148 pKd/pKi) | Soluble (1.000) | Non-hemolytic (0.015) | 1.84 | 1410 Da |
| 3 | Weak binding affinity (5.897 pKd/pKi) | Soluble (1.000) | Non-hemolytic (0.014) | 2.84 | 1531 Da |
| 4 | Weak binding affinity (4.842 pKd/pKi) | Soluble (1.000) | Non-hemolytic (0.027) | -0.06 | 1463 Da |
| 5 | Weak binding affinity (5.968 pKd/pKi) | Soluble (1.000) | Non-hemolytic (0.047) | 2.76 | 1507 Da |
Binder 1, the binder with the highest ipTM value, also has the strongest binding affinity. None of the binders are predicted to be hemolytic. Unsurprisingly, Binder 1 appears to have the best balance of predicted binding and therapeutic properties. I will use Binder 1 to complete Part 4.
Part 4: Generate Optimized Peptides with moPPIt
Unfortunately, although I attempted to run motif 17-23 with multiple times and consulted ChatGPT for guidance, I was unable to make the moPPit program work without errors.
Part B: RD4 Drug Discovery Platform Tutorial
Will come back and do this later :)
Part C: Final Project: L-Protein Mutants
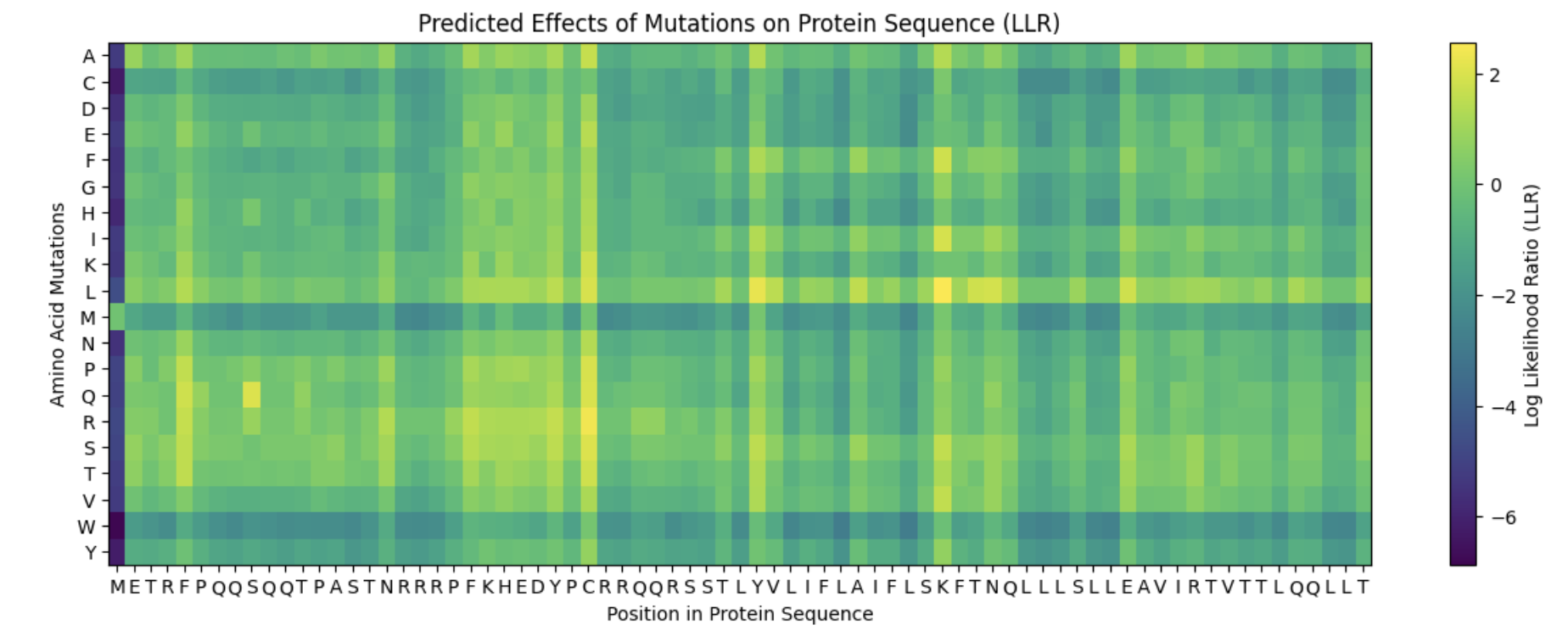
It appears that the L-protein mutants that result in a functional MS2 (1 in the “Lysis” column of the spreadsheet) correlate with the positive log likelihood ratio mutations on the heat map.
Proposed Mutations:
| Mutation | Domain | Reasoning | L-protein Multimer | Evaluation |
|---|---|---|---|---|
| F -> S at position 5 | Soluble Domain | When L-protein sequences was input into BLASTp, the F -> S mutation was observed in the third top hit in Emesvirus zinderi. Additionally, the F -> S mutation in position 5 appeared a yellowish-green on the heat map, indicating a “positive” mutation. | 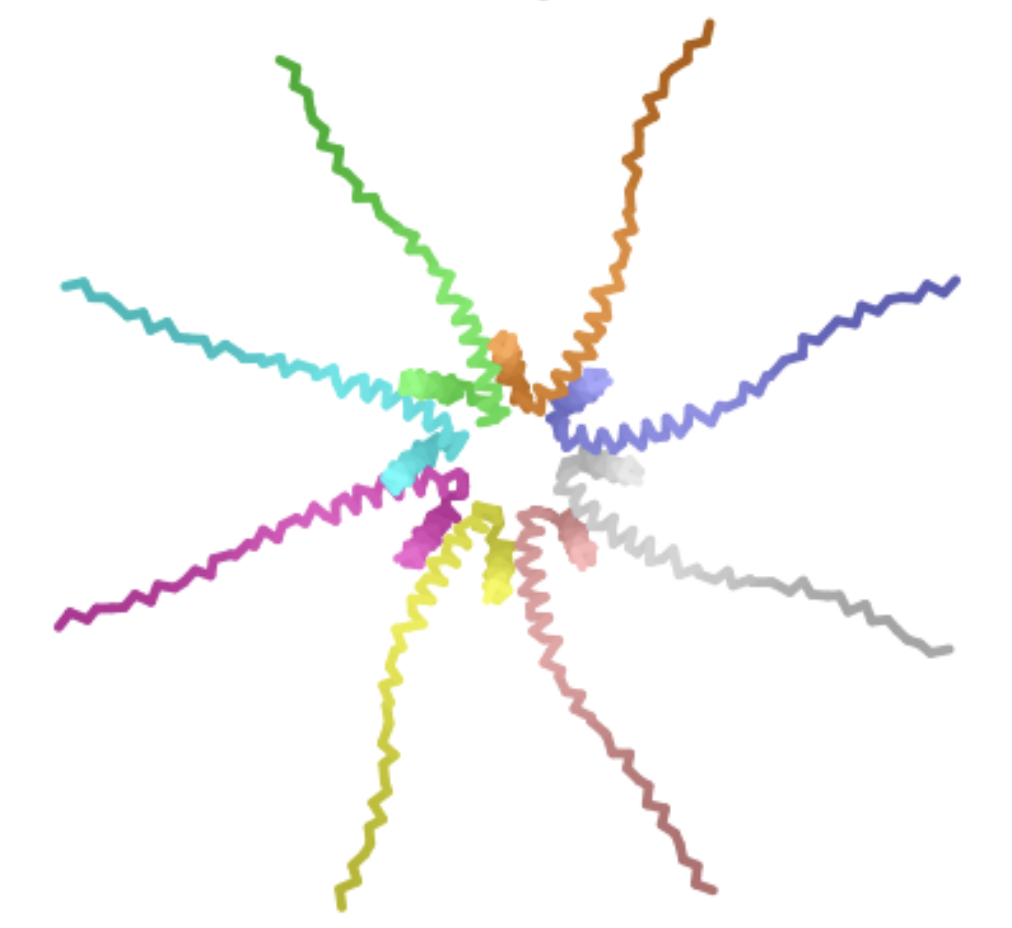
| The mutant octomer appears to form a pore-like quaternary structure. The ipTM score is 0.13, indicating low confidence that this is an accurate assembly of the mutant multimer. |
| R -> S at position 19 | Soluble Domain | In the table of L-protein mutants, this mutation has a score of 1 in the Lysis column, providing experimental evidence of a functional lysis protein. Additionally, mutations in the R amino acid at position 19 were identified in multiple top BLASTp hits. | 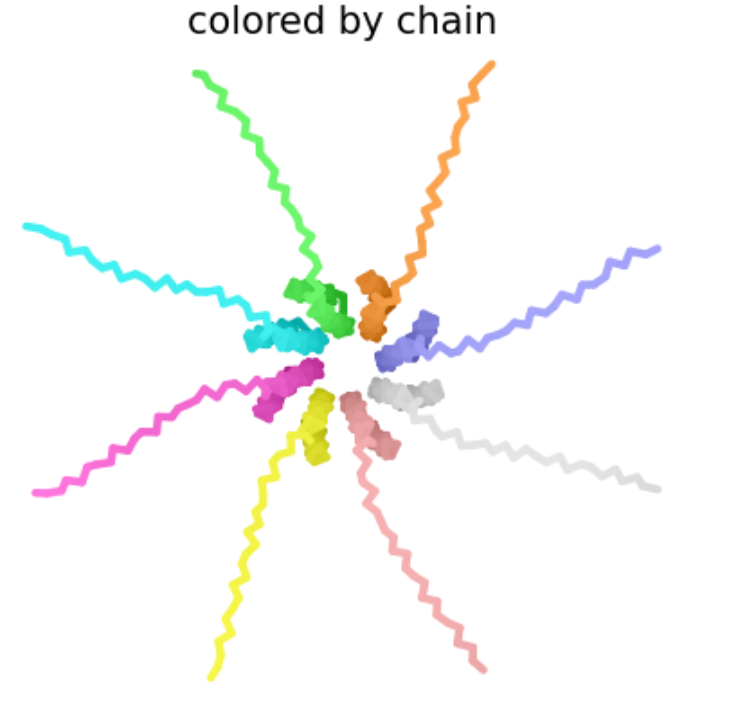
| Like the previous mutant, the Mutant 2 octomer forms a pore-like quaternary structure with an ipTM score of 0.14, indicating low confidence in this assembly. |
| R -> I at position 31 | Soluble Domain | This mutation is associated with a functional L protein on the “L-protein mutant” spreadsheet (1 in lysis column) and appears to have a positive log likelihood ratio on the heat map. | 
| The pore generated for this octomer appears to be tighter than Mutants 1 and 2, and although the ipTM score of 0.16 still indicates low confidence in the structure, it is slightly stronger than the previous two mutants. |
| A -> P at position 45 | Transmembrane Domain | Once again, this mutation was identified on the “L-protein mutant” spreadsheet as resulting in a function lysis protein. Although this mutation occurs in the transmembrane domain and therefore is less likely to result in a functional advantage to the MS2 phage, the functional lysis protein and low negative score on the heat map suggest it may be a better choice than other mutations in the transmembrane domain. | ||
| E -> S at position 61 | Transmembrane Domain | This mutation has a positive log likelihood ratio on the heat map, and a mutation in the E protein was identified in a BLASTp alignment with the L-protein for the MS12 phage. |