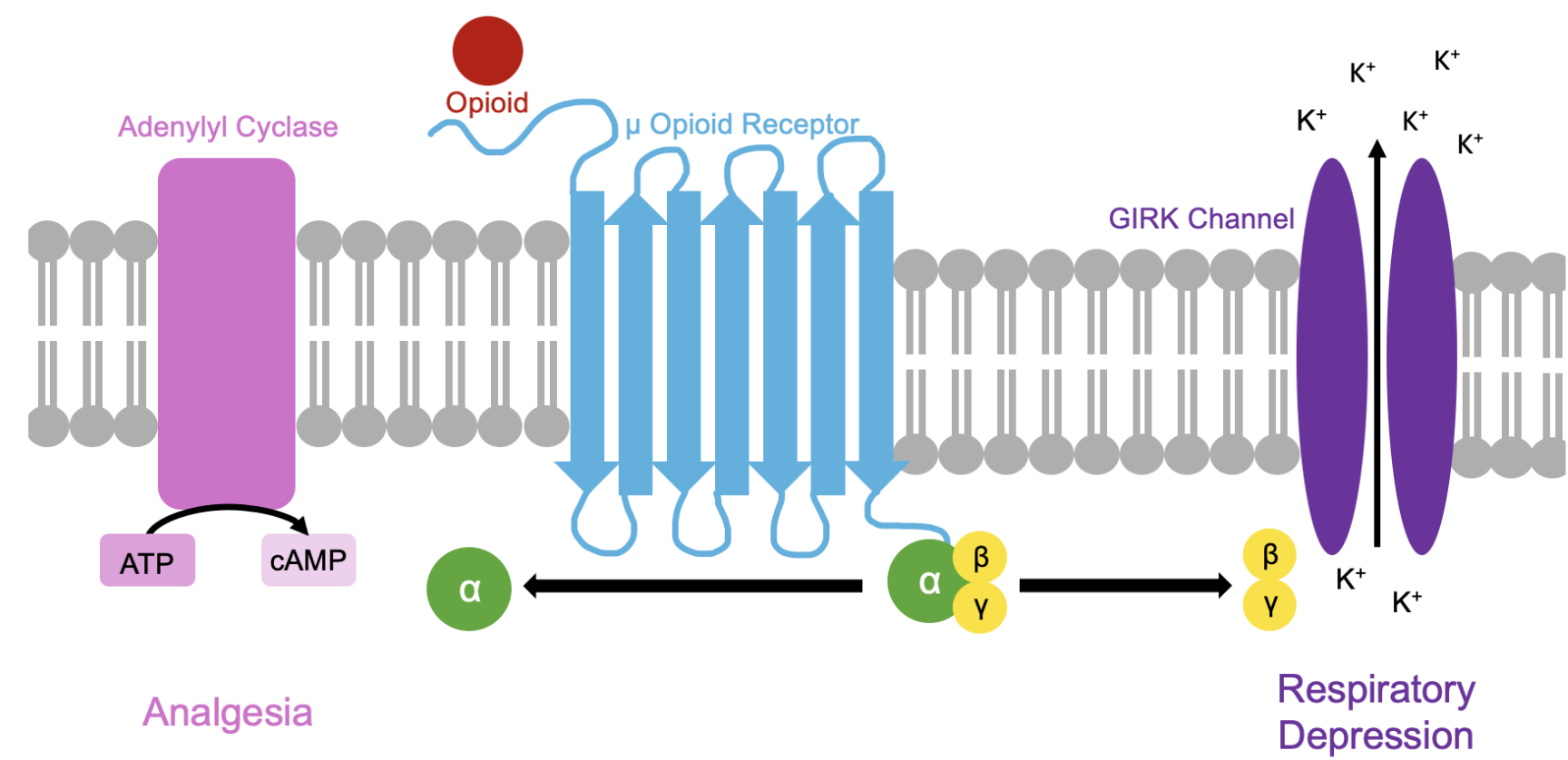
Section 1: Abstract Opioid-induced respiratory depression (OIRD) is responsible for thousands of overdose deaths annually. The μ-opioid receptor (MoR) mediates OIRD through heterotrimeric G-protein signaling, specifically via activation of Gβγ subunits, which subsequently modulate inwardly rectifying potassium channels (GIRK/Kir3) and other downstream effectors in the pre-Bötzinger complex, the brain’s primary respiratory control center. Current therapeutic approaches, such as naloxone, prevent OIRD by outcompeting an opioid to bind to the MoR ligand binding site, but because opioids are more fat soluble than naloxone, the risk of the opioid re-binding to the receptor and inducing delayed OIRD, known as renarcotization, is a serious risk without an existing therapeutic target. This has led to interest in developing a small molecule or protein capable of binding to the Gβγ subunits of the MoR rather than the ligand binding site to prevent OIRD without disrupting the analgesic effects of an opioid while also reducing the risk of renarcotization. This project aims to engineer a de novo peptide antagonist that selectively binds and inhibits Gβγ-mediated signaling, providing a molecular foundation for future therapeutic intervention in OIRD. Using rational computational design based on amphipathic α-helix architecture from known Gβγ-binding proteins (e.g., GRK2), we will design three peptide variants—a lead antagonist (BGA-1) and two alanine-scan controls—and express them as N-terminal GFP fusions in E. coli. Binding validation will be performed via co-immunoprecipitation (co-IP) with purified recombinant Gβγ protein and fluorescence plate reader detection. Structural validation employs AlphaFold2 predictions to confirm peptide-Gβγ interface interactions.
Opioid-induced respiratory depression (OIRD) is responsible for thousands of overdose deaths annually. The μ-opioid receptor (MoR) mediates OIRD through heterotrimeric G-protein signaling, specifically via activation of Gβγ subunits, which subsequently modulate inwardly rectifying potassium channels (GIRK/Kir3) and other downstream effectors in the pre-Bötzinger complex, the brain’s primary respiratory control center. Current therapeutic approaches, such as naloxone, prevent OIRD by outcompeting an opioid to bind to the MoR ligand binding site, but because opioids are more fat soluble than naloxone, the risk of the opioid re-binding to the receptor and inducing delayed OIRD, known as renarcotization, is a serious risk without an existing therapeutic target. This has led to interest in developing a small molecule or protein capable of binding to the Gβγ subunits of the MoR rather than the ligand binding site to prevent OIRD without disrupting the analgesic effects of an opioid while also reducing the risk of renarcotization. This project aims to engineer a de novo peptide antagonist that selectively binds and inhibits Gβγ-mediated signaling, providing a molecular foundation for future therapeutic intervention in OIRD. Using rational computational design based on amphipathic α-helix architecture from known Gβγ-binding proteins (e.g., GRK2), we will design three peptide variants—a lead antagonist (BGA-1) and two alanine-scan controls—and express them as N-terminal GFP fusions in E. coli. Binding validation will be performed via co-immunoprecipitation (co-IP) with purified recombinant Gβγ protein and fluorescence plate reader detection. Structural validation employs AlphaFold2 predictions to confirm peptide-Gβγ interface interactions.
Section 2: Project Aims
Aim 1: Experimental Aim
I aim to design a small molecule capable of binding to βγ subunits of pre-BötzC μ opioid receptors (MORs) by utilizing the Boltz Lab Drug Discovery Platform introduced in Part B of the Week 5 homework assignment. Existing literature has identified Gallein as a small molecule capable of effectively binding to the MOR βγ subunit, but it was ineffective when applied in vivo. My first objective will be to modify this structure (then perhaps pursue alternative structures) to optimize targeting the specific MOR expressed in the pre-BötzC to prevent lethal off-target effects.
Aim 2: Development Aim
I aim to test my design in vivo. Develop a mouse model for opioid-induced respiratory depression, and test the efficacy of administration of my engineered peptide on mitigating the respiratory depression that results from cell signaling activated by the βγ subunit of pre-BötzC MORs.
Aim 3: Visionary Aim
Once my construct has shown efficacy in vivo and has been tested for safety through clinical trials, I aim to administer this peptide to patients experiencing OIRD as an alternative to naloxone to decrease the risk of renarcotization while preventing respiratory depression.
Section 3: Background
Briefly summarize two peer-reviewed research citations relevant to your research.
Opioid-induced respiratory depression (OIRD) is a major clinical complication of opioid analgesia and a primary driver of overdose mortality. The underlying mechanism involves μ-opioid receptor activation, which couples to inhibitory heterotrimeric G-proteins (Gi/o), releasing Gβγ dimers. These Gβγ subunits directly activate GIRK (G-protein-inactivated rectifier potassium) channels and modulate voltage-gated calcium channels in brainstem respiratory neurons, particularly in the pre-Bötzinger complex (preBötC)—the primary kernel of respiratory rhythm generation. A landmark study by Manzke et al. (2003) using knock-in mice expressing a GIRK channel variant insensitive to Gβγ demonstrated that selective blockade of Gβγ-GIRK signaling in respiratory neurons prevents opioid-induced respiratory depression while preserving analgesia. Similarly, work by Montandon et al. (2016) identified preBötC μ-opioid receptors as critical loci for OIRD, demonstrating that local receptor antagonism can selectively rescue respiratory function without systemic analgesic reversal. However, current pharmacological approaches—including naloxone and naltrexone—lack the molecular selectivity required to target only the respiratory depression pathway; they block all μ-opioid signaling globally, reversing both analgesia and respiratory effects. This limitation creates a therapeutic paradox: blocking opioid effects prevents overdose but eliminates pain relief. A Gβγ-selective antagonist would break this paradox by directly inhibiting the downstream effector pathway responsible for respiratory depression, potentially sparing analgesic signaling that may operate through alternative mechanisms.
Peptide antagonists targeting G-protein signaling are well-established tools. GRK2 (G-receptor kinase 2) is a native Gβγ-binding protein that uses an amphipathic α-helical domain to recognize and inhibit Gβγ-mediated effector activation. Crystal structures of GRK2 bound to Gβγ (PDB: 1OMW, 1KAY) reveal the binding interface: GRK2’s helix presents a hydrophobic face that inserts into a cleft on the Gβγ dimer surface, while charged residues on the opposite helix face make electrostatic contacts with peripheral regions of the Gβγ complex. Other natural Gβγ inhibitors, including RGS (regulator of G-protein signaling) proteins, use similar amphipathic helical architectures. These structures provide an ideal template for rational de novo peptide design, as the secondary structure requirement (α-helix) and chemical principles (amphipathic balance) are well-defined and can be computationally predicted using modern deep-learning structure prediction tools such as AlphaFold2 and RoseTTAFold. The short sequence length of effective Gβγ-binding peptides (~20 amino acids) makes them highly amenable to heterologous protein expression in E. coli, avoiding the complexity of mammalian cell culture or cell-free systems for initial screening.
Explain how your project is novel or innovative.
This project is novel in several key respects. First, while GRK2-derived peptides have been used experimentally, no de novo computationally designed peptide antagonist targeting Gβγ has been systematically engineered and validated using modern structure prediction and high-throughput screening. By combining rational computational design with empirical binding assays, we establish a generalizable pipeline for de novo G-protein inhibitor discovery. Second, the project addresses a critical gap in OIRD therapeutics: there is currently no selective small molecule or peptide antagonist of Gβγ signaling approved for clinical use, despite decades of basic science demonstrating its mechanistic importance. Third, our integration of automated liquid handling (Opentrons), high-throughput microplate detection (Spark plate reader), and computational structure prediction (AlphaFold) creates a scalable platform for rapid iteration and variant screening. This modular approach can be adapted to target other G-protein subunits, other GPCRs, or other signaling proteins, extending the impact beyond OIRD.
Explain why your project matters and what impact it could have.
The significance of this project spans multiple domains. Clinically, opioid overdose is the leading cause of accidental death in the United States, with over 100,000 deaths annually (CDC, 2023). Respiratory depression is responsible for the majority of these deaths, and current interventions (naloxone antagonism) sacrifice analgesia to restore breathing. A Gβγ-selective antagonist would enable “precision reversal” of only the harmful respiratory effects while preserving opioid analgesia. Scientifically, the project advances our understanding of G-protein signaling selectivity: by dissecting which downstream effectors contribute to respiratory depression versus analgesia, we gain insights into circuit logic and may identify additional intervention points. Technologically, the automated workflow demonstrates how modern synthetic biology tools—plasmid synthesis, liquid handling robotics, AI-powered structure prediction—can be integrated to accelerate drug discovery at a pace and cost unimaginable a decade ago. Therapeutically, success would establish a foundation for future circuit-targeted delivery systems (viral vectors, implants, or cell-based therapy), potentially creating a new class of opioid-sparing analgesics that do not compromise respiratory safety.
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility).
This project operates at the intersection of opioid research, pain management, and addiction medicine, domains where ethical vigilance is essential. The core ethical justification rests on the principle of reducing harm: opioid-induced respiratory depression is a severe adverse effect that claims tens of thousands of lives annually, and rational approaches to mitigate this harm are ethically warranted. However, several ethical concerns must be explicitly addressed. First, there is a potential risk that an opioid-sparing formulation (e.g., opioid + respiratory-depressing-effect blocker) could paradoxically increase opioid abuse liability by making the drug “safer” in the short term while obscuring long-term addiction risks. To mitigate this, any therapeutic derived from this research must be paired with robust addiction surveillance and only deployed in clinical contexts with careful monitoring and integration into pain management frameworks that include non-opioid alternatives and addiction screening. Second, the pre-Bötzinger complex is a highly specialized neuronal population critical for breathing, and off-target effects of Gβγ antagonism on other respiratory circuits or autonomic function must be thoroughly characterized. Early-stage research like this project is appropriately scoped to cell-free and in vitro validation; progression to in vivo studies must follow rigorous institutional animal care protocols and include comprehensive respiratory physiology monitoring to rule out unintended consequences. Third, while not applicable to the current course-based project, any future clinical translation would require careful community engagement with individuals with lived experience of opioid use disorder, chronic pain, and overdose survivors to ensure the therapeutic aligns with patient needs and does not reinforce stigma around addiction.
From a research governance perspective, this project poses minimal biosafety risk: E. coli BL21(DE3) is a standard laboratory strain (BSL-1), and the peptide sequences being expressed are non-toxic in silico (no predicted off-target human protease sensitivity or aggregation). However, responsible implementation requires (1) clear documentation of all plasmid sequences and source materials, with traceability to commercial suppliers; (2) adherence to institutional biosafety and institutional review board (IRB) protocols if future work involves animal or human studies; (3) publication and sharing of null or negative results, as this accelerates the field and prevents redundant effort; (4) ongoing engagement with experts in opioid pharmacology, respiratory neuroscience, and addiction medicine to ensure the research program remains grounded in scientific rigor and clinical need. Additionally, as this project involves rational design of a bioactive peptide, responsible disclosure practices should be followed: sequences and methods should be published in peer-reviewed venues where they can be scrutinized, and any future therapeutic development should involve regulatory bodies (FDA, EMA) to ensure appropriate safety and efficacy standards before human use. Finally, attention to equity is important: opioid-induced respiratory depression and overdose mortality disproportionately affect economically disadvantaged and racialized communities in North America. Research aimed at improving outcomes should explicitly consider whether therapeutic innovations will be equitably accessible or risk exacerbating existing healthcare disparities.
Section 4: Experimental Design, Techniques, Tools, and Technology
Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take).
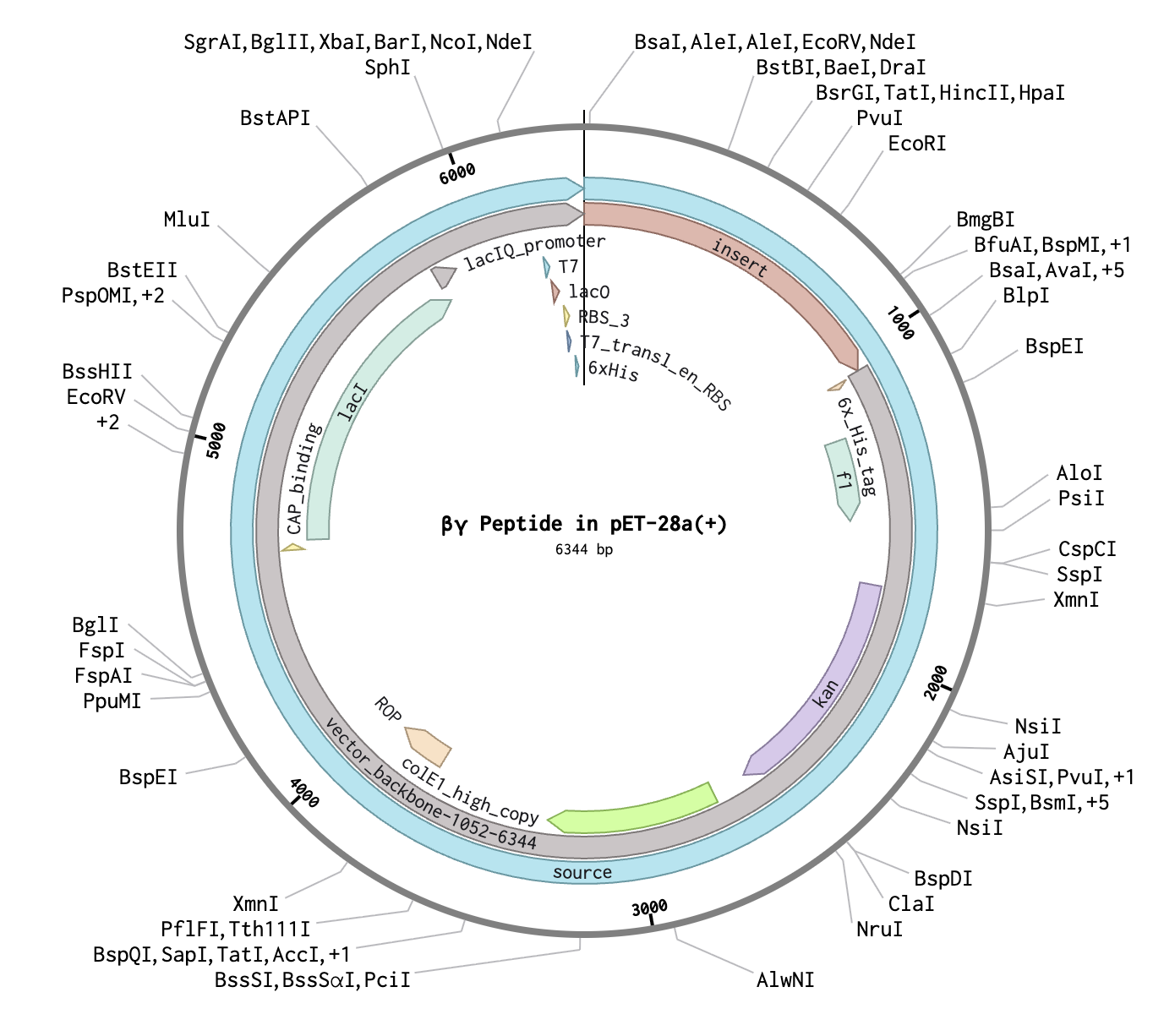
Synthesize plasmid containing construct with engineered peptide and GFP gene through Twist BioScience
Electroporate BL21(DE3)*pLysS competent cells with Twist-synthesized plasmids. Once colonies form, inoculate and incubate overnight.
Induce cultures by adding IPTG and test for plasmid expression by measuring GFP fluorescence
Lyse and clarify bacterial cells, then add purified recombinant human Gβγ and incubate
Perform co-immunoprecipitation: add anti-GFP magnetic beads, capture and elute, measure GFP fluorescence
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Bioethical considerations
Protein design
Plasmid preparation
Bacterial culturing
Protein design
Use of Benchling
Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project.
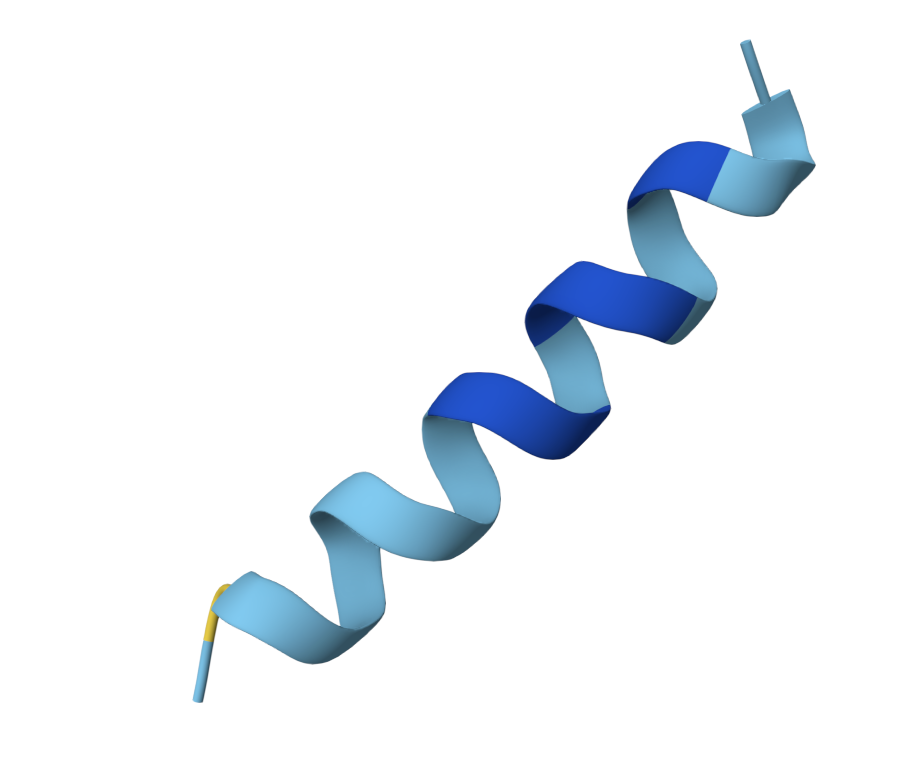
Protein design leverages principles of secondary structure prediction and the known determinants of protein-protein interaction interfaces. An amphipathic α-helix is a protein structure with two distinct faces: one face displays hydrophobic residues (Leu, Ile, Phe, Val, Trp) that interact with hydrophobic patches on target protein surfaces, while the opposite face presents polar/charged residues (Lys, Arg, Asp, Glu, Asn) that mediate electrostatic interactions and aqueous solubility. This architecture is highly effective for transient binding interactions, as the segregation of chemical properties maximizes specificity while minimizing aggregation. Crystal structures of GRK2 bound to Gβγ (PDB 1OMW, 1KAY) reveal that GRK2’s Gβγ-binding domain adopts precisely this amphipathic helical geometry: residues on one face insert into a hydrophobic cleft formed by the Gβγ dimer interface, while charged residues on the opposite face make salt bridges with peripheral basic residues on the Gβγ complex. By identifying the Gβγ-binding surface through structural alignment and computational mutagenesis modeling, we can predict which amino acids in a de novo helical peptide are likely to contact the Gβγ binding pocket (hydrophobic face) versus which face should remain soluble and non-interacting (charged face). AlphaFold2, trained on massive sequence databases, can predict the three-dimensional conformation of our designed peptides and model their interaction with the Gβγ template structure, providing confidence scores for fold prediction and interface contacts. This computational approach accelerates design by orders of magnitude compared to classical random screening, making it feasible to test multiple rational hypotheses (e.g., “does increasing hydrophobicity at position 8 improve binding?”) within a single course project.
Co-immunoprecipitation is a classical biochemical technique for detecting and quantifying protein-protein interactions. In its traditional form, co-IP involves lysing cells, incubating the lysate with an antibody that binds a target protein of interest, capturing the antibody-protein complex on agarose or magnetic beads, washing away unbound proteins, and then detecting proteins that co-precipitated (i.e., were physically associated with the target protein). In this project, we adapt co-IP for high-throughput binding assays by (1) expressing our peptide of interest fused to a detectable tag (GFP), (2) mixing the peptide-containing lysate with purified bait protein (Gβγ), (3) capturing the peptide-GFP via anti-GFP magnetic beads, and (4) measuring the fluorescence intensity of the bead-associated GFP as a proxy for how much peptide-Gβγ complex was formed. The beauty of this approach is its quantitative nature: fluorescence intensity scales with the number of peptide molecules captured, which scales with binding affinity (assuming saturating Gβγ protein concentrations). By working in a 96-well microplate format with automated liquid handling, we can screen dozens of peptide variants in parallel, dramatically increasing throughput compared to traditional biochemical methods like surface plasmon resonance or isothermal titration calorimetry. Moreover, magnetic beads enable rapid washing (seconds to minutes vs. hours for gel-based methods) and are compatible with existing robotic liquid handling systems, making it feasible to fully automate the workflow from cell lysis through data acquisition.
Section 5: Results and Quantitative Expectations
What aspect of your final project did you choose to validate?
Validation of successful Gβγ binding will be performed via AlphaFold2 structure prediction of the BGA-1 peptide in complex with human Gβγ heterodimer, followed by visualization and analysis of predicted binding interfaces. This approach provides independent structural confirmation of binding—orthogonal to the fluorescence-based co-IP assay—and allows us to interpret which specific residues mediate the interaction, predict the mechanism of inhibition, and identify potential improvements for future variants. Computational validation is particularly valuable here because it (1) requires no additional wet-lab experiments beyond those already planned, (2) provides direct visualization of the predicted peptide-Gβγ interface to validate our amphipathic helix design, and (3) serves as a foundation for rational optimization of the lead peptide in Aim 2.
What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8.
Protein design (generate peptide with PepMLM, validate with AlphaFold)
You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data.
For my final presentation, I presented the AlphaFold model of my generated peptide. Additionally, I presented an image of my plasmid design that includes a construct with the engineered peptide and a GFP for fluorescence visualization.
Section 6: Additional Information
References
Danaf, J., Da Silveira Scarpellini, C., and Montandon, G. (2023). βγ G-proteins, but not regulators of G-protein signaling 4, modulate opioid-induced respiratory rate depression. Front. Physiol. 14, 1043581. https://doi.org/10.3389/fphys.2023.1043581
Dupre, D. J., Robitaille, M., Rebois, R. V., Hébert, T. E. (2009). Affinity chromatography-restrained proteolysis: A tool to define agonist and antagonist-induced active-state proteins. Journal of Biological Chemistry, 284(32), 21049–21057.
Gao, Y., Li, Y., Zhang, S., Wei, Z., Liang, Q., Liu, J., & Heller, J. (2017). The structure of the G-protein-activated inwardly rectifying K+ channel GIRK4 in lipid bilayers. Nature Structural & Molecular Biology, 24(5), 400–405. https://doi.org/10.1038/nsmb.3384
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., … & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2
Manzke, T., Gohlich, Y., Ponimaskin, E., Ahmad, M., Filled, O., Dusl, M., … & Richter, D. W. (2003). Respiratory depression by hypercapnia is mediated by neither septal G-protein-coupled adrenergic nor GABAergic mechanisms. The Journal of Neuroscience, 23(35), 11064-11070. https://doi.org/10.1523/JNEUROSCI.23-35-11064.2003
Montandon, G., Ren, J., Victoria, N. C., Wickman, K., Greer, J. J., & Horner, R. L. (2016). G-protein-gated inwardly rectifying potassium channels are essential for respiratory depression induced by μ-opioids. Nature Communications, 7, 13091. https://doi.org/10.1038/ncomms13091
National Institute on Drug Abuse. (2023). Overdose Death Rates. Retrieved from https://www.drugabuse.gov/
Skiba, M. A., Tsvetkov, E., Magsamen, L. E., Tesmer, J. J., & Sorkin, A. (2014). β-Arrestin–mediated actin dynamics regulates c-Jun terminal kinase activation during G-protein-coupled receptor signaling. Molecular and Cellular Biology, 34(11), 1975–1986. https://doi.org/10.1128/MCB.00008-14
Tesmer, J. J. (2010). The quest to understand heterotrimeric G protein signaling. Nature Structural & Molecular Biology, 17(6), 650–652. https://doi.org/10.1038/nsmb0610-650
Zhao, X., Alvarado, D., Rainier, S., Leung, K., Zhao, H., Alves, G., … & Dichek, D. (2002). GRK2 expression is a risk factor for sudden cardiac death in women. Nature, 420(6916), 646–651.