I am a senior neuroscience major at William & Mary. My research project focuses on elucidating the roles of the tweety genes in the neural development of Xenopus laevis.
Governance Assignment Biological Engineering Application Immunotherapies are a promising avenue in cancer treatment as they leverage the immune system’s innate ability to recognize and target non-self structures. However, traditional immunotherapies often result in on-target off-tumor effects, particularly in solid tumors. Synthetic biology has enabled new avenues of discovery to minimize this immunotherapy-related toxicity: engineering immune cells to target tumor-associated antigens (TAAs) or engineering genetic circuits to detect cancer disease signatures (Zhu et al., 2024). For example, modifying the traditional Chimeric Antigen Receptor (CAR) T-cell immunotherapy approach with a synthetic Notch (synNotch) receptor has demonstrated the ability to suppress off-target cytotoxicity related to organ rejection (Reddy et al., 2024) and selectively target cancerous cells in the central nervous system of mice rather than elsewhere in the body (Simic et al., 2024). Yet, while synNotch-modified CAR-T therapies show promise in their ability to reduce immunotherapy-related toxicity, additional research is needed to effectively administer these bioengineered cell systems in patients beyond pre-clinical experimentation.
Part 1: Benchling and In-silico Gel Art Virtual restriction enzyme digest designed with DNA from the bacteriophage Kampy (isolated at W&M!) and the restriction enzymes BstXI, KpnI, and SfiI to resemble two bacteriophages. The chosen restriction enzymes were selected because they were in stock at William & Mary, had multiple cut sites in the Kampy DNA, and could be combined to make a design resembling a bacteriophage.
Python Script for Opentrons Artwork My Opentrons design is meant to resemble a frog because I use Xenopus laevis as my model organism in my honors thesis research at William & Mary.
Post-Lab Questions Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. In Sanders et al., 2022, the researchers use an Opentron robot to optimize a bacterial whole-genome sequencing (WGS) protocol for gut microbiota samples. The Opentron was used for DNA extraction and library preparation steps, reducing the overall cost of WGS by ~$10 per genome and eliminating the need for 16S rRNA gene-based screening.
Part A: Conceptual Questions Since 1 Da = 1 g/mol, 500 g of meat equates to about 5 mol of amino acids. The approximate number of amino acids within these 5 mols would be 3x10^24.
When humans eat, the macromolecules the beef are made of are broken down during digestion into monomers. These monomers are common to all life, and humans use them to build human-specific macromolecules.
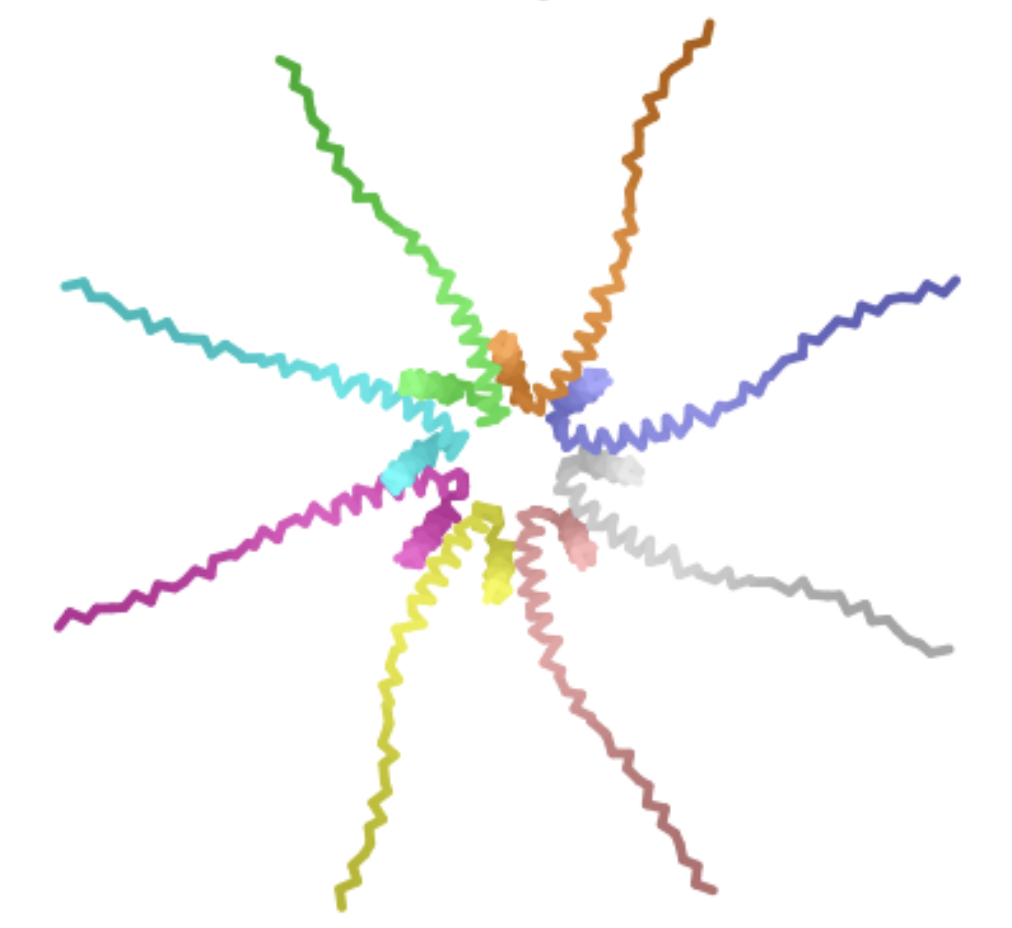
Part A: SOD1 Binder Peptide Design Part 1: Generate Binders with PepMLM SOD1_A4V Sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Peptide Perplexity Score WHYYVAVVRLGE 36.758428 WLYPPTAVAHKK 14.060910 WRYYPVALAHKK 11.940822 HRYPAVVVEHKE 16.230484 FLYRWLPSRRGG 20.635231 Part 2: Evaluate Binders with AlphaFold3 Binder ipTM Score Binding Site Evaluation 1 0.71 Binder 1 associates near the β barrel at the surface of the protein. 2 0.44 Like Binder 1, Binder 2 associates near the β barrel at the surface of the protein. However, this binder has a much lower ipTM score, meaning the confidence in this generated structure is much lower. 3 0.31 Binder 3 associates across the β barrel and disordered region at the surface of the SOD1 protein. 4 0.30 Binder 4 associates at the surface of the disordered region. 5 0.39 Binder 5 also associates at the surface of the disordered region. Binders 1 and 2 have stronger ipTM values than Binder 5, the known binder provided for this exercise. All binders associate near the surface of the SOD1 protein, not integrating into the protein interior. The ipTM value for Binder 1 is relatively strong (0.71), meaning there is high confidence in that association between SOD1 and the binder.
DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? DNA polymerase: uses dNTP monomers to synthesize new DNA strands dNTPs: monomers of new DNA strand Buffer: stabilizes the pH of the reaction for optimal enzymatic function MgCl2: Cofactor for the polymerase; optimizes enzymatic function and primer annealing What are some factors that determine primer annealing temperature during PCR? GC content (higher GC content = more hydrogen bonds = stronger primer annealing = higher annealing temperature) Primer length (longer primers = more hydrogen bonds = stronger primer annealing = higher annealing temperature) There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. PCR: Enzyme: DNA polymerase DNA polymerases bind to primers and synthesize complementary DNA to the template strand Purpose: amplify! Polymerases synthesize DNA by recognizing primers (designed to flank the specific region of interest) and incorporating dNTPs into a novel DNA strand When to use: To detect a specific sequence within a mixed sample To create more of a specific DNA sequence (amplify) Restriction enzyme: Enzyme: restriction endonuclease Restriction endonucleases recognize specific nucleotide sequences (4-8 bp) in double-stranded DNA and cut in a specific pattern (blunt or sticky ends, depending on the enzyme) Purpose: cut! Restriction enzymes cut existing template DNA, they do NOT amplify DNA fragments When to use: To linearize bacterial plasmids (ex. for in vitro transcription of capped mRNA for microinjection into X. laevis!) To cut DNA fragments to assemble/ligate together and transform into a plasmid How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? Primer design! The 5’ tail overhang of each primer should be identical to the adjacent DNA fragment the 3’ end of the primer should be complementary to the DNA fragment to which it will anneal Restriction Digest: Restriction enzyme cut sites should be avoided PCR: Primers should be designed that flank the overhang regions How does the plasmid DNA enter the E. coli cells during transformation? Heat shock transformation - briefly raising temperature increases the permeability of the bacterial cell wall, allowing the plasmid DNA to enter into the E. coli cell E. coli cell sample placed on ice for ~15-30 minutes Sample heated to 42ºC for ~15-30 seconds Sample then returned to ice for ~5 minutes Describe Golden Gate Assembly Golden Gate Assembly utilizes Type IIS restriction enzymes (such as BsaI) which recognize non-pallindromic sequences, cut outside the recognition site (to avoid damaging the DNA sequence of interest), and create sticky ends of variable lengths. First, template DNA is amplified using PCR with primers specified to include the TIIS recognition and cut sites. Next, a restriction digest with the TIIS enzyme is performed to create DNA fragments with sticky ends. The sticky ends of adjacent fragments should be complementary so the sequences can be ligated in the appropriate order. Following ligation of DNA fragments with a plasmid, the engineered plasmid can be transformed into a bacterial cell (ex. E. coli), and bacterial colonies containing the plasmid can be screened for furhter use. Image generated by ChatGPT
Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Variables input into Boolean functions are binary (either ON or OFF), whereas the variables input into IANNs can be continuous. Additionally, IANNs can input multiple variables at a time while Boolean functions (like a two-layer IANN) only integrate two variables.
General Homework Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis systems are advantageous for quick, high-throughput protein production. Cell-free protein expression is more time efficient than traditional in vivo methods because they do not require the cloning steps of cell-based systems. Additionally, cell-free systems can be modified to include non-canonical amino acids which enables numerous biotechnology and pharmaceutical applications. Cell-free systems also offer the advantage of ease of manipulating reaction conditions. Cell-free expression is more beneficial than cell production when evaluating proteins that are toxic to a cell or when a large yield of the desired protein is needed in a relatively short period of time.
Final Project For my final project, I will measure the binding affinity of my de novo peptide to the beta-gamma subunits of the human µ opioid receptor (MoR) using AlphaFold3. If I were to incorporate a wet lab component to this project, I would measure the GFP fluorescence of E. coli cells with my engineered plasmid (peptide+GFP) electroporated into them to confirm the genes of interest are being expressed then perform co-immunoprecipitation to measure the binding affinity of the de novo peptide to the human MoR Gβγ subunits.
Subsections of Homework
Week 1 HW: Principles and Practices
Governance Assignment
Biological Engineering Application
Immunotherapies are a promising avenue in cancer treatment as they leverage the immune system’s innate ability to recognize and target non-self structures. However, traditional immunotherapies often result in on-target off-tumor effects, particularly in solid tumors. Synthetic biology has enabled new avenues of discovery to minimize this immunotherapy-related toxicity: engineering immune cells to target tumor-associated antigens (TAAs) or engineering genetic circuits to detect cancer disease signatures (Zhu et al., 2024). For example, modifying the traditional Chimeric Antigen Receptor (CAR) T-cell immunotherapy approach with a synthetic Notch (synNotch) receptor has demonstrated the ability to suppress off-target cytotoxicity related to organ rejection (Reddy et al., 2024) and selectively target cancerous cells in the central nervous system of mice rather than elsewhere in the body (Simic et al., 2024). Yet, while synNotch-modified CAR-T therapies show promise in their ability to reduce immunotherapy-related toxicity, additional research is needed to effectively administer these bioengineered cell systems in patients beyond pre-clinical experimentation.
Governance Goals
Goal 1: Perform rigorous pre-clinical testing to ensure new immunotherapies meet safety and efficacy standards before introducing into human patients.
Goal 2: Design ethical clinical trials with standardized eligibility, safety, and efficacy protocols with consideration for unique patterns of patient progression or response to treatment.
Goal 3: Create policies and organizations to promote equitable access to cancer prevention, screening, diagnosis, and immunotherapy resources to patients from diverse backgrounds and socioeconomic statuses across the globe.
Governance Actions
Option 1: Create an international organization to create global standards for immunotherapy clinical trial design and safety measures
Purpose: Immunotherapy safety regulations are regulated at the national level, so efforts to promote global administration of novel cancer immunotherapies may experience roadblocks if national standards do not align. Establishing international safety and efficacy standards for immmunotherapy clinical trials will promote efficient administration of immunotherapy treatments across global lines.
Design: An international healthcare organization, such as the WHO, must establish and agree upon an international standard for efficacy and safety in immunotherapy clinical trials. Then, all countries that participate in this organization must agree to the international standards to ensure ease of treatment deployment across global lines.
Assumptions: This option assumes that international clinical trial standards will supercede any national guidelines, and that countries will be willing to adopt the international standard and/or deploy their immunotherapies in other countries.
Risks of Failure and “Successes”: Establishing international immunotherapy clinical trial safety and efficacy guidelines may “fail” if the organization lacks the power to enforce the adoption of these guidelines across its participating countries. However, “success” of this option may delay the time it takes to put immunotherapies into clinical trials if international standards are incredibly restrictive and difficult to meet.
Option 2: Establish an international database to upload immunotherapy pre-clinical and clinical trial data
Purpose: To create a centralized, accurate database with the safety and efficacy data for immunotherapies in pre-clinical and clinical trials adminsitered to diverse patient populations. This database will ultimately promote safer and more effective administration of immunotherapies as a large dataset is available for review.
Design: Either an international healthcare organization (ex. WHO) or an independent organization would oversee the funding for the database and ensure the uploaded data is both reliable and accurate.
Assumptions: This option would assume that immunotherapy administration can be standardized across diverse healthcare settings, particularly on the global scale. Additionally, creating a comprehensive and accessible database assumes that uploaded data is not subject to reportability bias.
Risks of Failure and “Successes”: This measure would fail if the uploaded data is skewed toward well-funded research programs and not local healthcare systems with patients who experience barriers to immunotherapy access because this would not be a comprehensive dataset. However, if this measure is “successful”, immunotherapies with lower overall efficacy according to database metrics but high efficacy in a small patient population may be deprioritized or defunded.
Option 3: Establish an independent organization to increase access to preventative cancer screenings, diagnostic tools, and long-term care
Purpose: Inadequate access to preventative cancer screenings (i.e. mammograms, pap smears, colonoscopies, etc.) as a result of financial, geographic, or other barriers leads to later diagnosis and poorer progonosis. As immunotherapies are most effective when treatment is begun at earlier stages of cancer progression, inadequate preventative measures undermine the innovative bioengineering design of novel immunotherapies. Establishing an organization to ensure equitable access to cancer screenings and diagnostic tools without financial barriers, both within the United States and abroad, will allow the advances of innovative immunotherapies to benefit more patients than just those with easy access to preventative measures.
Design: As this organization would be independent of government funding, it would require public or philanthropic funding to decrease the cost barriers to preventative cancer screenings for patients with financial concerns. Additionally, the efforts of this organization would need to be integrated with healthcare systems on both the local and international scales to establish sites to receive preventative screenings and the capability to receive long-term follow-up care.
Assumptions: This option relies on the assumption that patients whose cancer is detected by increased preventative measures will then be able to access the immunotherapy treatments that target their cancer.
Risks of Failure and “Successes”: Without sufficient funding, this independent organization could ultimately shut down and fail in its mission to increase equiable access to preventative cancer screenings. Additionally, if the organization does not have reliable connections to local healthcare systems, its efforts to reduce cost barriers for patients seeking cancer screenings will not be realized. If this organization were to be “successful”, the influx of patients who are identified by screening measures may cause strain on the healthcare system.
Scoring Governance Actions
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
1
• By helping respond
2
2
2
Foster Lab Safety
• By preventing incident
1
1
1
• By helping respond
2
1
1
Protect the environment
• By preventing incidents
1
1
1
• By helping respond
1
1
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
2
2
• Feasibility?
2
2
2
• Not impede research
2
3
3
• Promote constructive applications
3
3
3
Prioritization
I believe Option 3 should be prioritized. While innovative bioengineering applications to cancer immunotherapies are a promising avenue for decreasing cancer mortality both nationally and internationally, these efforts are in vain if they cannot be administered effectively in a large patient population. Increasing early detection of cancer by promoting equitable access to cancer screenings and diagnostic testing will ultimately increase the use of bioengineered immunotherapies as cancers detected at earlier stages are better candidates to be treated by immunotherapy approaches.
Week 2 Preparation
Professor Jacobson
The error rate for a polymerase is 1 in 106 (1,000,000) bases. The human genome is 3x109 (3,000,000,000) base pairs in length, leading to an estimated 3,000 errors per replication of a human cell. Biology has processes in place, such as mismatch repair, capable of recognizing and excising these errors, then synthesizing the correct nucleotide.
The genetic code is comprised of 64 distinct codons that encode 20 naturally-occuring amino acids. However, not all codons are used equally because each tRNA with an anticodon specified for a particular amino acid are not equally expressed in every organism.
Dr. LeProust
Currently, the most commonly used method for oligo synthesis is phosphoramidite chemistry.
Oligos longer than 200 bp have high error rates and high rates of decay.
As a result of the high error rate and rates of decay, a 2000 bp gene synthesized via direct oligo synthesis would no longer resemble the desired sequence and would not encode the desired protein.
George Church
The 10 essential amino acides are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. These amino acids are not naturally synthesized in the human body and therefore must be consumed through the diet. The “Lysine Contingency” from Jurassic Park describes a genetic mutation introduced into the dinosaur enzyme for lysine synthesis as a means of protection from dinosaurs getting off the island (if the dinosaurs cannot synthesize an essential amino acid like lysine, they will not survive long). However, considering lysine is one of the amino acids that is solely ingested through the diet, the “Lysine Contingency” would not have any affect on dinosaurs because they should not have an enzyme responsible for lysine synthesis.
Website Preparation
I completed the setup of my personal HTGAA website, including adding my biography, contact information, and cover photo to my homepage.
Week 2 HW: DNA Read, Write, and Edit
Part 1: Benchling and In-silico Gel Art
Virtual restriction enzyme digest designed with DNA from the bacteriophage Kampy (isolated at W&M!) and the restriction enzymes BstXI, KpnI, and SfiI to resemble two bacteriophages. The chosen restriction enzymes were selected because they were in stock at William & Mary, had multiple cut sites in the Kampy DNA, and could be combined to make a design resembling a bacteriophage.
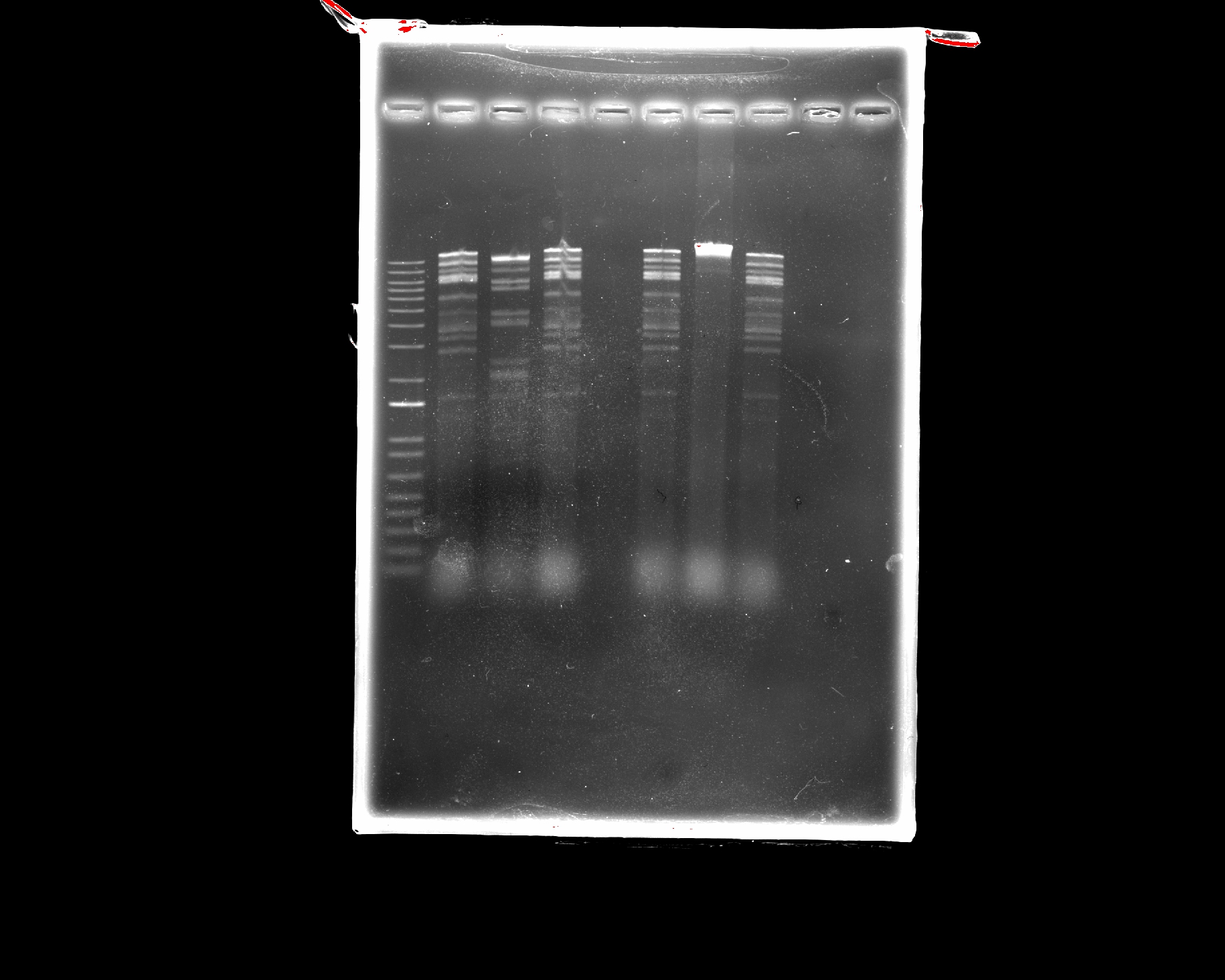
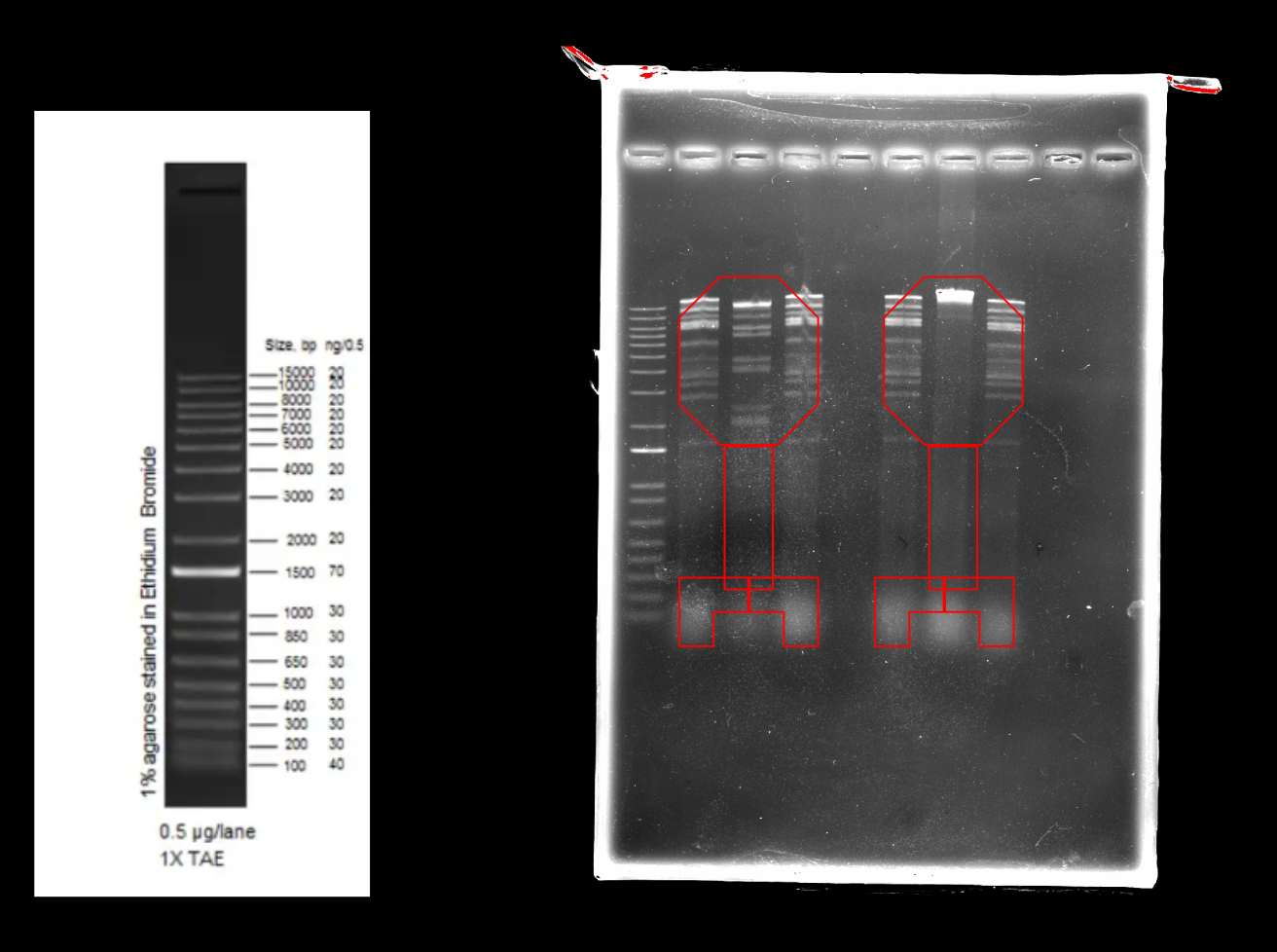
Part 2: Restriction Digests and Gel Electrophoresis
The imaged gel resembles the band lengths predicted by the virtual digest! While interpreting the gel image as two bacteriophages does require a bit of imagination, the digest was successful in that the true gel resembled the in silico prediction.
Part 3: DNA Design Challenge
3.1. Choose Your Protein
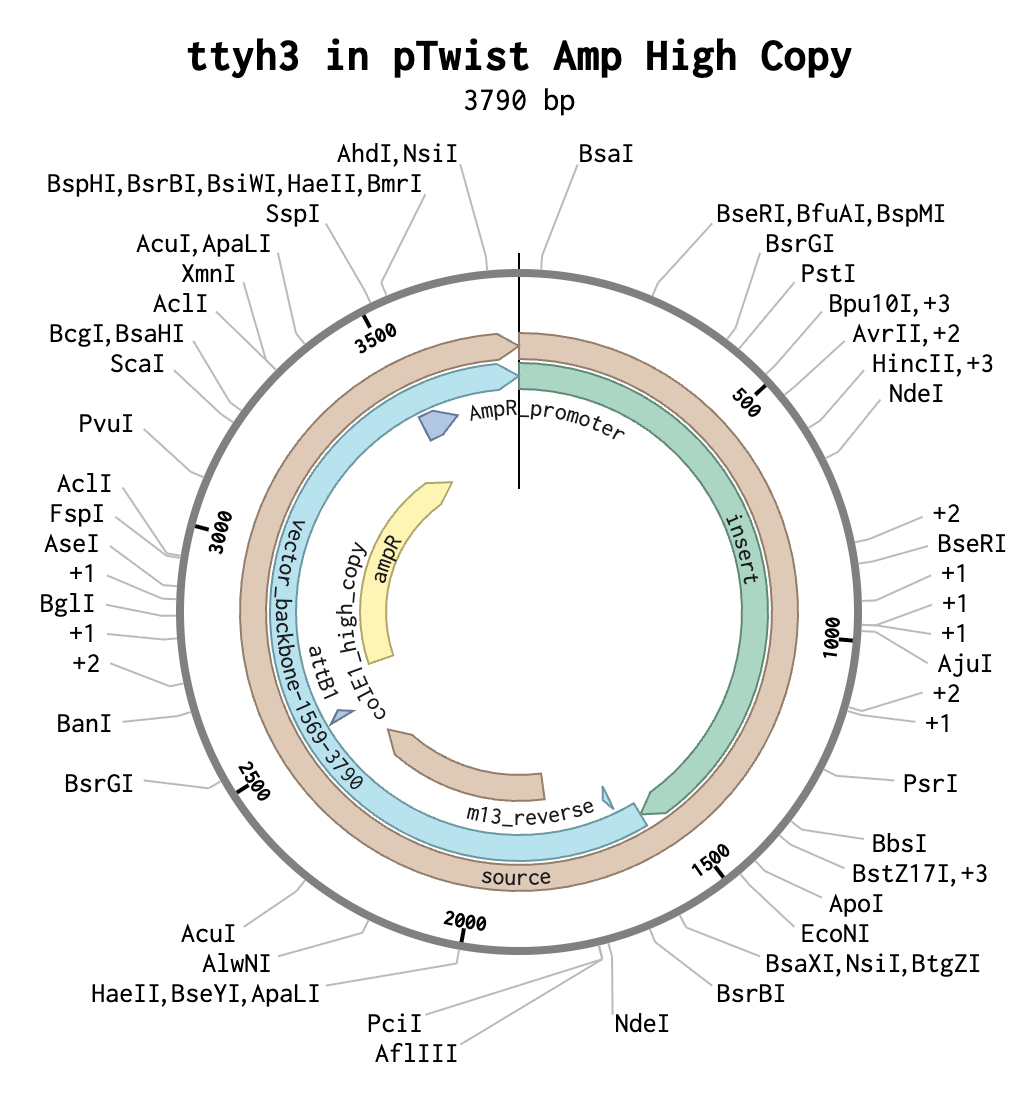
I chose the protein TTYH3 (tweety homolog 3) because it is the subject of my honors thesis research. ttyh3 encodes a calcium-dependent chloride channel and is a member of the tweety gene family- consisting of members ttyh1, ttyh2, and ttyh3- that is highly conserved across eukaryotes. ttyh3 is the least-characterized member of the tweety gene family, making it an intriguing subject of research. During neural development, the gene ttyh3 is primarily expressed in post-mitotic neurons. However, its precise function and role in neural development remain unknown. In my research, I aim to provide greater insight into the role of TTYH3 in neural development by overexpressing and knocking-out the ttyh3 gene in X. laevis and observing changes in expression of the downstream neural marker genes Sox2 and tubb2b.
Sequence from UniProt:
MAGVSYAAPWWVSLLHRLPHFDLSWEATSSQFRPEDTDYQQALLLLGAAALACLALDLLFLLFYSFWLCCRRRKSEEHLDADCCCTAWCVIIATLVCSAGIAVGFYGNGETSDGIHRATYSLRHANRTVAGVQDRVWDTAVGLNHTAEPSLQTLERQLAGRPEPLRAVQRLQGLLETLLGYTAAIPFWRNTAVSLEVLAEQVDLYDWYRWLGYLGLLLLDVIICLLVLVGLIRSSKGILVGVCLLGVLALVISWGALGLELAVSVGSSDFCVDPDAYVTKMVEEYSVLSGDILQYYLACSPRAANPFQQKLSGSHKALVEMQDVVAELLRTVPWEQPATKDPLLRVQEVLNGTEVNLQHLTALVDCRSLHLDYVQALTGFCYDGVEGLIYLALFSFVTALMFSSIVCSVPHTWQQKRGPDEDGEEEAAPGPRQAHDSLYRVHMPSLYSCGSSYGSETSIPAAAHTVSNAPVTEYMSQNANFQNPRCENTPLIGRESPPPSYTSSMRAKYLATSQPRPDSSGSH
3.2. Reverse Translate: Protein Sequence to DNA Sequence
Using the “reverse translate” tool on bioinformatics.org, I generated the following nucleotide sequence from the TTYH3 amino acid sequence:
atggcgggcgtgagctatgcggcgccgtggtgggtgagcctgctgcatcgcctgccgcattttgatctgagctgggaagcgaccagcagccagtttcgcccggaagataccgattatcagcaggcgctgctgctgctgggcgcggcggcgctggcgtgcctggcgctggatctgctgtttctgctgttttatagcttttggctgtgctgccgccgccgcaaaagcgaagaacatctggatgcggattgctgctgcaccgcgtggtgcgtgattattgcgaccctggtgtgcagcgcgggcattgcggtgggcttttatggcaacggcgaaaccagcgatggcattcatcgcgcgacctatagcctgcgccatgcgaaccgcaccgtggcgggcgtgcaggatcgcgtgtgggataccgcggtgggcctgaaccataccgcggaaccgagcctgcagaccctggaacgccagctggcgggccgcccggaaccgctgcgcgcggtgcagcgcctgcagggcctgctggaaaccctgctgggctataccgcggcgattccgttttggcgcaacaccgcggtgagcctggaagtgctggcggaacaggtggatctgtatgattggtatcgctggctgggctatctgggcctgctgctgctggatgtgattatttgcctgctggtgctggtgggcctgattcgcagcagcaaaggcattctggtgggcgtgtgcctgctgggcgtgctggcgctggtgattagctggggcgcgctgggcctggaactggcggtgagcgtgggcagcagcgatttttgcgtggatccggatgcgtatgtgaccaaaatggtggaagaatatagcgtgctgagcggcgatattctgcagtattatctggcgtgcagcccgcgcgcggcgaacccgtttcagcagaaactgagcggcagccataaagcgctggtggaaatgcaggatgtggtggcggaactgctgcgcaccgtgccgtgggaacagccggcgaccaaagatccgctgctgcgcgtgcaggaagtgctgaacggcaccgaagtgaacctgcagcatctgaccgcgctggtggattgccgcagcctgcatctggattatgtgcaggcgctgaccggcttttgctatgatggcgtggaaggcctgatttatctggcgctgtttagctttgtgaccgcgctgatgtttagcagcattgtgtgcagcgtgccgcatacctggcagcagaaacgcggcccggatgaagatggcgaagaagaagcggcgccgggcccgcgccaggcgcatgatagcctgtatcgcgtgcatatgccgagcctgtatagctgcggcagcagctatggcagcgaaaccagcattccggcggcggcgcataccgtgagcaacgcgccggtgaccgaatatatgagccagaacgcgaactttcagaacccgcgctgcgaaaacaccccgctgattggccgcgaaagcccgccgccgagctataccagcagcatgcgcgcgaaatatctggcgaccagccagccgcgcccggatagcagcggcagccat
3.3. Codon Optimization
Codon optimization is necessary because not all organisms use each codon with the same frequency due to differences in the abundance of various tRNAs, so optimizing the codons in the designed sequence will ideally increase the translational yield of my protein. I chose to optimize my sequence for X. laevis because that is the model organism in which I study the role of ttyh3 in neural development.
Using the Twist Bioscience Codon Optimization tool, I generated the following codon-optimized sequence:
ATGGCTGGTGTGTCTTATGCTGCTCCTTGGTGGGTCTCTTTATTACATCGGTTGCCACACTTCGACCTCTCCTGGGAAGCCACATCTAGTCAATTCCGACCAGAGGACACAGACTACCAACAAGCACTATTATTGCTAGGGGCTGCCGCTTTAGCTTGTTTGGCTCTTGACCTTCTCTTCCTTTTGTTCTACTCTTTCTGGTTATGTTGTAGAAGAAGGAAGTCAGAGGAGCACCTCGACGCAGACTGTTGTTGTACTGCTTGGTGTGTCATAATCGCTACTCTTGTATGTTCAGCAGGTATAGCAGTAGGATTCTACGGGAATGGTGAGACATCCGACGGAATCCACCGGGCAACTTACTCCCTCAGACACGCTAATAGAACTGTTGCTGGTGTACAAGACCGGGTATGGGACACTGCAGTAGGGTTGAATCACACAGCAGAGCCTTCATTACAAACTTTAGAGAGACAACTTGCTGGAAGACCTGAGCCACTTAGAGCTGTTCAAAGATTACAAGGATTGTTAGAGACGCTCCTAGGGTACACTGCAGCCATCCCATTCTGGCGAAATACTGCCGTATCCTTAGAGGTACTCGCAGAGCAAGTTGACCTCTACGACTGGTACCGATGGCTTGGATACCTTGGGTTGTTGTTGTTGGACGTTATCATATGTTTACTCGTATTAGTTGGACTCATCAGGTCATCTAAGGGAATACTTGTTGGGGTTTGTTTACTTGGGGTTCTTGCTCTCGTCATCTCTTGGGGAGCATTGGGTCTTGAGCTTGCTGTTTCAGTAGGGTCAAGTGACTTCTGTGTAGACCCCGACGCCTACGTCACAAAGATGGTCGAGGAGTACTCAGTTCTTAGTGGAGACATCTTACAATACTACCTCGCTTGTTCACCAAGGGCAGCTAATCCCTTCCAACAAAAGCTTTCAGGTTCTCACAAGGCACTCGTAGAGATGCAAGACGTTGTCGCAGAGTTGCTTAGAACAGTTCCTTGGGAGCAACCAGCAACAAAGGACCCATTGCTCAGAGTCCAAGAGGTCCTTAATGGAACTGAGGTTAATCTCCAACACCTAACAGCCCTTGTAGACTGTCGATCACTCCACTTGGACTACGTCCAAGCTTTGACAGGTTTCTGTTACGACGGAGTTGAGGGTCTAATATACCTCGCCCTTTTCTCCTTCGTTACAGCTCTAATGTTCTCCAGTATCGTTTGTTCTGTTCCCCACACTTGGCAACAAAAGAGAGGACCCGACGAGGACGGAGAGGAAGAGGCAGCACCCGGTCCCAGACAAGCACACGACTCTTTGTACCGGGTCCACATGCCAAGTTTGTACTCATGTGGGTCTTCTTACGGTAGTGAGACAAGTATACCAGCCGCTGCCCACACTGTTTCTAATGCCCCCGTTACAGAGTACATGTCCCAAAATGCAAATTTCCAAAATCCCCGATGTGAGAATACGCCTTTGATAGGACGGGAGAGTCCCCCACCTTCATACACATCATCAATGAGGGCAAAGTACCTTGCAACATCACAACCCCGACCCGACTCCAGTGGATCACAC
3.4. You have a sequence! Now what?
To synthesize the ttyh3 mRNA and protein, I would use an in vitro transcription and translation kit (such as the ones produced by ThermoFischer) to synthesize ttyh3 mRNA and TTYH3 protein, respectively.
3.5. How does it work in nature/biological systems?
Single genes can produce multiple proteins at the transcriptional level through the process of alternative splicing. In eukaryotic cells, transcription and mRNA processing occurs in the nucleus whereas translation occurs in the cytoplasm. After mRNA is synthesized via transcription, mRNA processing (5’ cap addition, poly(A) tail addition, and intron splicing) occurs and exons (which make the coding region of the protein) are annealed together after introns are removed. However, the combinations of different exons (alternative splicing) can produce different proteins from the same primary (immature) mRNA transcript.
Part 4: Prepare a Twist DNA Synthesis Order
Part 5: DNA Read/Write/Edit
5.1. DNA Read
What DNA would you want to sequence (e.g., read) and why?
I would like to sequence the DNA of a tumor sample from a cancer in which ttyh3 is observed to be misregulated (i.e. HCC, genitourinary cancers, colorectal cancer, etc.). I would be interested to observe which mutations in ttyh3 gene lead to functional changes that promote tumorigenesis, tumor invasiveness, etc. and which changes have little to no impact.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Illumina whole-genome sequencing (WGS), a second-generation sequencing method, because it is a relatively quick method of obtaining a base-by-base view of any single nucleotide variants (SNVs) in the genome which is essential information for understanding how mutations in the ttyh3 gene increase tumor proliferation, invaseiness, or aggression. The input for this sequencing is gDNA extracted from a tumor biopsy and prepared using the Illumina DNA Library Prep Kit. Following DNA extraction, the sample is sequenced using the “sequencing by synthesis” (SBS) technology which utilizes dNTPs bound to fluorescently-labeled reversible terminators to decode each base. The output of Illumina sequencing is a FASTQ file with quality scores.
5.2. DNA Write
What DNA would you want to synthesize (e.g., write) and why?
Completely unrelated to my work with X. laevis, I think it would be fascinating to design a phage with a novel gene (or perhaps an entirely synthetic genome!). The protein structure could be predicted with a tool like AlphaFold, then the amino acid sequence could be reverse transcribed into cDNA for the gene of interest and integrated into a plasmid. As advancements in phage therapy show promise in combating antibiotic resistance, it would be fascinating to design a protein that would increase phage DNA replication, phage entry, etc. to increase the efficacy of the treatment.
What technology or technologies would you use to perform this DNA synthesis and why?
First, and I don’t know if this is possible, I would create the desired 3D protein structure with a tool like AlphaFold and obtain the amino acid sequence. Then, similarly to this homework assignment, I would generate cDNA by reverse transcribing the AA sequence and order a plasmid from Twist (or elsewhere) with the gene of interest.
5.3. DNA Edit
What DNA would you want to edit and why?
I would like to develop a technology to perform somatic editing of the DNA of individuals with genetic diseases that result from mutations in a single gene (i.e. Huntington’s, cystic fibrosis, etc.). While many similar technologies already exist and some clinical trials have shown promise (i.e. sickle cell anemia!), I would like to expand these technologies to other genetic diseases.
What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR/Cas9-mediated homlogy directed repair to excise the mutated gene and replace it with a nonmutated, functional gene. This would involve design of the nonmutated gene and the sgRNA to direct the Cas9 enzyme to the appropriate cut site. The limitations of CRISPR/Cas9 technology are possible off-target effects (low precision) or the construct not being effectively delivered to all cells (low efficiency).
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
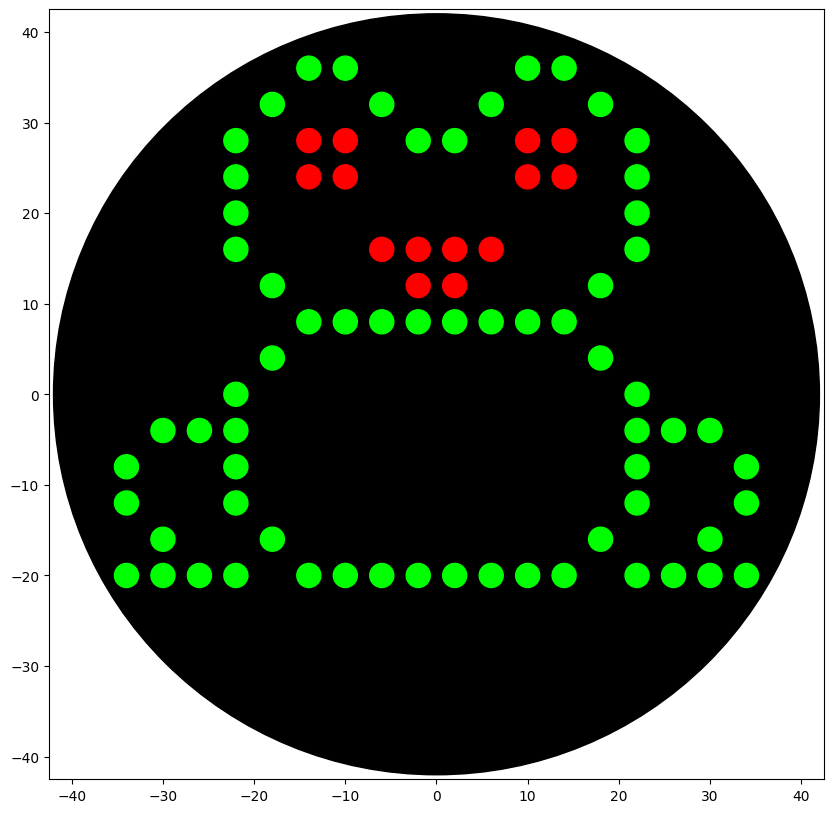
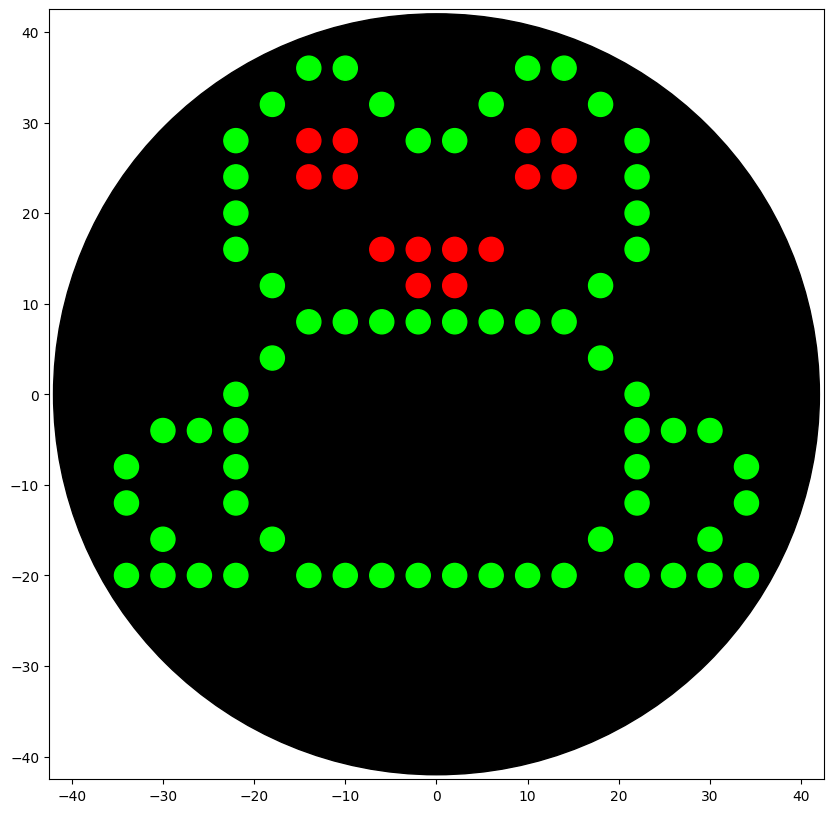
My Opentrons design is meant to resemble a frog because I use Xenopus laevis as my model organism in my honors thesis research at William & Mary.
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
In Sanders et al., 2022, the researchers use an Opentron robot to optimize a bacterial whole-genome sequencing (WGS) protocol for gut microbiota samples. The Opentron was used for DNA extraction and library preparation steps, reducing the overall cost of WGS by ~$10 per genome and eliminating the need for 16S rRNA gene-based screening.
Write a description about what you intend to do with automation tools for your final project.
My final project will likely involve the design of a genetic circuit whose expression is regulated by a synNotch system. As such, I could use the Opentron to automatize construction of my genetic circuit.
Final Project Ideas
For my final project, I am interested in engineering a synNotch-regulated circuit into Xenopus laevis, either to control cell-fate decisions or to control expression of a fluorescent protein to observe cell-cell communication during embryonic development. See my slide in the Committed Listener slide deck linked here.
Week 4 HW: Protein Design Part I
Part A: Conceptual Questions
Since 1 Da = 1 g/mol, 500 g of meat equates to about 5 mol of amino acids. The approximate number of amino acids within these 5 mols would be 3x10^24.
When humans eat, the macromolecules the beef are made of are broken down during digestion into monomers. These monomers are common to all life, and humans use them to build human-specific macromolecules.
These 20 amino acids are what evolution happened to select for. These 20 amino acids happen to be enough to build all the proteins that are necessary for life that has evolved on Earth. Theoretically, there could be more, but in our “system” of life, these 20 are enough.
Theoretically, yes. The R group can be anything, but only some will be functional with the 20 natural amino acids. Below are designs of new amino acids; I based them off of preexisting ones and added/removed atoms
new amino acids
Amino acids formed under natural conditions on Earth. One example is demonstrated by the Miller-Urey experiments where they demonstrated that atmospheric gases could form amino acids spontaneously when energy is put in in the form of lightning.
Left-handed because natural amino acids are L and create right-handed α-helixes. This means D-amino acids mirror L-amino acids, so the α-helix would also be mirrored.
Yes, you can discover new α-helices that already exist in proteins using tools/techniques like AlphaFold and X-ray crystallography. Discovering novel types of helices would require changing the chemistry of the amino acids, so discovering pre-existing ones would be unlikely. Creating new types of helices would be interesting, though.
This is because most biological molecules are D-enantiomers (right-handed), so they also create right-handed helices.
β-sheets are formed using hydrogen bonds across backbones. β-sheet backbones “want” to form more hydrogen bonds with another peptide backbone, and can do that by stacking with other β-sheets.
Many amyloid diseases form β-sheets because hydrogen bonding in β-sheets is extremely favorable, so if the protein is able to “misfold” into β-sheets, it will do that. β-sheets can be used as materials. In fact, silk is rich in β-sheets, which is why it has so much tensile strength.
Part B: Protein Analysis and Visualization
Part C: Using ML-Based Protein Design Tools
C1: Protein Language Modeling
C2: Protein Folding
C3: Protein Generation
Part D: Group Brainstorm on Bacteriophage Engineering
I collaborated with Heather Qian on this assignment!
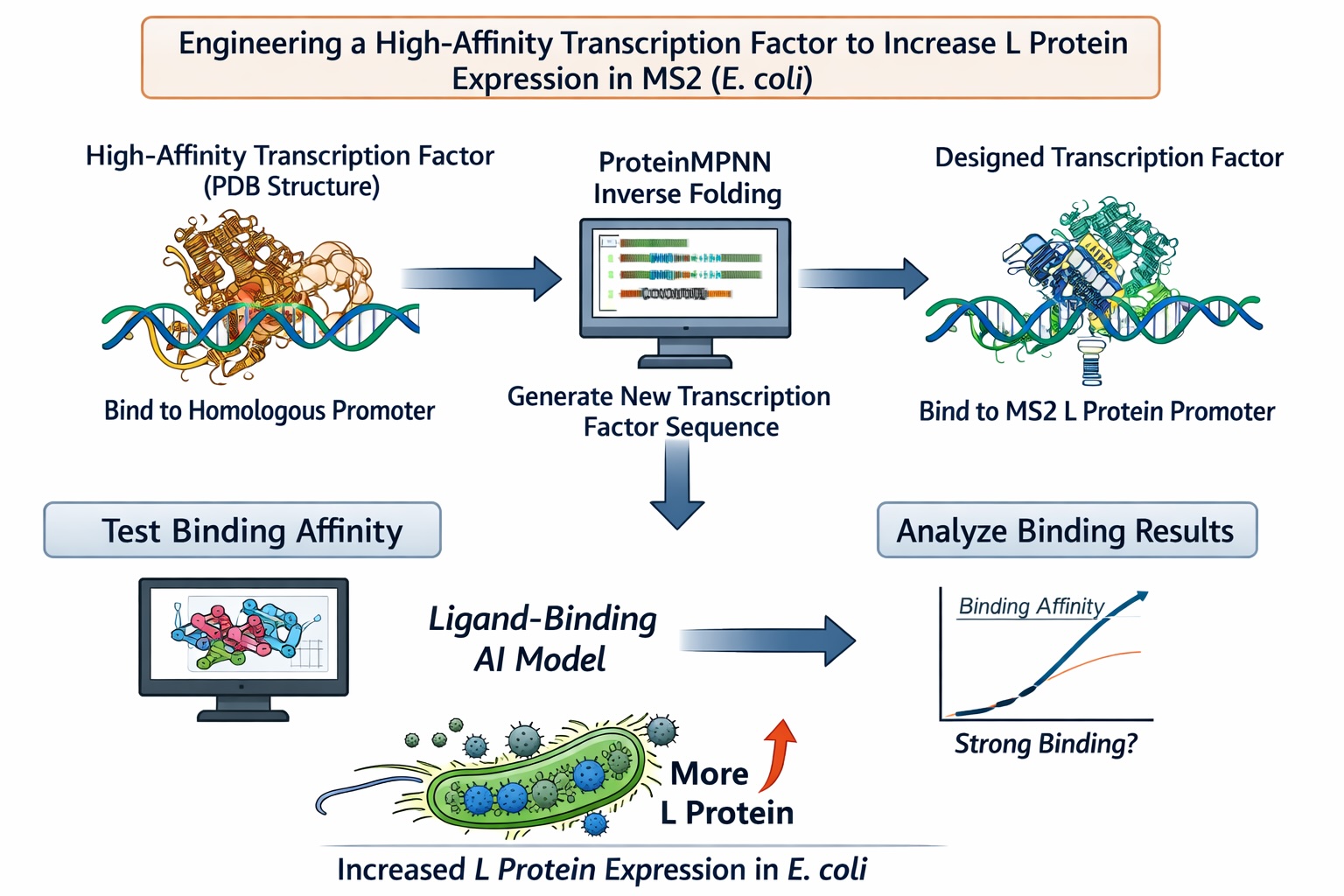
Computational Goal: We will attempt to increase the titer of the L protein expressed by MS2 in the E. coli host.
Overall solution: create a new transcription factor that binds very tightly to the promoter, increasing expression of the L protein
Inverse protein folding using ProteinMPNN
Use structure of a transcription factor (a PDB file) that binds with very high affinity to a promoter that is highly homologous to the L protein promoter
Input the structure of the aforementioned transcription factor into ProteinMPNN along with the amino acid sequence of a native transcription factor that binds to the L protein promoter
This will theoretically generate the sequence of a protein that is structurally similar to a transcription factor with high DNA-binding affinity but is specific to the L protein promoter
Confirm the binding affinity between our designed transcription factor and the MS2 L protein promoter with a ligand-binding AI model
Why will these tools accomplish our computational goal?
Protein MPNN is an inverse-folding algorithm. The sequence of the L protein promoter and other transcription factors that bind to this promoter are known. However, to generate a transcription factor with higher affinity for this promoter sequence than the native L protein transcription factors, we will model our inverse-folding after an existing transcription factor that behaves in the manner we envision (binds with high affinity) for our engineered transcription factor. As discussed in this week’s lecture, existing AI algorithms are good at designing structures similar to previously-characterized structures but less good at designing different (novel) structures. As such, our inverse-folding approach hinges on the existence of another high affinity transcription factor for a homologous promoter
Using a ligand-binding AI model will provide a computational indication of the success of our engineering without the need to use an in vitro or in vivo model
Possible pitfalls:
We are unable to find a transcription factor that binds to a highly homologous promoter sequence with high affinity :(
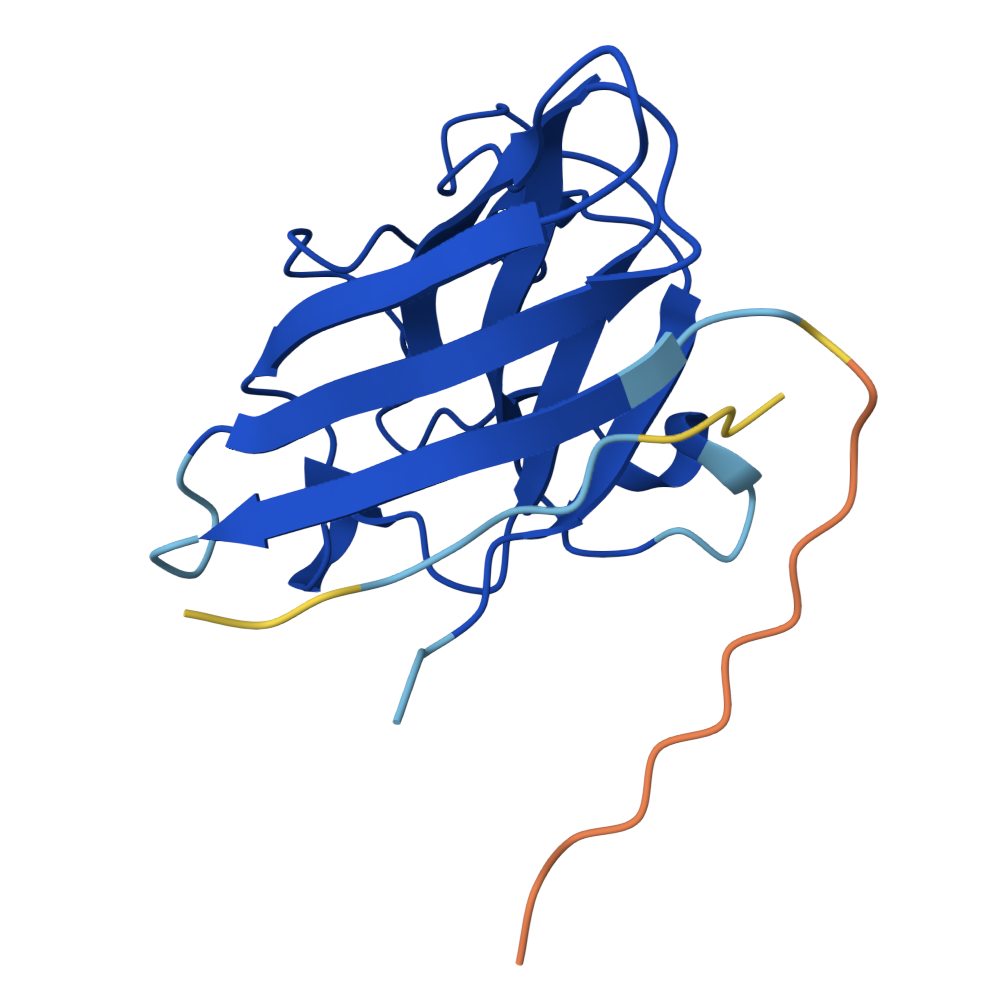
Binder 1 associates near the β barrel at the surface of the protein.
2
0.44
Like Binder 1, Binder 2 associates near the β barrel at the surface of the protein. However, this binder has a much lower ipTM score, meaning the confidence in this generated structure is much lower.
3
0.31
Binder 3 associates across the β barrel and disordered region at the surface of the SOD1 protein.
4
0.30
Binder 4 associates at the surface of the disordered region.
5
0.39
Binder 5 also associates at the surface of the disordered region.
Binders 1 and 2 have stronger ipTM values than Binder 5, the known binder provided for this exercise. All binders associate near the surface of the SOD1 protein, not integrating into the protein interior. The ipTM value for Binder 1 is relatively strong (0.71), meaning there is high confidence in that association between SOD1 and the binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Binder
Binding Affinity
Solubility
Hemolysis Probability
Net Charge
Molecular Weight
1
Weak binding affinity (6.150 pKd/pKi)
Soluble (1.000)
Non-hemolytic (0.095)
-0.15
1491 Da
2
Weak binding affinity (5.148 pKd/pKi)
Soluble (1.000)
Non-hemolytic (0.015)
1.84
1410 Da
3
Weak binding affinity (5.897 pKd/pKi)
Soluble (1.000)
Non-hemolytic (0.014)
2.84
1531 Da
4
Weak binding affinity (4.842 pKd/pKi)
Soluble (1.000)
Non-hemolytic (0.027)
-0.06
1463 Da
5
Weak binding affinity (5.968 pKd/pKi)
Soluble (1.000)
Non-hemolytic (0.047)
2.76
1507 Da
Binder 1, the binder with the highest ipTM value, also has the strongest binding affinity. None of the binders are predicted to be hemolytic. Unsurprisingly, Binder 1 appears to have the best balance of predicted binding and therapeutic properties. I will use Binder 1 to complete Part 4.
Part 4: Generate Optimized Peptides with moPPIt
Unfortunately, although I attempted to run motif 17-23 with multiple times and consulted ChatGPT for guidance, I was unable to make the moPPit program work without errors.
Part B: RD4 Drug Discovery Platform Tutorial
Will come back and do this later :)
Part C: Final Project: L-Protein Mutants
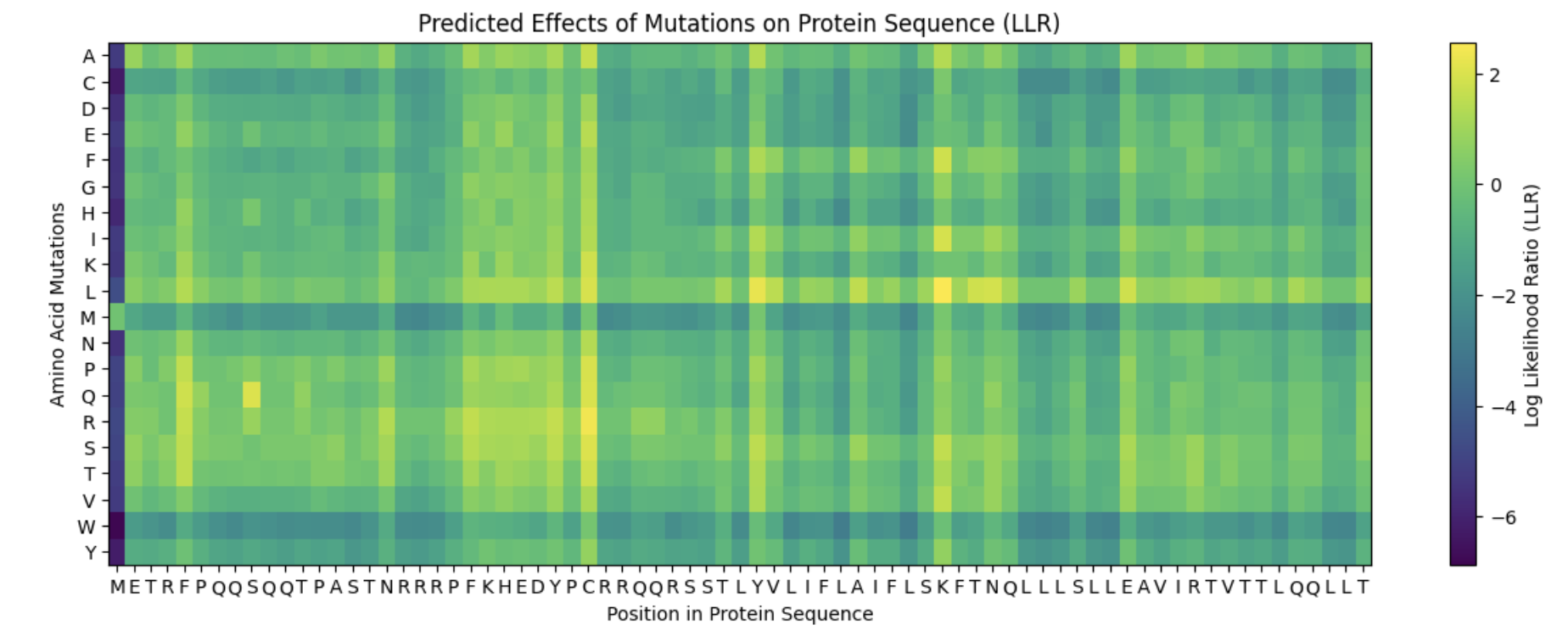
It appears that the L-protein mutants that result in a functional MS2 (1 in the “Lysis” column of the spreadsheet) correlate with the positive log likelihood ratio mutations on the heat map.
Proposed Mutations:
Mutation
Domain
Reasoning
L-protein Multimer
Evaluation
F -> S at position 5
Soluble Domain
When L-protein sequences was input into BLASTp, the F -> S mutation was observed in the third top hit in Emesvirus zinderi. Additionally, the F -> S mutation in position 5 appeared a yellowish-green on the heat map, indicating a “positive” mutation.
The mutant octomer appears to form a pore-like quaternary structure. The ipTM score is 0.13, indicating low confidence that this is an accurate assembly of the mutant multimer.
R -> S at position 19
Soluble Domain
In the table of L-protein mutants, this mutation has a score of 1 in the Lysis column, providing experimental evidence of a functional lysis protein. Additionally, mutations in the R amino acid at position 19 were identified in multiple top BLASTp hits.
Like the previous mutant, the Mutant 2 octomer forms a pore-like quaternary structure with an ipTM score of 0.14, indicating low confidence in this assembly.
R -> I at position 31
Soluble Domain
This mutation is associated with a functional L protein on the “L-protein mutant” spreadsheet (1 in lysis column) and appears to have a positive log likelihood ratio on the heat map.
The pore generated for this octomer appears to be tighter than Mutants 1 and 2, and although the ipTM score of 0.16 still indicates low confidence in the structure, it is slightly stronger than the previous two mutants.
A -> P at position 45
Transmembrane Domain
Once again, this mutation was identified on the “L-protein mutant” spreadsheet as resulting in a function lysis protein. Although this mutation occurs in the transmembrane domain and therefore is less likely to result in a functional advantage to the MS2 phage, the functional lysis protein and low negative score on the heat map suggest it may be a better choice than other mutations in the transmembrane domain.
E -> S at position 61
Transmembrane Domain
This mutation has a positive log likelihood ratio on the heat map, and a mutation in the E protein was identified in a BLASTp alignment with the L-protein for the MS12 phage.
Week 6 HW: Genetic Circuits Part I
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
DNA polymerase: uses dNTP monomers to synthesize new DNA strands
dNTPs: monomers of new DNA strand
Buffer: stabilizes the pH of the reaction for optimal enzymatic function
MgCl2: Cofactor for the polymerase; optimizes enzymatic function and primer annealing
What are some factors that determine primer annealing temperature during PCR?
GC content (higher GC content = more hydrogen bonds = stronger primer annealing = higher annealing temperature)
Primer length (longer primers = more hydrogen bonds = stronger primer annealing = higher annealing temperature)
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR:
Enzyme: DNA polymerase
DNA polymerases bind to primers and synthesize complementary DNA to the template strand
Purpose: amplify!
Polymerases synthesize DNA by recognizing primers (designed to flank the specific region of interest) and incorporating dNTPs into a novel DNA strand
When to use:
To detect a specific sequence within a mixed sample
To create more of a specific DNA sequence (amplify)
Restriction enzyme:
Enzyme: restriction endonuclease
Restriction endonucleases recognize specific nucleotide sequences (4-8 bp) in double-stranded DNA and cut in a specific pattern (blunt or sticky ends, depending on the enzyme)
Purpose: cut!
Restriction enzymes cut existing template DNA, they do NOT amplify DNA fragments
When to use:
To linearize bacterial plasmids (ex. for in vitro transcription of capped mRNA for microinjection into X. laevis!)
To cut DNA fragments to assemble/ligate together and transform into a plasmid
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Primer design!
The 5’ tail overhang of each primer should be identical to the adjacent DNA fragment
the 3’ end of the primer should be complementary to the DNA fragment to which it will anneal
Restriction Digest:
Restriction enzyme cut sites should be avoided
PCR:
Primers should be designed that flank the overhang regions
How does the plasmid DNA enter the E. coli cells during transformation?
Heat shock transformation - briefly raising temperature increases the permeability of the bacterial cell wall, allowing the plasmid DNA to enter into the E. coli cell
E. coli cell sample placed on ice for ~15-30 minutes
Sample heated to 42ºC for ~15-30 seconds
Sample then returned to ice for ~5 minutes
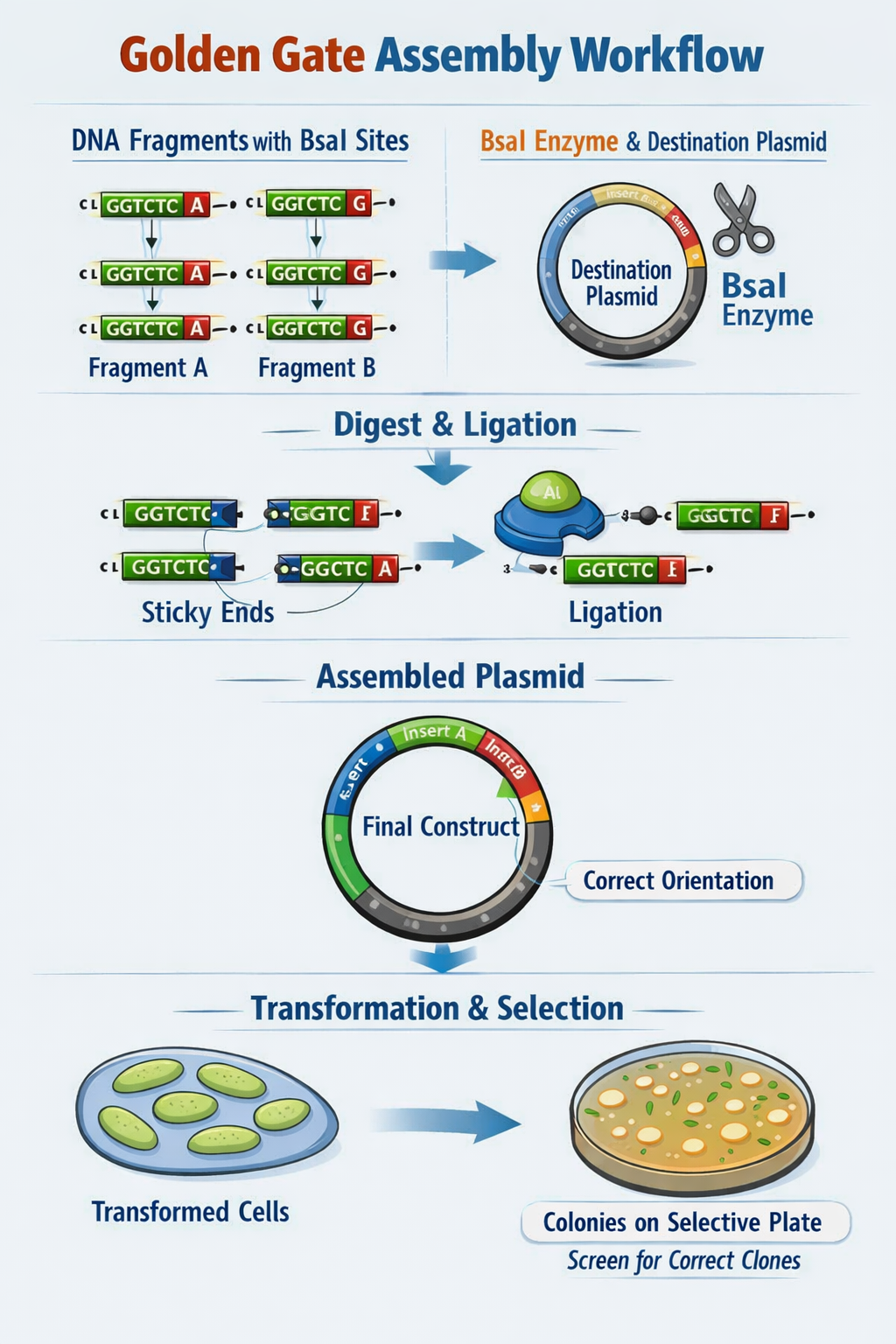
Describe Golden Gate Assembly
Golden Gate Assembly utilizes Type IIS restriction enzymes (such as BsaI) which recognize non-pallindromic sequences, cut outside the recognition site (to avoid damaging the DNA sequence of interest), and create sticky ends of variable lengths. First, template DNA is amplified using PCR with primers specified to include the TIIS recognition and cut sites. Next, a restriction digest with the TIIS enzyme is performed to create DNA fragments with sticky ends. The sticky ends of adjacent fragments should be complementary so the sequences can be ligated in the appropriate order. Following ligation of DNA fragments with a plasmid, the engineered plasmid can be transformed into a bacterial cell (ex. E. coli), and bacterial colonies containing the plasmid can be screened for furhter use.
Image generated by ChatGPT
Asimov Kernel
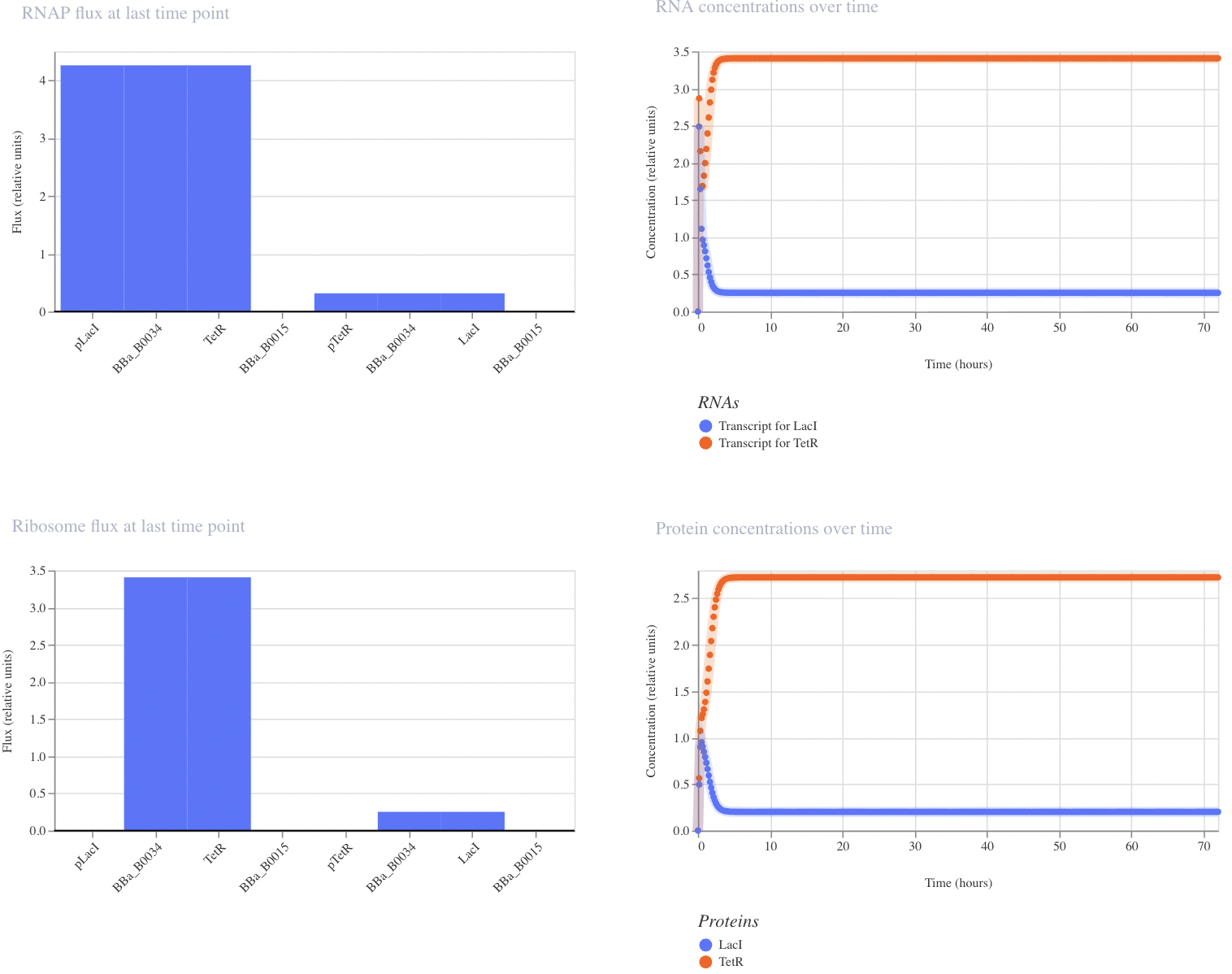
Repressilator
The protein concentrations generated from this recreation of the Repressilator meet my expectations in that they oscillate over time. However, unlike the in vivo system, the oscillations do not decay over time in this computational model.
Construct 1: Negative Feedback Loop
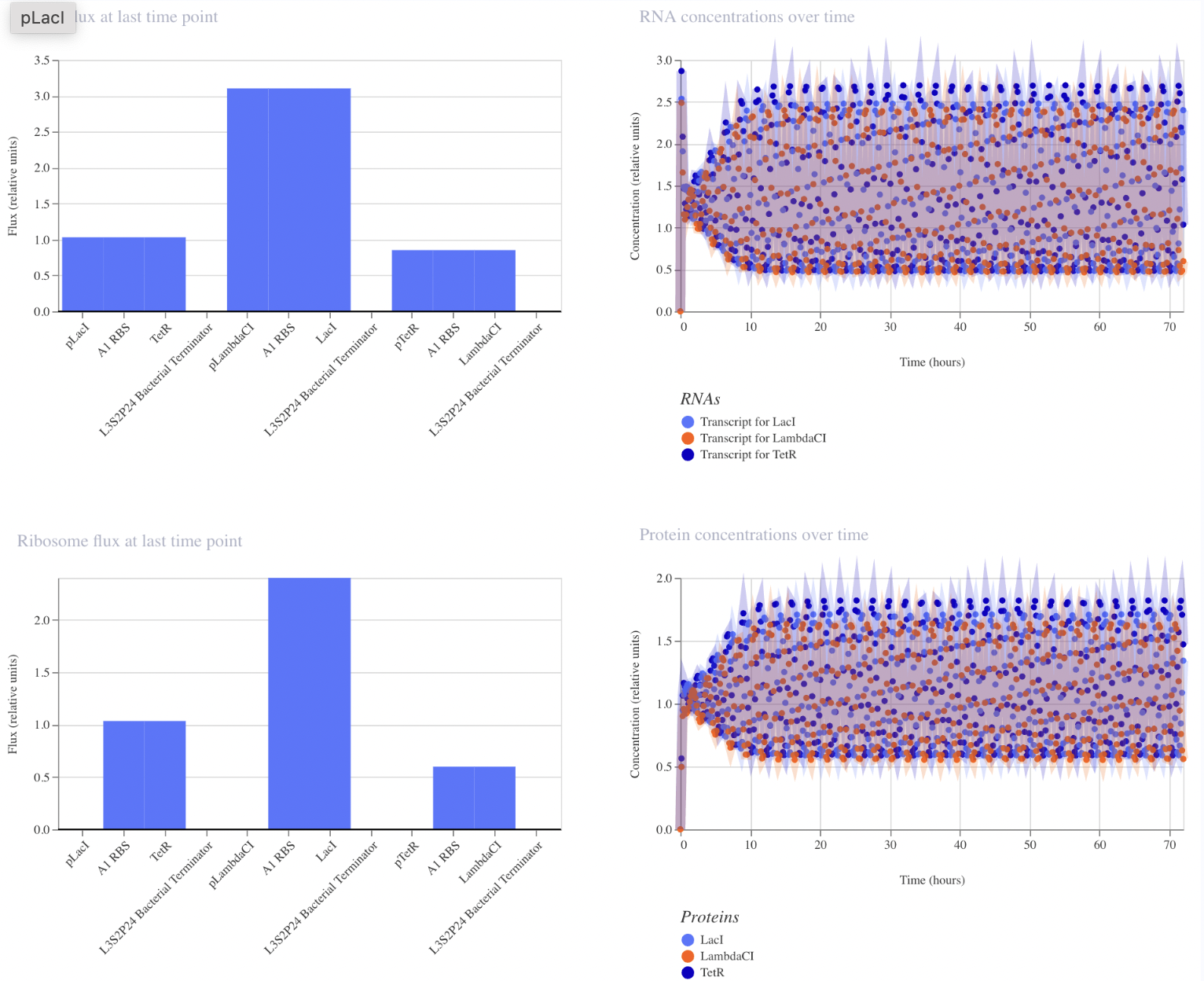
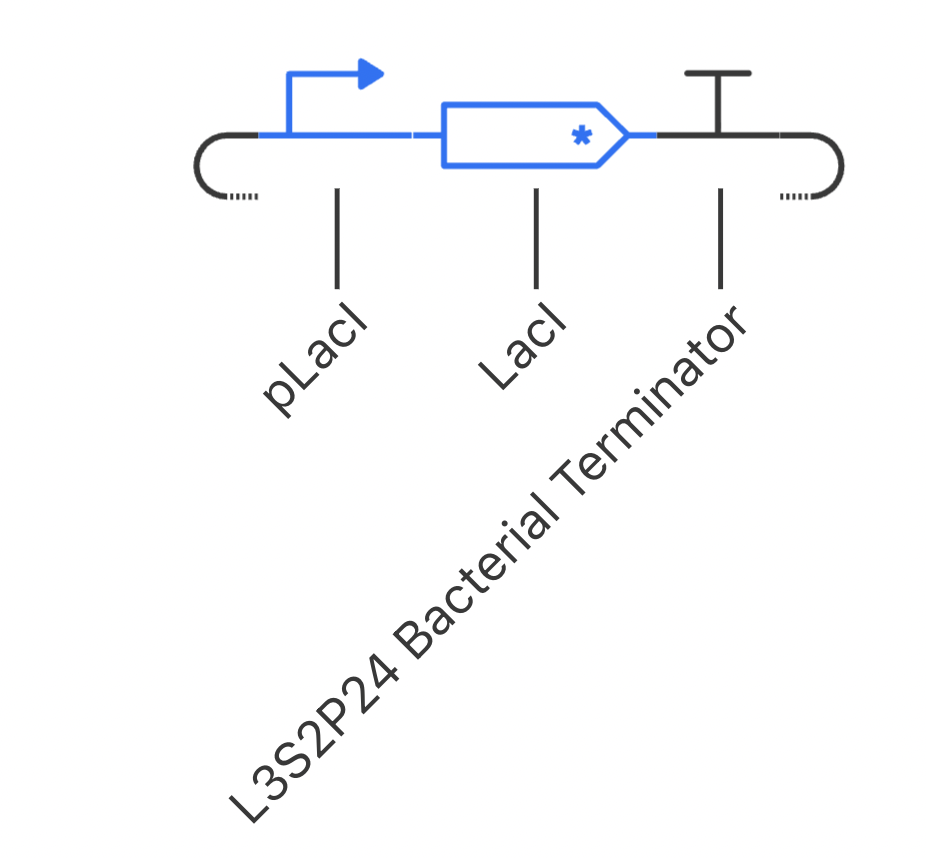
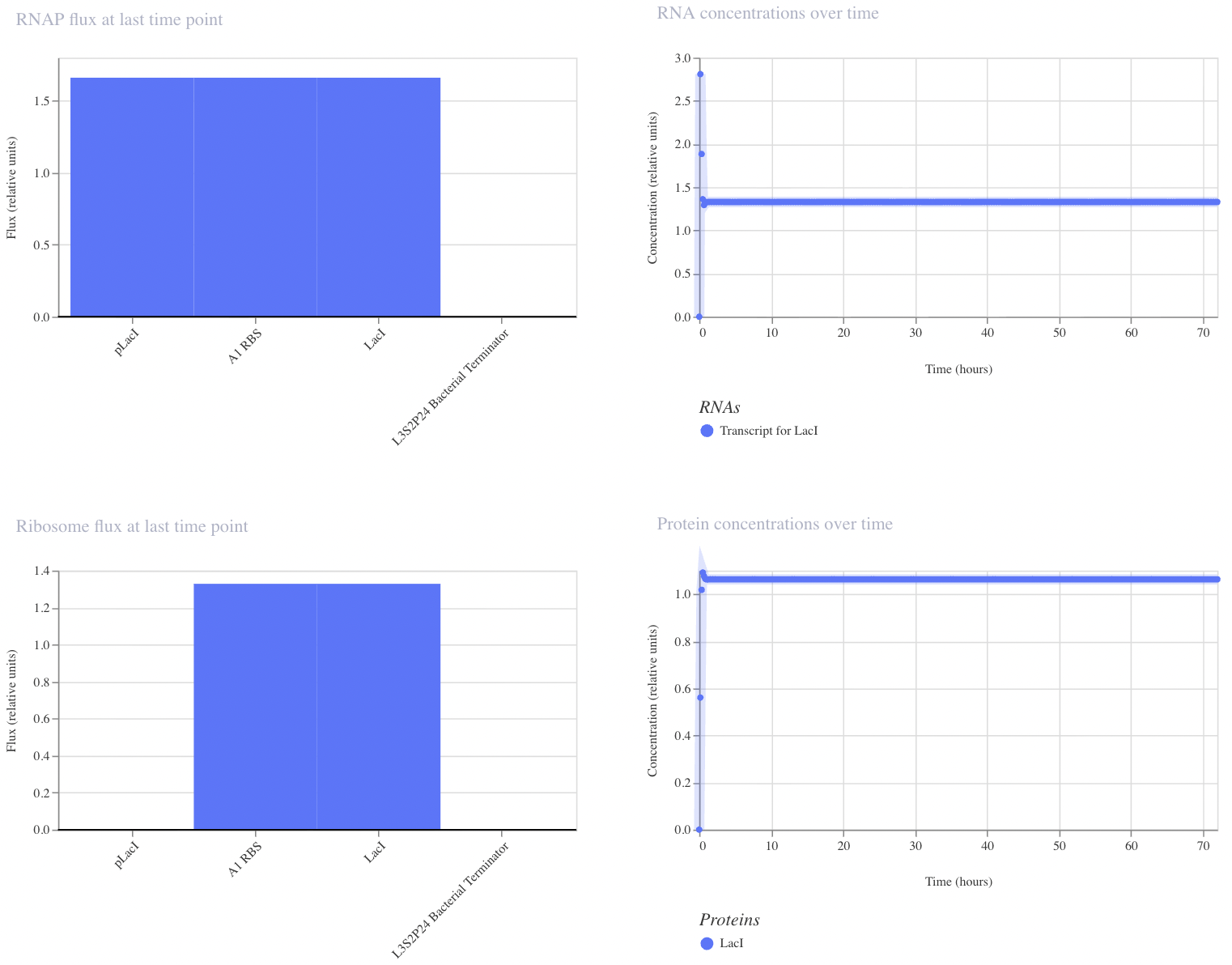
I have designed a LacI genetic circuit in which the LacI gene should bind to its own promoter and inhibit its own expression. I would expect the RNA and protein concentration to fluctate slightly, then reach steady state.
My system immediately reaches steady state, which is not what I predicted. However, I was unable to add an operator sequence in this circuit, so perhaps that is my issue.
Construct 2: Toggle Switch
Modeled after the classical toggle switch genetic circuit of mutual repression, I expect this construct to reach a bistable state. If I were able to add an inducer to this system, I would expect to be able to switch which bistable state is selected by the system. However, in this instance, my system reached a single steady state.
My system behaves the way I predicted it would!
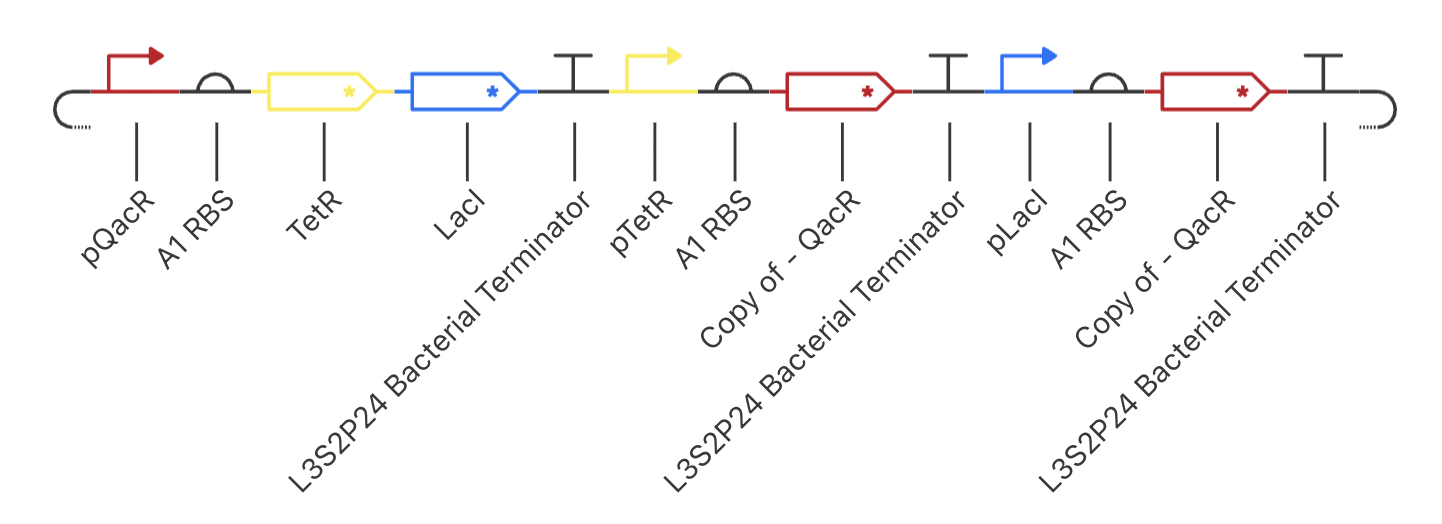
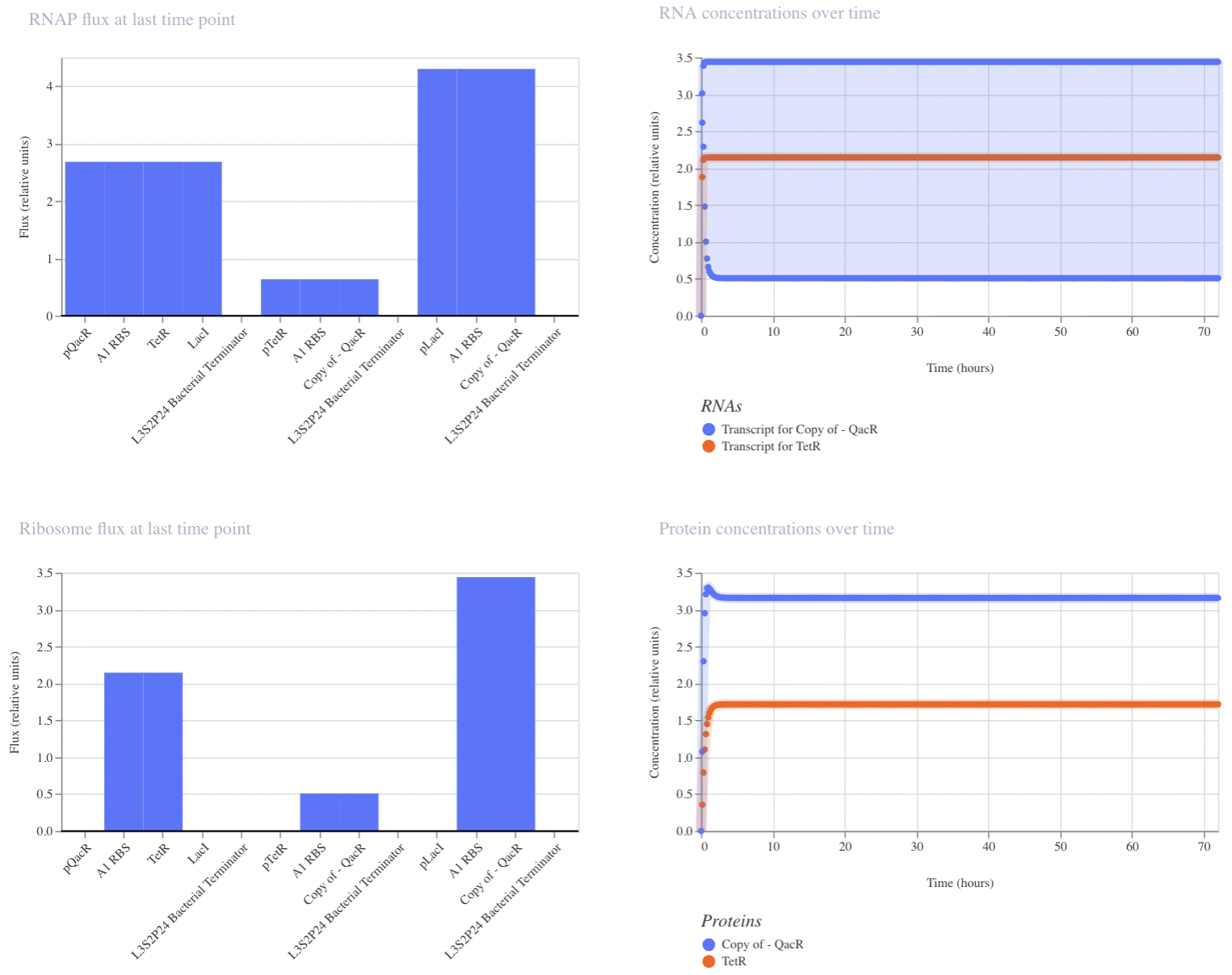
Construct 3: Dual Repressors
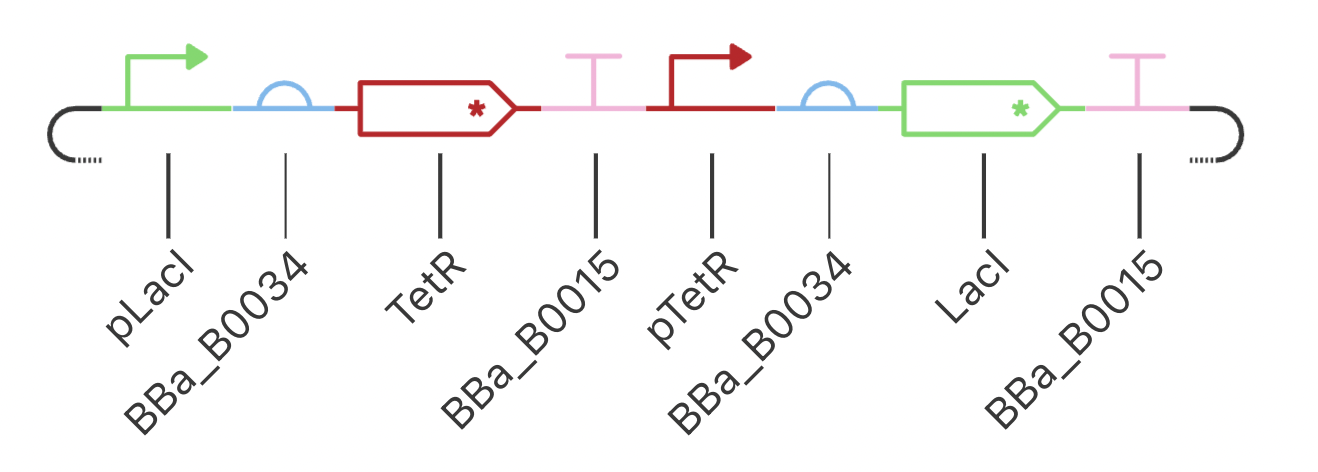
My final circuit contains two repressors, LacI and TetR, controlled by the QacR promoter. Additionally, the expression of QacR is controlled by the expression of either LacI or tetR (two separate QacR genes). I expect the system to reach a single steady state. If I was able to design an inducible promoter, I could be able to switch between stable states.
My system behaved as predicted :)
Week 7 HW: Genetic Circuits Part II
Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Variables input into Boolean functions are binary (either ON or OFF), whereas the variables input into IANNs can be continuous. Additionally, IANNs can input multiple variables at a time while Boolean functions (like a two-layer IANN) only integrate two variables.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
You could use an IANN to model cell fate decisions during embryonic development. The network could consider intracellular factors—such as gene expression—and extracellular influences—such as cell density and external signaling from morphogens or canonical signaling pathways—to predict the cell fate of a pluripotent or hemipotent cell during development. IANNs would provide a more representative in silico cell fate model than a Boolean function because, unlike the Boolean function, the IANN would be able to incorporate multiple influences (mentioned previously) that influence fate rather than one factor at a time. One limitation of the IANN in this application might be treating all cells in a developing embryo as homogeneous rather than incorporating noise and stochasticity into its fate predictions.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
[[ADD]]
Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Hinneberg et al. (2025) provide a recent review of existing fungal materials and exciting avenues for future innovation. Some examples of fungal material from the article include biodegradable packing, leather alternatives, and various strucutral components. Fungal materials are a sustainable alternative to many manufactured goods; however, the scalability of fungal material production is limited, and the process is time-consuming.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would be interested in engineering a fungus to detect pH changes or other environmental changes in the water of the tanks where I keep my Xenopus laevis frogs for my research here at William & Mary. It would be interesting to have a biosensor that can demonstrate when the environment the frogs are kept in is outside of the standard physiological range so it can be adjusted for their health and well-being.
Unlike bacteria, fungi are eukaryotic, meaning they are a better model of the intracellular complexity of human cells than bacteria. In particular, protein folding in fungi follows the same processing (chromatin remodeling, endomembrane system, RNA processing such as post-translational modifications, etc.) as proteins in human cells. Additionally, as fungi are eukaryotic (multicellular), they are capable of responding to signaling from neighboring cells or external influences in a similar manner to human cells.
Final Project!
Project Title
Engineering a small molecule to target the βγ subunits of pre-Bötzinger Complex μ opioid receptors
Final Project Description
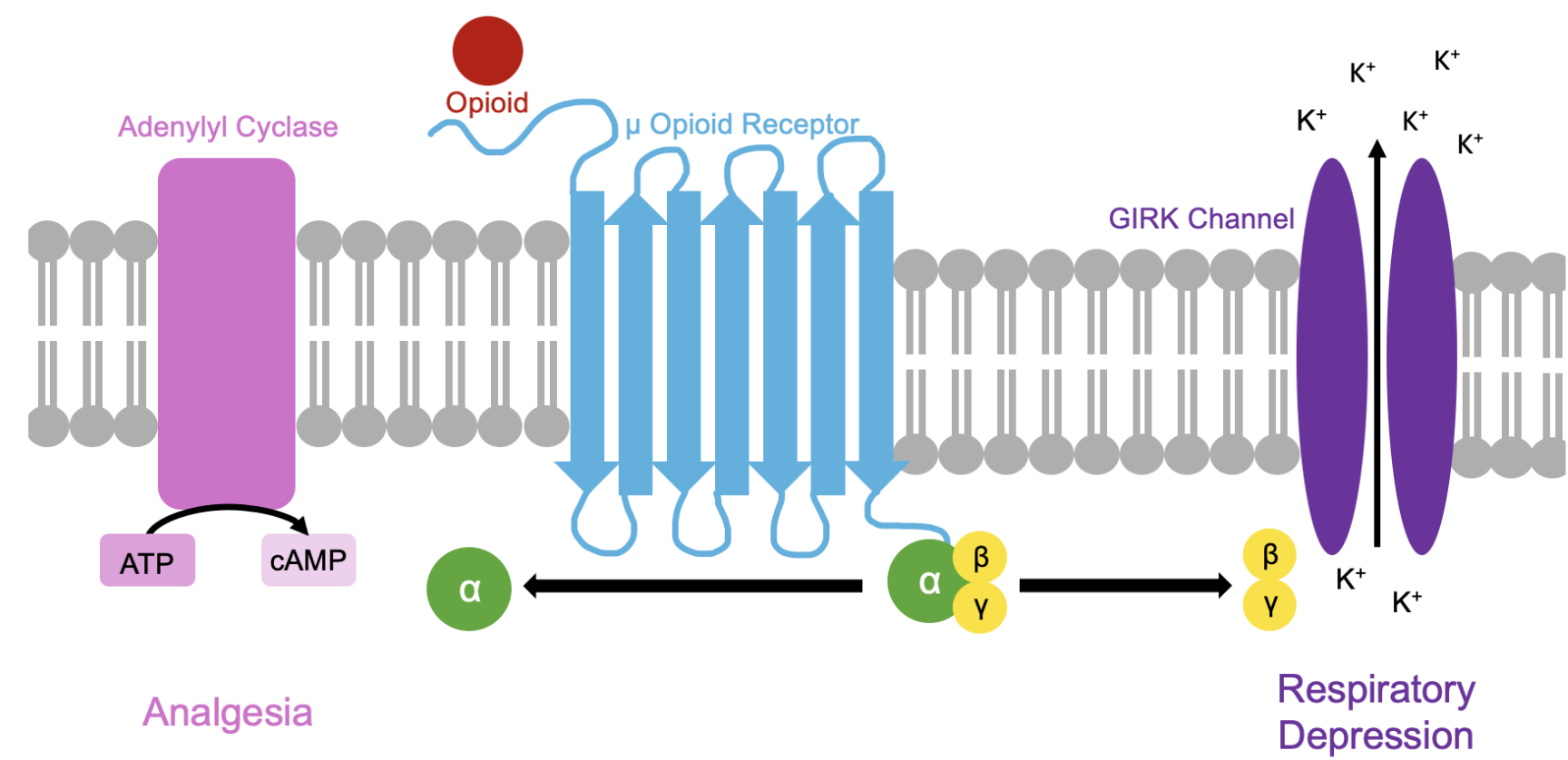
Renarcotization is a delayed consequence of naloxone administration after opioid consumption that occurs when an opioid re-binds to a receptor following release of naloxone from the μ opioid receptors (MOR) active site, resulting in delayed respiratory depression. Existing literature has identified the βγ subunits of the MOR as responsible for regulating the respiratory depression associated with opioid binding to MOR. In this project, I aim to design a small molecule that will bind to the βγ subunits of the MORs in the pre-Bötzinger complex (pre-BötzC) of M. musculus to target the respiratory depression associated with opioid administration without disrupting the analgesic effects associted with activation of the MOR α subunit.
Final Project Aim 1
The first aim of my final project is to design a small molecule capable of binding to βγ subunits of pre-BötzC MORs by utilizing the Boltz Lab Drug Discovery Platform introduced in Part B of the Week 5 homework assignment. Existing literature has identified Gallein as a small molecule capable of effectively binding to the MOR βγ subunit, but it was ineffective when applied in vivo. My first objective will be to modify this structure (then perhaps pursue alternative structures) to optimize targeting the specific MOR expressed in the pre-BötzC to prevent lethal off-target effects.
Week 9 HW: Cell-Free Systems
General Homework Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis systems are advantageous for quick, high-throughput protein production. Cell-free protein expression is more time efficient than traditional in vivo methods because they do not require the cloning steps of cell-based systems. Additionally, cell-free systems can be modified to include non-canonical amino acids which enables numerous biotechnology and pharmaceutical applications. Cell-free systems also offer the advantage of ease of manipulating reaction conditions. Cell-free expression is more beneficial than cell production when evaluating proteins that are toxic to a cell or when a large yield of the desired protein is needed in a relatively short period of time.
Describe the main components of a cell-free expression system and explain the role of each component.
Genetic template encoding the desired protein (DNA or mRNA)
Energy source (ATP)
RNA polymerase and nucleotides (if DNA template, for mRNA synthesis)
tRNA, amino acids, and ribosomes (for translation)
Enzymatic cofactors
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision is critical in cell-free systems because the energy source (ATP) powers both transcription and translation. One method that could be used to ensure a continuous supply of ATP in a cell-free experiment is the “protein synthesis using recombinant elements” (“PURE”) system which generates acetyl phosphate from pyruvate, phosphate, and oxygen, which is used to rephosphorylate ATP.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free expression systems are ideal for simple, high-yield protein synthesis with limited post-translational modifications. Eukaryotic systems are lower yield and higher cost, but they support the production of more complex proteins that undergo processing through the eukaryotic endomembrane system. An ideal protein for prokaryotic cell-free protein expression might be an enzyme such as beta-galactosidase which supports E coli metabolism by breaking lactose into glucose and galactose monomers. An example of a protein that may be produced in a eukaryotic cell-free system would be an antibody because antibodies require proper folding and assembly to be functional.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
I would work with a eukaryotic cell-free expression system because membrane proteins require complex folding which is more suited for a eukaryotic system. One challenge in this experiment might be aggregation of hydrophobic proteins (the exterior of a protein’s transmembrane domain is often hydrophobic) which may inhibit proper folding.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
The promoter and RBS are not well-suited to the expression system - change to a promoter of different strength
Insufficient energy regeneration - adjust energy regeneration system to ensure adequate ATP is produced to carry out cell-free protein production
Protein misfolding or aggregation - modify reaction conditions or incorporate molecular chaperone (or other proteins that facilitate proper folding) into the system
Homework question from Kate Adamala
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
My synthetic cell would detect a cancer-associated molecular signal and respond by producing a cytotoxic protein. The input would be a protein associated with tumorigenesis and the output would be a cytotoxic protein, such as DTA.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
A cell-free system could be used to express DTA, but without a cell membrane the reaction would not be spatially contained and the DTA toxin could diffuse everywhere (to non-tumor cells).
Could this function be realized by genetically modified natural cell?
Yes, engineered cells (ex. CAR-T cells) can detect tumor markers and express cytotoxic proteins.
Describe the desired outcome of your synthetic cell operation.
The circuit in the synthetic cell would not be activated until a tumor-associated molecular signature is detected. Once activated, the cell would produce and release DTA to target neighboring tumor cells.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
The membrane would be comprised of lipids that compose a standard cell (phospholipids, cholesterol, etc).
What would you encapsulate inside? Enzymes, small molecules.
The plasmid containing the input-responsive sensor and DTA gene, cell-free system components (see problem above for complete list), and the necessary buffers and co-factors for enzymatic function.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?
Some mammalian lysate that is genetically siilar enough to mimic the environment and support the folding of human proteins.
How will your synthetic cell communicate with the environment?
The circuit within the synthetic cell will be sensitive to tumor-associated proteins within the cellular environment and will release the cytotoxic DTA in a controlled manner into the cellular environment.
Experimental details.
List all lipids and genes.
Lipids: phospholipids, cholesterol
Genes: sensor gene (sensitive to tumor-associated protein) and DTA gene
How will you measure the function of your system?
I could incorporate a reporter fluorescence gene or measure the concentration of DTA in the cellular environment.
Homework question from Peter Nguyen
Write a one-sentence summary pitch sentence describing your concept.
A freeze-dried system that can coat the barriers of aquatic environments (i.e. aquarium displays, fish tanks) to visually signal when environmental conditions become hazardous (i.e. pH becomes too acidic or basic to support plant and animal life, algal bloom, etc.).
How will the idea work, in more detail? Write 3-4 sentences or more.
The coating consists of a thin hydrogel layer embedded with micro‑domains of freeze‑dried cell‑free reactions. Each micro‑domain contains a pH‑responsive genetic circuit that activates expression of a visible chromoprotein when the surrounding water becomes too acidic or too basic. When the aquarium water contacts the surface, the cell‑free spots rehydrate and begin sensing; if the pH crosses a preset threshold, the chromoprotein is produced and the coating changes color. Different regions can be tuned to different pH ranges, creating a spatial “map” of water quality across the structure.
What societal challenge or market need will this address?
Aquariums, aquaculture systems, and aquatic research facilities rely on stable water chemistry to keep organisms healthy, but existing monitoring tools can be expensive, technical, or require constant calibration. A passive, visually intuitive biosensing surface would make water‑quality monitoring more accessible for hobbyists, educators, and public installations. This reduces the risk of unnoticed pH drift, which is a major cause of stress and mortality in aquatic organisms.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
This system is intentionally water-activated (designed for aquatic environments). To promote stability, protective components will be embedded in the hydrogel matrix to protect against light damage or oxidation. To protect against one-time use, the hydogel film could be multi-layered or coated on a replacable panel.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting.
Long‑duration spaceflight increases the risk of nutrient deficiencies and immune changes because astronauts have limited access to fresh food and experience altered metabolism in microgravity. Vitamin D pathways are especially affected due to minimal UV exposure and changes in bone physiology. Transporting large quantities of supplements is mass‑limited, so an on‑demand system for producing beneficial proteins would improve crew health and mission resilience.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches.
A DNA construct encoding vitamin D–binding protein (DBP) fused to a fluorescent reporter, enabling simultaneous production and visualization of a nutritionally relevant protein.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses.
Vitamin D metabolism is disrupted in space due to reduced UV exposure and altered calcium homeostasis, contributing to bone loss and immune changes. Producing DBP in situ serves as a model for generating nutritional or therapeutic proteins from dry‑stored DNA during long missions. The fluorescent fusion allows astronauts to directly monitor expression using the P51 fluorescence viewer, making it easy to compare performance in microgravity versus Earth. This target links a concrete health concern to a generalizable strategy for on‑demand biomanufacturing in space.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (max 150 words)
Freeze‑dried cell‑free reactions can reliably express a functional DBP–fluorescent fusion protein under spaceflight conditions, demonstrating the feasibility of on‑demand production of beneficial proteins during long‑duration missions. Microgravity and space radiation may influence reaction kinetics, folding efficiency, or stability, but that properly formulated cell‑free reactions will retain sufficient activity for practical use. The research goal is to quantitatively compare expression yield and fluorescence intensity of the DBP fusion protein in space versus matched ground controls. Establishing robust expression in microgravity would support future development of compact “protein pharmacy” kits for astronauts, enabling flexible nutritional supplementation or rapid production of medically relevant proteins without relying solely on pre‑packaged supplies.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc.
Freeze‑dried cell‑free reactions containing the DBP–fluorescent fusion plasmid will be prepared alongside negative (no DNA) and positive (standard fluorescent protein) controls. Astronauts will rehydrate reactions with buffer, incubate them at ambient ISS temperature, and measure fluorescence using the P51 fluorescence viewer. Identical ground controls will be run with the same lots and timing. Data collected will include fluorescence intensity over time and qualitative observations of reaction color. Comparing space and Earth results will reveal how microgravity and storage affect cell‑free protein expression.
Week 10 HW: Imaging and Measurement
Final Project
For my final project, I will measure the binding affinity of my de novo peptide to the beta-gamma subunits of the human µ opioid receptor (MoR) using AlphaFold3. If I were to incorporate a wet lab component to this project, I would measure the GFP fluorescence of E. coli cells with my engineered plasmid (peptide+GFP) electroporated into them to confirm the genes of interest are being expressed then perform co-immunoprecipitation to measure the binding affinity of the de novo peptide to the human MoR Gβγ subunits.
Part I: Molecular Weight
The theoretical molecular weight of the eGFP protein (without the linker and histidine tag) is 26941.48 Da. With the linker and histidine tag, the molecular weight is 28006.60 Da.
Charge state approach:
z = 32.09
MW = 27823 Da
Accuracy –> 0.65% off from true value
Perhaps if I had a higher resolution image where I could more accurately read the peaks, but with the image provided, no.
Part II: Secondary/Tertiary Structure
Native state proteins are stabilized by intermolecular forces (i.e. hydrogen bonds, disulfide bonds, VDW interactions, hydrophobic interactions, etc) with well-ordered and low free energy (ΔG). Denaturated proteins are prone to aggregation (of hydrophobic regions) or protonation (of basic regions) as amino acid residues that would be insulated on the protein interior in the native state are exposed to the surrounding environment. A denatured protein will have more peaks than a native state protein due to greater numbers of charges.
The charge state appears to be around +4.6. This is calculated by z = 1/(2799.6365-2799.4199)
Part III: Peptide Mapping - Primary Structure
20 lysine, 6 arginine
19 peptides
Yes, the number of peaks (~20) is approximately the same as the number of peptides
One more peak than expected
Part IV: Oligomers
7FU Decamer: Intensity ~15 (at 3.4 MDa)
8FU Didecamer: Intensity ~170 (at 8 MDa)
8FU 3-Decamer: Intensity ~50 (at 12 MDa)
8FU 4-Decamer: Intensity close to 0 (at 16 MDa)
Part V: Did I Make GFP?
Our lab did not have work done at the Wates Immerse Lab, so this is the table from the screenshot data:
Opentrons Design Opentrons CoLab Code ### Green cursor = center_location.move(types.Point(x=-18, y = 12)) pipette_20ul.pick_up_tip() pipette_20ul.aspirate(15, location_of_color('Green')) dispense_and_detach(pipette_20ul, 3, cursor) cursor = cursor.move(types.Point(x=-4, y=4)) for i in range(4): dispense_and_detach(pipette_20ul, 3, cursor) if i!=3: cursor = cursor.move(types.Point(y=4)) pipette_20ul.aspirate(15, location_of_color('Green')) for i in range(2): cursor = cursor.move(types.Point(x=4, y=4)) dispense_and_detach(pipette_20ul, 3, cursor) cursor = cursor.move(types.Point(x=4)) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=4, y=-4)) pipette_20ul.aspirate(18, location_of_color('Green')) cursor = cursor.move(types.Point(x=4)) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=4, y=4)) cursor = cursor.move(types.Point(x=4)) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=4, y=-4)) pipette_20ul.aspirate(15, location_of_color('Green')) cursor = cursor.move(types.Point(y=-4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(y=-4)) cursor = cursor.move(types.Point(y=-4)) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=-4, y=-4)) cursor = cursor.move(types.Point(x=-4)) pipette_20ul.aspirate(18, location_of_color('Green')) for i in range(6): dispense_and_detach(pipette_20ul, 3, cursor) if i!=5: cursor = cursor.move(types.Point(x=-4)) cursor = cursor.move(types.Point(x=-4)) pipette_20ul.aspirate(15, location_of_color('Green')) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=-4, y=-4)) cursor = cursor.move(types.Point(y=-4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(y=-4)) cursor = cursor.move(types.Point(y=-4)) pipette_20ul.aspirate(15, location_of_color('Green')) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=4, y=-4)) cursor = cursor.move(types.Point(x=-12, y=16)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=-4)) cursor = cursor.move(types.Point(x=-4, y=-4)) pipette_20ul.aspirate(9, location_of_color('Green')) dispense_and_detach(pipette_20ul, 3, cursor) cursor = cursor.move(types.Point(y=-4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=4, y=-4)) cursor = cursor.move(types.Point(x=-4, y=-4)) pipette_20ul.aspirate(12, location_of_color('Green')) for i in range(4): dispense_and_detach(pipette_20ul, 3, cursor) if i!=3: cursor = cursor.move(types.Point(x=4)) cursor = cursor.move(types.Point(x=12)) pipette_20ul.aspirate(18, location_of_color('Green')) for i in range(6): dispense_and_detach(pipette_20ul, 3, cursor) if i!=5: cursor = cursor.move(types.Point(x=4)) cursor = cursor.move(types.Point(x=4)) pipette_20ul.aspirate(15, location_of_color('Green')) for i in range(3): dispense_and_detach(pipette_20ul, 3, cursor) if i!=2: cursor = cursor.move(types.Point(x=4, y=4)) cursor = cursor.move(types.Point(y=4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(y=4)) cursor = cursor.move(types.Point(y=4)) pipette_20ul.aspirate(15, location_of_color('Green')) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=-4, y=4)) cursor = cursor.move(types.Point(x=8, y=-8)) dispense_and_detach(pipette_20ul, 3, cursor) cursor = cursor.move(types.Point(x=4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=4, y=-4)) cursor = cursor.move(types.Point(y=-4)) pipette_20ul.aspirate(18, location_of_color('Green')) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=-4, y=-4)) cursor = cursor.move(types.Point(x=4, y=-4)) for i in range(4): dispense_and_detach(pipette_20ul, 3, cursor) if i!=3: cursor = cursor.move(types.Point(x=-4)) pipette_20ul.drop_tip() pipette_20ul.pick_up_tip() ### Red cursor = cursor.move(types.Point(x=-8, y=44)) pipette_20ul.aspirate(12, location_of_color('Red')) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=-4)) cursor = cursor.move(types.Point(y=4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=4)) cursor = cursor.move(types.Point(x=-24)) pipette_20ul.aspirate(12, location_of_color('Red')) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=-4)) cursor = cursor.move(types.Point(y=-4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=4)) cursor = cursor.move(types.Point(x=4, y=-8)) pipette_20ul.aspirate(18, location_of_color('Red')) for i in range(4): dispense_and_detach(pipette_20ul, 3, cursor) if i!=3: cursor = cursor.move(types.Point(x=4)) cursor = cursor.move(types.Point(x=-4, y=-4)) for i in range(2): dispense_and_detach(pipette_20ul, 3, cursor) if i!=1: cursor = cursor.move(types.Point(x=-4)) pipette_20ul.drop_tip()
Subsections of Labs
Week 1 Lab: Pipetting
Week 2 Lab: DNA Gel Art
Benchling Gel Design
Restriction Digest Protocol
Gel Preparation Protocol
Gel Run and Imaging Protocol
Final Gel Image
Week 3 Lab: Opentrons Art
Opentrons Design
Opentrons CoLab Code
### Green
cursor = center_location.move(types.Point(x=-18, y = 12))
pipette_20ul.pick_up_tip()
pipette_20ul.aspirate(15, location_of_color('Green'))
dispense_and_detach(pipette_20ul, 3, cursor)
cursor = cursor.move(types.Point(x=-4, y=4))
for i in range(4):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=3:
cursor = cursor.move(types.Point(y=4))
pipette_20ul.aspirate(15, location_of_color('Green'))
for i in range(2):
cursor = cursor.move(types.Point(x=4, y=4))
dispense_and_detach(pipette_20ul, 3, cursor)
cursor = cursor.move(types.Point(x=4))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=4, y=-4))
pipette_20ul.aspirate(18, location_of_color('Green'))
cursor = cursor.move(types.Point(x=4))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=4, y=4))
cursor = cursor.move(types.Point(x=4))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=4, y=-4))
pipette_20ul.aspirate(15, location_of_color('Green'))
cursor = cursor.move(types.Point(y=-4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(y=-4))
cursor = cursor.move(types.Point(y=-4))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=-4, y=-4))
cursor = cursor.move(types.Point(x=-4))
pipette_20ul.aspirate(18, location_of_color('Green'))
for i in range(6):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=5:
cursor = cursor.move(types.Point(x=-4))
cursor = cursor.move(types.Point(x=-4))
pipette_20ul.aspirate(15, location_of_color('Green'))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=-4, y=-4))
cursor = cursor.move(types.Point(y=-4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(y=-4))
cursor = cursor.move(types.Point(y=-4))
pipette_20ul.aspirate(15, location_of_color('Green'))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=4, y=-4))
cursor = cursor.move(types.Point(x=-12, y=16))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=-4))
cursor = cursor.move(types.Point(x=-4, y=-4))
pipette_20ul.aspirate(9, location_of_color('Green'))
dispense_and_detach(pipette_20ul, 3, cursor)
cursor = cursor.move(types.Point(y=-4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=4, y=-4))
cursor = cursor.move(types.Point(x=-4, y=-4))
pipette_20ul.aspirate(12, location_of_color('Green'))
for i in range(4):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=3:
cursor = cursor.move(types.Point(x=4))
cursor = cursor.move(types.Point(x=12))
pipette_20ul.aspirate(18, location_of_color('Green'))
for i in range(6):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=5:
cursor = cursor.move(types.Point(x=4))
cursor = cursor.move(types.Point(x=4))
pipette_20ul.aspirate(15, location_of_color('Green'))
for i in range(3):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=2:
cursor = cursor.move(types.Point(x=4, y=4))
cursor = cursor.move(types.Point(y=4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(y=4))
cursor = cursor.move(types.Point(y=4))
pipette_20ul.aspirate(15, location_of_color('Green'))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=-4, y=4))
cursor = cursor.move(types.Point(x=8, y=-8))
dispense_and_detach(pipette_20ul, 3, cursor)
cursor = cursor.move(types.Point(x=4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=4, y=-4))
cursor = cursor.move(types.Point(y=-4))
pipette_20ul.aspirate(18, location_of_color('Green'))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=-4, y=-4))
cursor = cursor.move(types.Point(x=4, y=-4))
for i in range(4):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=3:
cursor = cursor.move(types.Point(x=-4))
pipette_20ul.drop_tip()
pipette_20ul.pick_up_tip()
### Red
cursor = cursor.move(types.Point(x=-8, y=44))
pipette_20ul.aspirate(12, location_of_color('Red'))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=-4))
cursor = cursor.move(types.Point(y=4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=4))
cursor = cursor.move(types.Point(x=-24))
pipette_20ul.aspirate(12, location_of_color('Red'))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=-4))
cursor = cursor.move(types.Point(y=-4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=4))
cursor = cursor.move(types.Point(x=4, y=-8))
pipette_20ul.aspirate(18, location_of_color('Red'))
for i in range(4):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=3:
cursor = cursor.move(types.Point(x=4))
cursor = cursor.move(types.Point(x=-4, y=-4))
for i in range(2):
dispense_and_detach(pipette_20ul, 3, cursor)
if i!=1:
cursor = cursor.move(types.Point(x=-4))
pipette_20ul.drop_tip()
Section 1: Abstract Opioid-induced respiratory depression (OIRD) is responsible for thousands of overdose deaths annually. The μ-opioid receptor (MoR) mediates OIRD through heterotrimeric G-protein signaling, specifically via activation of Gβγ subunits, which subsequently modulate inwardly rectifying potassium channels (GIRK/Kir3) and other downstream effectors in the pre-Bötzinger complex, the brain’s primary respiratory control center. Current therapeutic approaches, such as naloxone, prevent OIRD by outcompeting an opioid to bind to the MoR ligand binding site, but because opioids are more fat soluble than naloxone, the risk of the opioid re-binding to the receptor and inducing delayed OIRD, known as renarcotization, is a serious risk without an existing therapeutic target. This has led to interest in developing a small molecule or protein capable of binding to the Gβγ subunits of the MoR rather than the ligand binding site to prevent OIRD without disrupting the analgesic effects of an opioid while also reducing the risk of renarcotization. This project aims to engineer a de novo peptide antagonist that selectively binds and inhibits Gβγ-mediated signaling, providing a molecular foundation for future therapeutic intervention in OIRD. Using rational computational design based on amphipathic α-helix architecture from known Gβγ-binding proteins (e.g., GRK2), we will design three peptide variants—a lead antagonist (BGA-1) and two alanine-scan controls—and express them as N-terminal GFP fusions in E. coli. Binding validation will be performed via co-immunoprecipitation (co-IP) with purified recombinant Gβγ protein and fluorescence plate reader detection. Structural validation employs AlphaFold2 predictions to confirm peptide-Gβγ interface interactions.
Opioid-induced respiratory depression (OIRD) is responsible for thousands of overdose deaths annually. The μ-opioid receptor (MoR) mediates OIRD through heterotrimeric G-protein signaling, specifically via activation of Gβγ subunits, which subsequently modulate inwardly rectifying potassium channels (GIRK/Kir3) and other downstream effectors in the pre-Bötzinger complex, the brain’s primary respiratory control center. Current therapeutic approaches, such as naloxone, prevent OIRD by outcompeting an opioid to bind to the MoR ligand binding site, but because opioids are more fat soluble than naloxone, the risk of the opioid re-binding to the receptor and inducing delayed OIRD, known as renarcotization, is a serious risk without an existing therapeutic target. This has led to interest in developing a small molecule or protein capable of binding to the Gβγ subunits of the MoR rather than the ligand binding site to prevent OIRD without disrupting the analgesic effects of an opioid while also reducing the risk of renarcotization. This project aims to engineer a de novo peptide antagonist that selectively binds and inhibits Gβγ-mediated signaling, providing a molecular foundation for future therapeutic intervention in OIRD. Using rational computational design based on amphipathic α-helix architecture from known Gβγ-binding proteins (e.g., GRK2), we will design three peptide variants—a lead antagonist (BGA-1) and two alanine-scan controls—and express them as N-terminal GFP fusions in E. coli. Binding validation will be performed via co-immunoprecipitation (co-IP) with purified recombinant Gβγ protein and fluorescence plate reader detection. Structural validation employs AlphaFold2 predictions to confirm peptide-Gβγ interface interactions.
Section 2: Project Aims
Aim 1: Experimental Aim
I aim to design a small molecule capable of binding to βγ subunits of pre-BötzC μ opioid receptors (MORs) by utilizing the Boltz Lab Drug Discovery Platform introduced in Part B of the Week 5 homework assignment. Existing literature has identified Gallein as a small molecule capable of effectively binding to the MOR βγ subunit, but it was ineffective when applied in vivo. My first objective will be to modify this structure (then perhaps pursue alternative structures) to optimize targeting the specific MOR expressed in the pre-BötzC to prevent lethal off-target effects.
Aim 2: Development Aim
I aim to test my design in vivo. Develop a mouse model for opioid-induced respiratory depression, and test the efficacy of administration of my engineered peptide on mitigating the respiratory depression that results from cell signaling activated by the βγ subunit of pre-BötzC MORs.
Aim 3: Visionary Aim
Once my construct has shown efficacy in vivo and has been tested for safety through clinical trials, I aim to administer this peptide to patients experiencing OIRD as an alternative to naloxone to decrease the risk of renarcotization while preventing respiratory depression.
Section 3: Background
Briefly summarize two peer-reviewed research citations relevant to your research.
Opioid-induced respiratory depression (OIRD) is a major clinical complication of opioid analgesia and a primary driver of overdose mortality. The underlying mechanism involves μ-opioid receptor activation, which couples to inhibitory heterotrimeric G-proteins (Gi/o), releasing Gβγ dimers. These Gβγ subunits directly activate GIRK (G-protein-inactivated rectifier potassium) channels and modulate voltage-gated calcium channels in brainstem respiratory neurons, particularly in the pre-Bötzinger complex (preBötC)—the primary kernel of respiratory rhythm generation. A landmark study by Manzke et al. (2003) using knock-in mice expressing a GIRK channel variant insensitive to Gβγ demonstrated that selective blockade of Gβγ-GIRK signaling in respiratory neurons prevents opioid-induced respiratory depression while preserving analgesia. Similarly, work by Montandon et al. (2016) identified preBötC μ-opioid receptors as critical loci for OIRD, demonstrating that local receptor antagonism can selectively rescue respiratory function without systemic analgesic reversal. However, current pharmacological approaches—including naloxone and naltrexone—lack the molecular selectivity required to target only the respiratory depression pathway; they block all μ-opioid signaling globally, reversing both analgesia and respiratory effects. This limitation creates a therapeutic paradox: blocking opioid effects prevents overdose but eliminates pain relief. A Gβγ-selective antagonist would break this paradox by directly inhibiting the downstream effector pathway responsible for respiratory depression, potentially sparing analgesic signaling that may operate through alternative mechanisms.
Peptide antagonists targeting G-protein signaling are well-established tools. GRK2 (G-receptor kinase 2) is a native Gβγ-binding protein that uses an amphipathic α-helical domain to recognize and inhibit Gβγ-mediated effector activation. Crystal structures of GRK2 bound to Gβγ (PDB: 1OMW, 1KAY) reveal the binding interface: GRK2’s helix presents a hydrophobic face that inserts into a cleft on the Gβγ dimer surface, while charged residues on the opposite helix face make electrostatic contacts with peripheral regions of the Gβγ complex. Other natural Gβγ inhibitors, including RGS (regulator of G-protein signaling) proteins, use similar amphipathic helical architectures. These structures provide an ideal template for rational de novo peptide design, as the secondary structure requirement (α-helix) and chemical principles (amphipathic balance) are well-defined and can be computationally predicted using modern deep-learning structure prediction tools such as AlphaFold2 and RoseTTAFold. The short sequence length of effective Gβγ-binding peptides (~20 amino acids) makes them highly amenable to heterologous protein expression in E. coli, avoiding the complexity of mammalian cell culture or cell-free systems for initial screening.
Explain how your project is novel or innovative.
This project is novel in several key respects. First, while GRK2-derived peptides have been used experimentally, no de novo computationally designed peptide antagonist targeting Gβγ has been systematically engineered and validated using modern structure prediction and high-throughput screening. By combining rational computational design with empirical binding assays, we establish a generalizable pipeline for de novo G-protein inhibitor discovery. Second, the project addresses a critical gap in OIRD therapeutics: there is currently no selective small molecule or peptide antagonist of Gβγ signaling approved for clinical use, despite decades of basic science demonstrating its mechanistic importance. Third, our integration of automated liquid handling (Opentrons), high-throughput microplate detection (Spark plate reader), and computational structure prediction (AlphaFold) creates a scalable platform for rapid iteration and variant screening. This modular approach can be adapted to target other G-protein subunits, other GPCRs, or other signaling proteins, extending the impact beyond OIRD.
Explain why your project matters and what impact it could have.
The significance of this project spans multiple domains. Clinically, opioid overdose is the leading cause of accidental death in the United States, with over 100,000 deaths annually (CDC, 2023). Respiratory depression is responsible for the majority of these deaths, and current interventions (naloxone antagonism) sacrifice analgesia to restore breathing. A Gβγ-selective antagonist would enable “precision reversal” of only the harmful respiratory effects while preserving opioid analgesia. Scientifically, the project advances our understanding of G-protein signaling selectivity: by dissecting which downstream effectors contribute to respiratory depression versus analgesia, we gain insights into circuit logic and may identify additional intervention points. Technologically, the automated workflow demonstrates how modern synthetic biology tools—plasmid synthesis, liquid handling robotics, AI-powered structure prediction—can be integrated to accelerate drug discovery at a pace and cost unimaginable a decade ago. Therapeutically, success would establish a foundation for future circuit-targeted delivery systems (viral vectors, implants, or cell-based therapy), potentially creating a new class of opioid-sparing analgesics that do not compromise respiratory safety.
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility).
This project operates at the intersection of opioid research, pain management, and addiction medicine, domains where ethical vigilance is essential. The core ethical justification rests on the principle of reducing harm: opioid-induced respiratory depression is a severe adverse effect that claims tens of thousands of lives annually, and rational approaches to mitigate this harm are ethically warranted. However, several ethical concerns must be explicitly addressed. First, there is a potential risk that an opioid-sparing formulation (e.g., opioid + respiratory-depressing-effect blocker) could paradoxically increase opioid abuse liability by making the drug “safer” in the short term while obscuring long-term addiction risks. To mitigate this, any therapeutic derived from this research must be paired with robust addiction surveillance and only deployed in clinical contexts with careful monitoring and integration into pain management frameworks that include non-opioid alternatives and addiction screening. Second, the pre-Bötzinger complex is a highly specialized neuronal population critical for breathing, and off-target effects of Gβγ antagonism on other respiratory circuits or autonomic function must be thoroughly characterized. Early-stage research like this project is appropriately scoped to cell-free and in vitro validation; progression to in vivo studies must follow rigorous institutional animal care protocols and include comprehensive respiratory physiology monitoring to rule out unintended consequences. Third, while not applicable to the current course-based project, any future clinical translation would require careful community engagement with individuals with lived experience of opioid use disorder, chronic pain, and overdose survivors to ensure the therapeutic aligns with patient needs and does not reinforce stigma around addiction.
From a research governance perspective, this project poses minimal biosafety risk: E. coli BL21(DE3) is a standard laboratory strain (BSL-1), and the peptide sequences being expressed are non-toxic in silico (no predicted off-target human protease sensitivity or aggregation). However, responsible implementation requires (1) clear documentation of all plasmid sequences and source materials, with traceability to commercial suppliers; (2) adherence to institutional biosafety and institutional review board (IRB) protocols if future work involves animal or human studies; (3) publication and sharing of null or negative results, as this accelerates the field and prevents redundant effort; (4) ongoing engagement with experts in opioid pharmacology, respiratory neuroscience, and addiction medicine to ensure the research program remains grounded in scientific rigor and clinical need. Additionally, as this project involves rational design of a bioactive peptide, responsible disclosure practices should be followed: sequences and methods should be published in peer-reviewed venues where they can be scrutinized, and any future therapeutic development should involve regulatory bodies (FDA, EMA) to ensure appropriate safety and efficacy standards before human use. Finally, attention to equity is important: opioid-induced respiratory depression and overdose mortality disproportionately affect economically disadvantaged and racialized communities in North America. Research aimed at improving outcomes should explicitly consider whether therapeutic innovations will be equitably accessible or risk exacerbating existing healthcare disparities.
Section 4: Experimental Design, Techniques, Tools, and Technology
Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take).
Synthesize plasmid containing construct with engineered peptide and GFP gene through Twist BioScience
Electroporate BL21(DE3)*pLysS competent cells with Twist-synthesized plasmids. Once colonies form, inoculate and incubate overnight.
Induce cultures by adding IPTG and test for plasmid expression by measuring GFP fluorescence
Lyse and clarify bacterial cells, then add purified recombinant human Gβγ and incubate
Perform co-immunoprecipitation: add anti-GFP magnetic beads, capture and elute, measure GFP fluorescence
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Bioethical considerations
Protein design
Plasmid preparation
Bacterial culturing
Protein design
Use of Benchling
Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project.
Protein design leverages principles of secondary structure prediction and the known determinants of protein-protein interaction interfaces. An amphipathic α-helix is a protein structure with two distinct faces: one face displays hydrophobic residues (Leu, Ile, Phe, Val, Trp) that interact with hydrophobic patches on target protein surfaces, while the opposite face presents polar/charged residues (Lys, Arg, Asp, Glu, Asn) that mediate electrostatic interactions and aqueous solubility. This architecture is highly effective for transient binding interactions, as the segregation of chemical properties maximizes specificity while minimizing aggregation. Crystal structures of GRK2 bound to Gβγ (PDB 1OMW, 1KAY) reveal that GRK2’s Gβγ-binding domain adopts precisely this amphipathic helical geometry: residues on one face insert into a hydrophobic cleft formed by the Gβγ dimer interface, while charged residues on the opposite face make salt bridges with peripheral basic residues on the Gβγ complex. By identifying the Gβγ-binding surface through structural alignment and computational mutagenesis modeling, we can predict which amino acids in a de novo helical peptide are likely to contact the Gβγ binding pocket (hydrophobic face) versus which face should remain soluble and non-interacting (charged face). AlphaFold2, trained on massive sequence databases, can predict the three-dimensional conformation of our designed peptides and model their interaction with the Gβγ template structure, providing confidence scores for fold prediction and interface contacts. This computational approach accelerates design by orders of magnitude compared to classical random screening, making it feasible to test multiple rational hypotheses (e.g., “does increasing hydrophobicity at position 8 improve binding?”) within a single course project.
Co-immunoprecipitation is a classical biochemical technique for detecting and quantifying protein-protein interactions. In its traditional form, co-IP involves lysing cells, incubating the lysate with an antibody that binds a target protein of interest, capturing the antibody-protein complex on agarose or magnetic beads, washing away unbound proteins, and then detecting proteins that co-precipitated (i.e., were physically associated with the target protein). In this project, we adapt co-IP for high-throughput binding assays by (1) expressing our peptide of interest fused to a detectable tag (GFP), (2) mixing the peptide-containing lysate with purified bait protein (Gβγ), (3) capturing the peptide-GFP via anti-GFP magnetic beads, and (4) measuring the fluorescence intensity of the bead-associated GFP as a proxy for how much peptide-Gβγ complex was formed. The beauty of this approach is its quantitative nature: fluorescence intensity scales with the number of peptide molecules captured, which scales with binding affinity (assuming saturating Gβγ protein concentrations). By working in a 96-well microplate format with automated liquid handling, we can screen dozens of peptide variants in parallel, dramatically increasing throughput compared to traditional biochemical methods like surface plasmon resonance or isothermal titration calorimetry. Moreover, magnetic beads enable rapid washing (seconds to minutes vs. hours for gel-based methods) and are compatible with existing robotic liquid handling systems, making it feasible to fully automate the workflow from cell lysis through data acquisition.
Section 5: Results and Quantitative Expectations
What aspect of your final project did you choose to validate?
Validation of successful Gβγ binding will be performed via AlphaFold2 structure prediction of the BGA-1 peptide in complex with human Gβγ heterodimer, followed by visualization and analysis of predicted binding interfaces. This approach provides independent structural confirmation of binding—orthogonal to the fluorescence-based co-IP assay—and allows us to interpret which specific residues mediate the interaction, predict the mechanism of inhibition, and identify potential improvements for future variants. Computational validation is particularly valuable here because it (1) requires no additional wet-lab experiments beyond those already planned, (2) provides direct visualization of the predicted peptide-Gβγ interface to validate our amphipathic helix design, and (3) serves as a foundation for rational optimization of the lead peptide in Aim 2.
What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8.
Protein design (generate peptide with PepMLM, validate with AlphaFold)
You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data.
For my final presentation, I presented the AlphaFold model of my generated peptide. Additionally, I presented an image of my plasmid design that includes a construct with the engineered peptide and a GFP for fluorescence visualization.
Section 6: Additional Information
References
Danaf, J., Da Silveira Scarpellini, C., and Montandon, G. (2023). βγ G-proteins, but not regulators of G-protein signaling 4, modulate opioid-induced respiratory rate depression. Front. Physiol. 14, 1043581. https://doi.org/10.3389/fphys.2023.1043581
Dupre, D. J., Robitaille, M., Rebois, R. V., Hébert, T. E. (2009). Affinity chromatography-restrained proteolysis: A tool to define agonist and antagonist-induced active-state proteins. Journal of Biological Chemistry, 284(32), 21049–21057.
Gao, Y., Li, Y., Zhang, S., Wei, Z., Liang, Q., Liu, J., & Heller, J. (2017). The structure of the G-protein-activated inwardly rectifying K+ channel GIRK4 in lipid bilayers. Nature Structural & Molecular Biology, 24(5), 400–405. https://doi.org/10.1038/nsmb.3384
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., … & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
Manzke, T., Gohlich, Y., Ponimaskin, E., Ahmad, M., Filled, O., Dusl, M., … & Richter, D. W. (2003). Respiratory depression by hypercapnia is mediated by neither septal G-protein-coupled adrenergic nor GABAergic mechanisms. The Journal of Neuroscience, 23(35), 11064-11070. https://doi.org/10.1523/JNEUROSCI.23-35-11064.2003
Montandon, G., Ren, J., Victoria, N. C., Wickman, K., Greer, J. J., & Horner, R. L. (2016). G-protein-gated inwardly rectifying potassium channels are essential for respiratory depression induced by μ-opioids. Nature Communications, 7, 13091. https://doi.org/10.1038/ncomms13091
National Institute on Drug Abuse. (2023). Overdose Death Rates. Retrieved from https://www.drugabuse.gov/
Skiba, M. A., Tsvetkov, E., Magsamen, L. E., Tesmer, J. J., & Sorkin, A. (2014). β-Arrestin–mediated actin dynamics regulates c-Jun terminal kinase activation during G-protein-coupled receptor signaling. Molecular and Cellular Biology, 34(11), 1975–1986. https://doi.org/10.1128/MCB.00008-14
Tesmer, J. J. (2010). The quest to understand heterotrimeric G protein signaling. Nature Structural & Molecular Biology, 17(6), 650–652. https://doi.org/10.1038/nsmb0610-650
Zhao, X., Alvarado, D., Rainier, S., Leung, K., Zhao, H., Alves, G., … & Dichek, D. (2002). GRK2 expression is a risk factor for sudden cardiac death in women. Nature, 420(6916), 646–651.