Biological Engineering Application & Research Proposal Agnostic Biosurveillance in the Age of Generative AI Important HTGAA | APPLICATION Describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
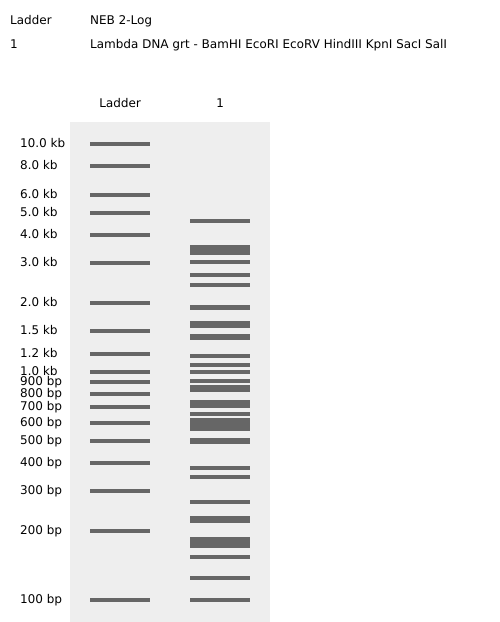
Part 1: Benchling & In-silico Gel Art Simulación de restricción enzimática mediante Benchling
1.1 Enzimas utilizadas para la simulación EcoRI HindIII BamHI KpnI EcoRV SacI SalI Part 3: DNA Design Challenge 3.1 Choose Your Protein Protein: Envelope small membrane protein (Gene: E) Organism: Severe acute respiratory syndrome coronavirus 2 (2019-nCoV) (SARS-CoV-2) Metadata: Length: 75 amino acids Mass: 8,365 Da Amino Acid Sequence: MYSFVSEETGTLIVNSVLLFLAFVVFLLVTLAILTALRLCAYCCNIVNVSLVKPSFYVYSRVKNLNSSRVPDLLV
Important HTGAA | Question Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Workflow for the testing of SARS-CoV-2 using Opentrons OT-2 robots
Villanueva-Cañas et al. (PLOS ONE, 2021) describe the setup of a full SARS-CoV-2 RT-qPCR testing workflow using Opentrons OT-2 robots. This allowed them to automate what would otherwise be manual pipetting. Instead, they automated the critical steps and connected them like a lab “production line”: multiple OT-2 units to prepare plates and reagents, a system to extract RNA, and then a thermocycler to run the PCR.
Important HTGAA | Part A: Protein Analysis and Visualization Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:.
Briefly describe the protein you selected and why you selected it
How long is it? What is the most frequent amino acid?.
Important HTGAA | Part A: SOD1 Binder Peptide Design (From Dr Pranam Chatterjee) Retrieve the human SOD1 sequence (P00441) from UniProt, introduce the A4V mutation, use the PepMLM Colab to generate four 12–amino acid peptides conditioned on the mutant sequence, include the known binder FLYRWLPSRRGG for comparison, and record the resulting perplexity scores to assess model confidence.
Subsections of Homework
Week 1 HW: Principles and Practices
Biological Engineering Application & Research Proposal
Agnostic Biosurveillance in the Age of Generative AI
Important
HTGAA | APPLICATION
Describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
The Landscape of Risk
As AI capabilities grow, concerns about biosecurity risks posed by frontier large language models (LLMs) have grown significantly. Current reports based on surveys of experts in biosecurity and biology, indicate that if AI reaches certain performance levels, the annual risk of a human-caused epidemic (with more than 100,000 deaths) could increase from 0.3% to 1.5%.
Given this landscape, my proposal is to develop a detection system capable of identifying any genetically modified organism, even if it is completely new and does not exist in any database.
Focus on how it was made, not on what it is
As I understand, the problem with current biosecurity is that it functions reactively, similar to a computer “antivirus” that only recognizes threats it has seen before. If someone creates a new pathogen from scratch using AI, our current tools might not detect it because they would not recognize the specific sequence.
My proposal, instead does not focus on the identity of the organism but on its creation process.
The “Human Accent” Hypothesis
The central idea is that human-manufactured DNA possesses an “accent” or statistical pattern distinct from that of natural DNA. This occurs because humans use specific tools to design, optimize, and assemble genetic code. My idea proposes to use AI models (specifically genomic language models) to learn this “human accent” systematically.
Mechanism and Vision
Once the AI identifies an artificially designed sequence, it would be coupled with an enhanced CRISPR-Cas13 system to trigger a biological alarm. Ultimately, I seek to create a tool that makes any genetic engineering intervention “visible,” ensuring that we can anticipate and mitigate the growing risks posed by generative AI applied to biology.
References of Interest
Linder, J., et al. (2024) | Nature Machine Intelligence Validates the use of AI architectures to identify statistical biases in synthetic DNA.
Gootenberg, J. S., et al. (2023) | Science Establishes the technical viability of CRISPR-Cas13 sensors for PCR-free biodefense applications.
Zhou et al. (2024) | Eliyon Confirms that CRISPR-Cas13a systems achieve exceptional sensitivity with the capacity for single-copy detection.
Forecasting LLM-enabled biorisk and the efficacy of safeguards (2025)Forecast Research Institute.
Important
HTGAA | ETHICS & GOVERNANCE
Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.**
Goal 1: Preventing Misuse of the System (Security)
Since this tool can have a wide reach, so would its implications; therefore, we must prevent anyone from using it to learn how to manufacture threats that “deceive” detection.
Controlled Access (API): Do not release the AI code openly. Instead, allow it to be used under supervision so that no one can use it to “train” new invisible pathogens.
Attack Simulations (Red-Teaming): Assemble a panel of experts to constantly try to break or deceive the system. In this way, the goal is to stay ahead of those who wish to use AI to cause harm.
Goal 2: Fair Access for Everyone (Equity)
Biological security should not be a luxury for rich countries; it must protect everyone.
Accessible CRISPR Technology: Although the AI remains private for security reasons, the instructions to manufacture the biological sensor (CRISPR) must be free and low-cost so that any country can use them.
Solidarity-based Global Alert: Create a network where, if a country detects something unusual, it can warn the rest of the world efficiently.
Goal 3: Protecting Privacy and Legitimate Science (Ethics)
The system must be capable of distinguishing between a real threat and a medical breakthrough or a natural mutation.
Clear Boundaries: Create rules that separate what is a dangerous pathogen from what is medicine (such as gene therapy), in order to avoid slowing down science that actually helps people.
Human Oversight: Design safeguards and the corresponding alignment so that the AI does not make extreme decisions on its own. If the system detects something, a group of experts must review the situation before launching a panic-inducing alarm.
Additional References
International AI Safety Report (2026) | A Comprehensive review of latest scientific research on the capabilities and risks of general-purpose AI systems.
Important
HTGAA | STRATEGIC ACTIONS
Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)**
Action 1: An International “Alert System”
Technical Strategy
Purpose: Currently, threat detection is isolated and slow. This proposal could eventually encourage the creation of a global network of shared sensors that use AI to report genetic anomalies in real-time.
Design: Academic researchers and hospitals install low-cost CRISPR biosensors. Data is sent to a centralized platform (similar to the financial system that detects bank fraud).
Assumptions: We assume that countries will be willing to share health data for the common good.
Risks: If the system is “successful,” we could have too many alerts (false positives). If it fails, a real threat could go unnoticed if sensors are not well distributed geographically.
Action 2: “Black Box” Licenses for AI
Rule / Requirement
Purpose: To prevent the AI that detects the “human accent” from being used by criminals to learn how to erase that trace.
Design: Restrictions must be established so that technology companies do not release the model’s source code openly. Only verified users (governments and certified scientists) will be able to access the tool using special keys.
Assumptions: We believe we can maintain technological secrecy and that no one will manage to “hack” or replicate the AI privately.
Risks: The “success” of this rule could slow down scientific innovation if researchers cannot freely access the tool. Failure would be the emergence of a black market for “unrestricted” versions of this AI.
Action 3: “Clean DNA” Certification
Incentive
Purpose: Today, DNA synthesis companies cannot always detect if an order is dangerous if it is a new design.
Design: DNA synthesis companies receive a certification or security seal if they run their orders through this AI filter before sending them to the customer. A regulatory entity validates that these companies comply with the standard.
Assumptions: We assume that companies will seek certification and that customers will not go to illegal providers.
Risks: If successful, designing biopathogens becomes almost impossible to print legally. A risk of success is that biotechnology becomes slower or more expensive. A failure would be that malicious actors simply buy their own “DNA printers” to bypass control.
Important
HTGAA | SCORES
Score each of your governance actions against your rubric of policy goals
🧭 POLICY GOVERNANCE | EVALUATION MATRIX
STRATEGIC ACTION
SECURITY 🛡️
EQUITY 🌍
EFFICACY ⚡
STRATEGIC NOTES
01. Global Alert System
● ● ●
● ● ●
● ● ○
Best for universal protection. Requires heavy coordination.
02. Black Box Licenses
● ● ●
● ○ ○
● ○ ○
High security. Risks excluding Global South & slowing science.
03. Clean DNA Certification
● ● ○
● ● ○
● ● ●
High industry flow. Relies on voluntary company adoption.
Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.**
Strategic Priority: Academic-Industry Alliance
I recommend that MIT lead a joint task force with the International Gene Synthesis Consortium (IGSC) to implement a combination of Action 1 (Alert System) and Action 3 (Clean DNA Certification).
Why this combination? This alliance allows MIT’s cutting-edge research to become an industrial standard. While MIT provides the AI to detect the “human accent,” IGSC companies act as the first global filter, ensuring that no suspicious sequence is synthesized without oversight. This maximizes Security and Efficacy without relying solely on slow-moving government regulations.
Trade-offs
Security vs. Industrial Speed: Implementing an AI filter on every DNA order might make synthesis slightly slower. However, the cost of not doing so (an artificial pandemic) is much higher.
Transparency vs. Intellectual Property: Companies must allow an external AI to analyze their orders. To protect their trade secrets, this proposal points toward a system where the AI processes data in an encrypted manner (Privacy-Preserving AI).
Assumptions and Uncertainties
Universal Adoption: We assume that companies outside the consortium will not offer “no-questions-asked” services to attract malicious actors.
False Positives: There is uncertainty regarding whether the AI will halt legitimate synthetic biology experiments, which would require a fast and expert human review process.
Note
Ethical Reflections & New Actions
Regarding key concerns about the scientist’s responsibility:
The Duality of Detection: The same system that detects engineering could be used to teach others how to hide it.
Proposed Action: Establish a “Black Box” protocol where the source code for detection is only accessible to certified biosecurity auditors.
Technological Justice: We do not want this technology to create a gap where only MIT and large companies are protected.
Proposed Action: Create an Open Biosurveillance fund to donate low-cost CRISPR sensors to laboratories in countries of the Global South.
Conclusion for MIT and the IGSC
Biological security in the age of AI should not be solved by a single institution. By combining MIT’s intelligence with the IGSC’s infrastructure, the proposal aims to create an ecosystem where genetic engineering could be visible and accountable.
🧬 Q&A: Professor Jacobson
Polymerase Error Rate
10-4 to 10-5 (1 error per 10,000–100,000 bases).
Genome Comparison
With 3.2 x 10^9 bp, lack of repair would cause ~100,000 mutations per division, making life impossible.
Biological Solution
A three-tier system (Selectivity – Proofreading – Mismatch Repair) reduces error to < 1 mutation per genome.
Coding Possibilities
~400 amino acids can be encoded by 10^191 different DNA sequences.
Functional Constraints
Not all codes work due to:
tRNA availability
Folding kinetics
mRNA stability
Splicing signals
Reference: Alberts et al. (2022). Molecular Biology of the Cell.
Q&A: Professor LeProust
Oligo Synthesis: Methods and Limits
1. Most Common Method
The standard method is Phosphoramidite synthesis. It is a chemical, solid-phase process that adds nucleotides one by one to a growing chain.
2. Why is it hard to exceed 200nt?
It is a matter of cumulative yield. Even with a 99% efficiency per step, errors add up exponentially:
At 100nt, ~36% of the final molecules are correct.
At 200nt, only ~13% are correct.
Beyond 200nt, the mixture contains too many “short” or damaged sequences, making it nearly impossible to isolate the right one.
3. Why not a 2,000bp gene directly?
Zero Yield: At 2,000 bases, the math (0.99^2000) results in essentially zero correct molecules.
DNA Damage: The chemicals used in each cycle eventually degrade the DNA chain before it can reach that length.
As a solution, to make a 2,000bp gene, companies synthesize multiple small 60-100nt fragments (oligos) and then “stitch” them together using biological methods like Assembly PCR or Gibson Assembly.
Q&A: Professor Church
The 10 dietary essential amino acids are:
The 10 dietary essential amino acids are:
Arg, His, Ile, Leu, Lys, Met, Phe, Thr, Trp, Val
The “lysine contingency” from Jurassic Park doesn’t work well because animals already can’t make lysine on their own. They normally get it from food. So if one escaped, it could still find lysine in nature, which makes this a weak safety control.
Week 2 — DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
Simulación de restricción enzimática mediante Benchling
1.1 Enzimas utilizadas para la simulación
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Part 3: DNA Design Challenge
3.1 Choose Your Protein
Protein: Envelope small membrane protein (Gene: E)
Organism: Severe acute respiratory syndrome coronavirus 2 (2019-nCoV) (SARS-CoV-2)
I am interested in the SARS-CoV-2 Envelope (E) protein from a biosurveillance standpoint. This protein is essential for viral assembly and release; consequently, it cannot undergo significant mutations without halting these vital functions.
Given this, primers for the E gene should tend to be more stable over time, unlike those for the Spike (S) protein, which constantly mutates to evade vaccines and immune responses. Therefore, the E protein can be considered a more reliable and “secure” target for viral detection and monitoring.
3.2 Reverse Translation
Conversion of Protein (amino acid) sequence to DNA (nucleotide) sequence.
Description: Reverse translation of sample sequence to a 226-base sequence of most likely codons.
Codon optimization is basically a synthetic biology hack to get the most out of gene expression in a specific organism.
Since the genetic code is degenerate, different species have their own “favorite” codons to code for the same amino acid.
This technique tweaks the genetic sequence to match the tRNA availability of the host, clearing out any bottlenecks during translation.
By swapping “rare” codons for “frequent” ones, we keep the ribosome from stalling, ensuring we get a steady and efficient protein production.
3.4. You have a sequence! Now what?
Question: What technologies could be used to produce this protein from your DNA? Describe in your words how the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
To produce this Protein E, we can use different technologies. For example, bacterial expression systems, mammalian cell cultures, or insect cell systems (infected with baculoviruses). Cell-free protein synthesis technologies also exist.
For the case of Protein E, I have decided to use bacterial systems technology, as it is the fastest and lowest in cost. Although it does not include post-translational modifications (PTMs), for the objectives of this production, this does not represent an issue.
Workflow for Protein E Production: To produce my Protein E, I need the following workflow. I will use E. coli for production, which requires two major components: 1) a plasmid and 2) the host cell.
The Plasmid: This is the vehicle that allows us to integrate our gene of interest into an expression system.
A recommended model is the pET plasmid system.
It primarily consists of a promotor (T7) that is activated in a regulated manner (by adding IPTG).
I will use restriction enzymes for cloning or Gibson Assembly technology to insert my sequence into the plasmid.
To purify this protein, specific labels are used, such as a His-tag.
The Cell (E. coli): For my gene to be converted into Protein E within E. coli, it first needs to be transcribed into mRNA.
This process utilizes a T7 RNA polymerase, which reads the sequences and generates a complementary strand (mRNA).
Codon optimization will ensure that the message structure is stable and efficient.
Next is the translation of mRNA into protein, which occurs in the ribosomes.
The ribosome attaches to the mRNA and reads its optimized codons.
tRNAs (transfer RNA) bring the corresponding amino acids (aa), and the ribosome links them together, and then builds the amino acid chain of Protein E.
Finally: Culture and Purification: Once the bacteria has produced the protein, it must be extracted.
Using bioreactors or shake flasks, the bacteria are grown in nutrient-rich media such as LB.
To release the protein, we must sonicate to disrupt the bacterial cells and use detergents to extract it from the membrane and keep it stable.
Finally, using affinity chromatography techniques, we can purify it by using the His-tag.
This part (from 4.1 to 4.6) was done succesfully and the plasmid is shown below.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I want to sequence the E gene in hospitalized patients for more reliable biosurveillance. Unlike the Spike (S) protein, which mutates rapidly to evade immunity, the E protein is structurally essential and highly conserved. Its lower mutation rate makes it a more stable anchor for detection, ensuring consistent monitoring and fewer false negatives over time.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
To monitor the E protein gene, I’ve chosen Illumina technology due to its high accuracy and ability to process multiple patient samples simultaneously. It would allow us not only to detect its presence but also to identify specific mutations, track variants, and confirm that our “anchor” (the E gene) remains stable.
Generation
This is a second-generation technology (Next-Generation Sequencing ), as it utilizes massive amplification of fragments on a solid surface and detection via fluorescence.
Input and Preparation
The input is viral genomic DNA. Since SARS-CoV-2 is an RNA virus, we must first perform Reverse Transcription to convert its RNA into complementary DNA (cDNA), as sequencing technologies like Illumina only work with DNA.
Regarding the samples, we use nasopharyngeal swabs (where the viral load is highest) or blood samples from hospitalized patients to monitor systemic presence.
Essential Preparation Steps:
Fragmentation: Cutting the DNA into small pieces.
Adapter Ligation: Attaching sequences to the ends to fix them to the flow cell.
Indexing: Adding “barcodes” to identify each individual patient.
Enrichment PCR: Amplifying the library to ensure a detectable signal.
Essential Steps and Base Calling
Bridge PCR (Amplification): Creates “clusters” of identical copies on the flow cell.
Sequencing by Synthesis: Nucleotides with fluorescent terminators are incorporated one by one.
Base Calling: A camera captures the color of each cluster; the software translates that color (e.g., Blue = C) into a nitrogenous base and assigns a quality score.
Output
The final results are FASTQ files, which contain the sequences or reads and their respective quality values (Q-scores) for subsequent bioinformatic analysis.
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
I want to synthesize the codon-optimized gene for the SARS-CoV-2 Envelope (E) protein. While reading or sequencing allows us to monitor the virus, writing the DNA enables us to develop tools for detection.
Why synthesize the E gene?
Synthesizing this specific sequence is critical for biosurveillance for three main reasons:
Safe Diagnostics: It allows for the creation of synthetic positive controls to validate PCR kits and biosensors without the need to handle the live, infectious virus.
Immune Response Monitoring: The synthesized protein serves as a stable antigen for serological assays, helping identify if patients are developing antibodies against the most conserved regions of the virus
Analysis of Structural Stability: Having the pure protein enables structural studies (such as cryo-EM) to predict how potential mutations might impact viral integrity.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
To produce the Protein E gene, I would use silicon microchip-based DNA synthesis, mainly because this technology allows for the simultaneous writing of thousands of genes with high precision.
Its essential Steps are:
Digital Design: Fragmentation of the gene E sequence into shorter oligonucleotides.
Chemical Printing: Deposition of phosphoramidites via inkjet printing onto the silicon chip.
Elongation Cycle: Base-by-base construction (Coupling, Oxidation, Deprotection).
Assembly: Joining short fragments using PCR or Gibson Assembly to form the full-length gene.
Verification: Final quality control using NGS to guarantee the sequence is 100% accurate.
Limitations:
Speed: Delivery times can extend to weeks due to logistics and rigorous quality control steps.
Accuracy: The risk of errors increases proportionally with sequence length, requiring costly error-correction processes.
Scalability: Excellent for massive projects, but less efficient in terms of cost and time if only a single, isolated gene is required.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would like to edit the gene encoding the Cas13a protein to optimize its affinity for the guide RNA (gRNA), thereby creating a hypersensitive variant with enhanced detection capabilities.
gRNA Source: The gRNAs used in this system will be derived from a preliminary scan of suspicious sequences, specifically those exhibiting signatures of genetic engineering or a “human accent” (such as the use of artificially optimized codons). This allows the detection to be targeted toward synthetic threats created by malicious actors.
Why: In biosecurity, it is important to detect these sequences at extremely low concentrations. By editing Cas13a, it would improve its binding capacity and the collateral cleavage signal, enabling rapid detection upon gRNA recognition.
Application: This would facilitate the development of a rapid diagnostic kit to identify modified pathogens or synthetic threats in the field, eliminating the need for complex laboratory infrastructure.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use Prime Editing to perform these DNA edits. This technology is often referred to as a “genetic word processor” because it allows for high-precision search-and-replace capabilities without requiring double-strand breaks.
How it works and Essential Steps:
Prime Editing uses a specialized protein complex and a unique guide RNA to rewrite the target sequence:
Targeting: The pegRNA (prime editing guide RNA) directs the nCas9-RT (a fusion of nickase Cas9 and Reverse Transcriptase) to the specific site in the Cas13a gene.
Nicking: The nCas9 nicks only one strand of the DNA.
Reverse Transcription: The Prime Editor uses the "tail" of the pegRNA as a template to synthesize new DNA (containing the desired mutation) directly into the target site.
Flap Resolution: The cell’s natural repair machinery removes the old DNA strand and incorporates the new edited sequence, which permanently changes the Cas13a gene.
Preparation and Input:
Design Steps: The process involves designing a specific pegRNA that contains both the targeting sequence and the search-and-replace template for the desired Cas13a mutations. A secondary nick-gRNA is also designed to increase editing efficiency.
Input:
Plasmid/mRNA: Encoding the Prime Editor protein (nCas9-RT fusion).
pegRNA & nick-gRNA: Synthesized RNA or DNA templates for their expression.
Host Cells: For example, a production strain of E. coli or a yeast system where the modified Cas13a gene will be hosted and expressed.
Limitations:
Efficiency: Prime Editing can be less efficient (lower percentage of successfully edited cells) than traditional CRISPR-Cas9, particularly in certain cell types.
Complexity: Designing effective pegRNAs is more complex than designing standard gRNAs and often requires multiple rounds of optimization to achieve the desired hypersensitivity in the protein.
Week 3 HW: Lab Automation
Important
HTGAA | Question
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Workflow for the testing of SARS-CoV-2 using Opentrons OT-2 robots
Villanueva-Cañas et al. (PLOS ONE, 2021) describe the setup of a full SARS-CoV-2 RT-qPCR testing workflow using Opentrons OT-2 robots. This allowed them to automate what would otherwise be manual pipetting. Instead, they automated the critical steps and connected them like a lab “production line”: multiple OT-2 units to prepare plates and reagents, a system to extract RNA, and then a thermocycler to run the PCR.
What’s valuable (and novel) is that beyond simply “putting a robot in charge of pipetting,” they developed open-source Python software to make the process robust and traceable (including smart liquid handling, pipetting height control, volume control, and logging), and they show it can process 96-sample plates in a few hours with results comparable to commercial platforms. In short: they demonstrate that accessible, open automation can support a real, high-impact biological application—clinical diagnostics at scale.
Reference: Villanueva-Cañas et al. (2021) Implementation of an open-source robotic
platform for SARS-CoV-2 testing by real-time RT-PCR. PLoS ONE 16(7):e0252509
Important
HTGAA | Question
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
An Automated Agnostic Platform for the Detection of Synthetic DNA Signatures via Genomic Foundational Models and E-CRISPR Biosensors
My proposal is focused on the design of a biosecurity platform that integrates Artificial Intelligence and CRISPR biosensors to detect human-engineered DNA sequences in the environment. Its goal is to identify synthetic biological threats based on engineering patterns, even if the pathogen is entirely new or unknown.
Below are two methodological steps of the workflow that could be implemented using Lab Automation:
Evasion Stress-Test (via Ginkgo Bioworks)
This involves utilizing Ginkgo’s DNA “foundries” to create synthetic sequences specifically designed to attempt to deceive the AI. By manufacturing these genomic “disguises,” we can train the model to be significantly more resilient against potential adversarial bypass attempts.
Diagnostic Robotization (via Opentrons)
This stage focuses on programming a robot to prepare wastewater samples and perform biosensor assays automatically (this is the component where, if synthetic sequences are detected, a field diagnostic system is deployed). This eliminates human error and ensures the system can detect minute DNA concentrations (less than 100 copies) with consistent precision.
Environmental Sample Workflow for Synthetic Signature Detection
The following describes a potential automated workflow for processing environmental samples to identify synthetic signatures or sequences:
Reaction Preparation (Echo 525 & Bravo):
The Echo 525 transfers ultra-low volumes (nanoliters) of the 10 specific gRNAs and activators designed in Phase 2 into the destination plate.
Then, the Bravo performs the “stamping” of the Master Mix containing the Cas13a enzyme and RPA pre-amplification components into all wells of a 384-well plate.
Detection Initiation (Multiflo & PlateLoc):
The Multiflo rapidly dispenses the pre-processed environmental sample extract into all wells to initiate the molecular recognition reaction.
Immediately, the PlateLoc thermally seals the plate to prevent micro-droplet evaporation and cross-contamination from DNA aerosols.
Incubation and Activation (Inheco):
The plate is transferred to the Inheco module for a controlled incubation at 37°C. In this stage, the Cas13a—upon identifying an “engineering signature”—is activated and begins cleaving the molecular reporters.
Readout and Confirmation (XPeel & PHERAstar):
Following incubation, the XPeel automatically removes the seal.
Finally, the PHERAstar FSX performs a fluorescence readout (or electrochemical signal if the sensor is integrated) to quantify the presence of synthetic sequences. Data is sent directly to the foundational model to confirm if the signal corresponds to a real threat.
Week 4 HW: Protein Design Part I
Important
HTGAA | Part A: Protein Analysis and Visualization
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:.
Briefly describe the protein you selected and why you selected it
How long is it? What is the most frequent amino acid?.
SOD1 Description
Characteristic
Number
AA Length
53
Most Frequent AA
Glycine (25X)
Protein Homologs
250
The SOD1 family
The protein Superoxide dismutase [Cu–Zn] (SOD1), UniProt P00441, belongs to the Cu/Zn superoxide dismutase family, a group of antioxidant enzymes widely conserved across species. Members of this family are characterized by a Greek-key β-barrel fold, the coordination of copper and zinc ions at the active site, and a functional homodimeric structure. Their primary role is to protect cells from oxidative stress by catalyzing the conversion of superoxide radicals into molecular oxygen and hydrogen peroxide.
Important
HTGAA | Part D. Group Brainstorm on Bacteriophage Engineering
One-page engineering proposal outlining your computational toolkit, its strategic justification, potential functional pitfalls, and a structured pipeline schematic:
L PROTEIN ENGINEERING PROPOSAL
Two goals addressed in this proposal: increased stability and higher lysis toxicity. These are not treated as separate objectives, they are coupled and pursued through a unified strategy.
The central hypothesis is that of conditional stability: the L protein can be engineered to be more resistant to host proteolytic degradation before membrane insertion, while at the same time more efficient at cooperative oligomerization after insertion. A more stable pre-insertion L survives longer in the bacterial cytoplasm; a better-oligomerizing post-insertion L generates larger membrane lesions more rapidly. Both goals are therefore addressed by the same logic.
Biological Background
A brief summary of what makes L protein unusual, and what makes it difficult to engineer:
L is not a typical lysis protein. Most phage lysis proteins work by blocking bacterial cell wall construction. L does not do that. Instead, it inserts into the inner membrane and assembles into clusters that physically tear it apart (Mezhyrova et al., 2023). The mechanism is closer to a membrane demolition event than a biochemical inhibition.
A bacterial helper protein is required. The host chaperone DnaJ is obligatorily required for L-mediated lysis. When DnaJ carries the P330Q mutation, lysis is completely blocked (Chamakura et al., 2017). The region of L that interacts with DnaJ is therefore treated as a hard design constraint, it cannot be altered.
The gene encodes three proteins simultaneously. The DNA sequence of L overlaps with the coat protein gene (in the +1 reading frame) and with the start of the replicase gene (in frame 0). This means most nucleotide changes that would be desirable in L would simultaneously break one of the other two proteins. Only a small subset of mutations is experimentally accessible.
Computational Approach
The goal of this proposal is to use a series of AI-based computational tools to design improved variants of L before any experimental work be carried out.
Step 1. Structural reference model
The goal of this step is to establish a reliable 3D model of L in its membrane context. AlphaFold3 and Boltz-1 will be used to predict the structure of L in membrane configuration. Per-residue confidence scores (pLDDT) will be evaluated to distinguish structurally reliable regions (the N-terminal cytoplasmic domain: aa ~1–35) from uncertain regions, the C-terminal transmembrane domain (aa ~36–75). Only high-confidence regions will be used as fixed design constraints. Low-confidence regions will be treated as designable positions.
Step 2. Evolutionary landscape mining
The goal of this step is to learn which parts of L can be changed and which cannot. FoldSeek will be used to identify structural homologs of L across Leviviridae and Alloleviviridae phage families. These homologs will then be processed by SaProt, a protein language model that encodes both sequence and structural information. The result is a per-domain map of conserved positions (those functionally critical, not to be mutated) versus variable positions (candidates for redesign). SaProt is preferred over sequence-only models such as ESM-2 for this task because it captures structural context, which is particularly important for transmembrane regions.
Step 3. Generative sequence design
The goal of this step is to generate thousands of candidate L variants that satisfy all design constraints. ProteinMPNN is applied in two parallel layers:
Layer A (N-terminal domain): sequences will be designed to increase thermodynamic stability in solution, with the DnaJ-binding interface and the LS (leu-ser) motif masked as immutable constraints.
Layer B (transmembrane domain): sequences will be designed with symmetry constraints (C₂/C₃) to maximize cooperative oligomerization interfaces in a lipid bilayer environment, since oligomerization is the direct cause of membrane disruption and cell death.
All candidates are then passed through a dual-codon overlap filter, i. e. a dynamic programming algorithm that retains only those nucleotide substitutions that produce the desired amino acid change in L (frame +1) while leaving the coat protein sequence unchanged (frame 0) and preserving the replicase reading frame. Candidates that fail this filter will be discarded regardless of their predicted fitness.
Step 4. Multi-metric fitness scoring
The goal of this step is to rank surviving candidates by three independent fitness metrics:
ESM-2 log-likelihood: a global evolutionary plausibility score
SaProt fitness score: the primary metric for the transmembrane domain, incorporating structural context
Evo (Arc Institute, 2024): a genomic language model trained on millions of phage genomes, will be used to score the full MS2 ssRNA genome after each mutation, penalizing any disruption to the replicase operator, CP initiation site, ribosomal pause sites, or RNA packaging signals
Only candidates ranking in the top 5% across all three metrics simultaneously will be advanced.
Step 5. DnaJ interface validation
The goal of this step is to confirm that top candidates preserve the obligatory interaction with DnaJ. Boltz-2 will be used to model the L::DnaJ complex and predict binding affinity for each candidate. Candidates will be retained only if their predicted binding affinity does not decrease by more than 0.5 kcal/mol relative to wild-type L, and if their interface confidence score (ipTM) remains at or above 90% of the wild-type value.
Potential Pitfalls
*Pitfall 1. Increased stability may reduce membrane insertion efficiency.
A more thermodynamically stable L protein may present a higher energy barrier for membrane insertion, potentially reducing rather than increasing lytic activity. This is an major risk because the tools used to predict folding stability (RosettaΔΔG, ThermoMPNN) model stability in aqueous solution, not insertion energy into a lipid bilayer. This risk will be mitigated by restricting stability design to the N-terminal soluble domain only, leaving the transmembrane domain free to insert. Molecular dynamics simulations in an explicit lipid bilayer (POPE:POPG 3:1, CHARMM-GUI/GROMACS) are planned for top candidates to validate insertion energy before any synthesis is ordered.
*Pitfall 2. The genomic overlap constraint drastically reduces the accessible design space.
Because the L gene is encoded in the same DNA as the coat protein and replicase, most amino acid changes that would be desirable in L are simply not achievable without breaking another gene. In practice, the dual-codon overlap filter is expected to eliminate the vast majority of computationally generated candidates. potentially leaving only a few dozen experimentally accessible variants out of tens of thousands generated. This is an inherent limitation of the MS2 genome architecture, not a failure of the computational approach. It is addressed by applying the overlap filter as early n the pipeline to avoid scoring inaccessible designs.
Week 5 HW: Protein Design Part II
Important
HTGAA | Part A: SOD1 Binder Peptide Design (From Dr Pranam Chatterjee)
Retrieve the human SOD1 sequence (P00441) from UniProt, introduce the A4V mutation, use the PepMLM Colab to generate four 12–amino acid peptides conditioned on the mutant sequence, include the known binder FLYRWLPSRRGG for comparison, and record the resulting perplexity scores to assess model confidence.
Peptides generated using PepMLM (Target Sequence-Conditioned Generation of Peptide Binders via Masked Language Modeling)
Binder
Pseudo Perplexity
HHSPAVAAEHGK (B1)
11.3666
WLYPATAAALKE (B2)
9.7318
WHYGPVGARHKX (B3)
9.0100
WHYGPVAVEWWE (B4)
18.0932
FLYRWLPSRRGG (Control)
For Comparison
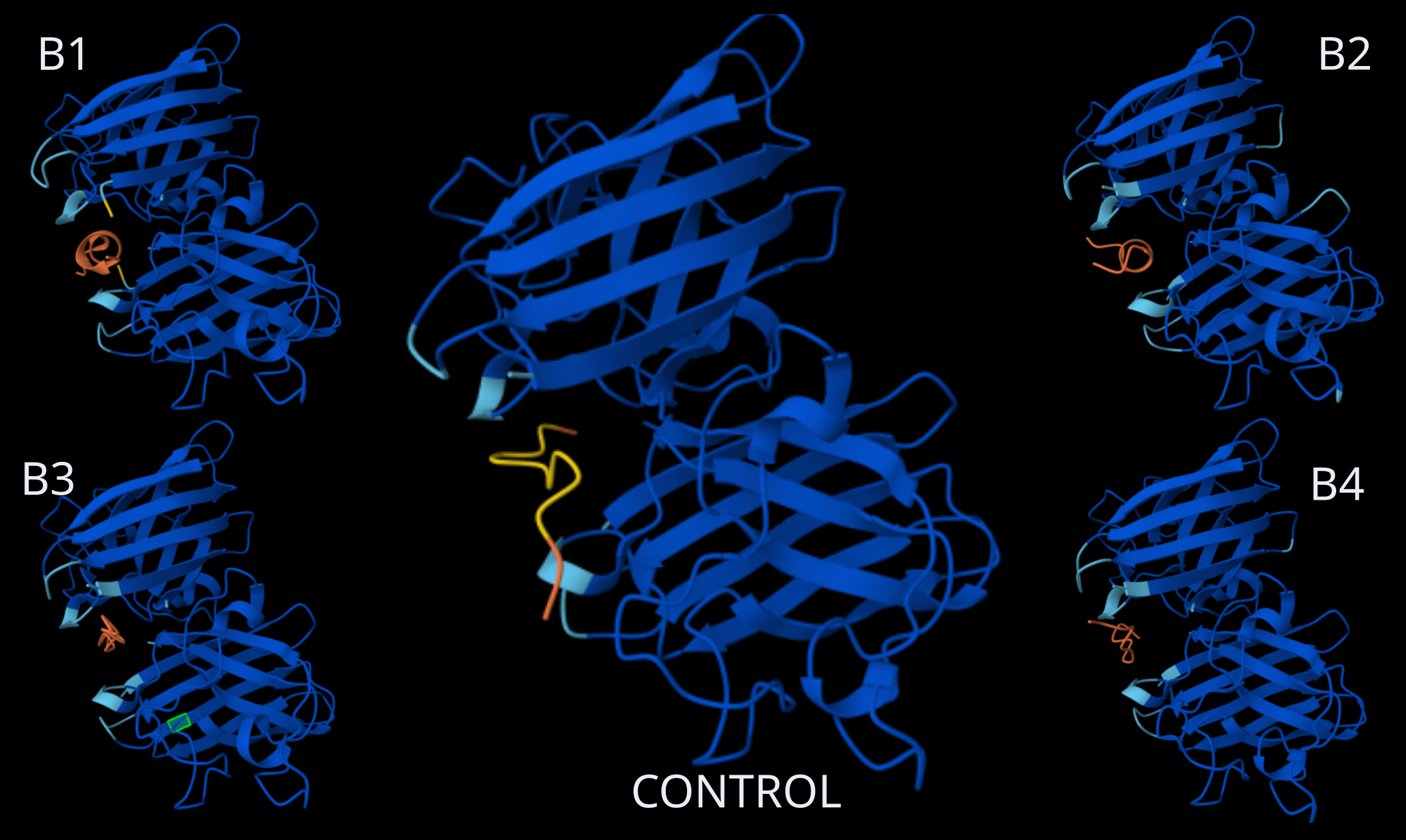
Homodimer Sod1 + Binder Peptides, including Control Peptide (Source: AlphaFold)
Binding Peptides ipTM scores
Binder
ipTM score
HHSPAVAAEHGK (B1)
0.87
WLYPATAAALKE (B2)
0.77
WHYGPVGARHKX (B3)
0.86
WHYGPVAVEWWE (B4)
0.82
FLYRWLPSRRGG (Control)
0.89
Interaction Analysis and Binding Sites in SOD1
Looking at the AlphaFold3 structural predictions (see image above) for the five binder peptides (B1-B4 and the Control) and analyzing their confidence metrics (ipTM), all of the 12-amino acid peptides show a strong tendency to converge on the exact same binding pocket.
Where does the peptide appear to bind?
All the peptides (in orange, with the control in yellow) position themselves right in the central cleft or groove formed between the two SOD1 monomers (in blue). This binding site is highly conserved across all five predictions, suggesting a strong structural affinity for this specific pocket.
Does it localize near the N-terminus where A4V sits?
Yes. In the SOD1 structure, the N-terminus (where the Alanine to Valine mutation occurs at position 4) is a critical component of the dimerization interface. Since the peptides anchor right at the epicenter of the junction between the two monomers, they are located in the immediate vicinity of both the N-terminus and the A4V mutation site.
Does it engage the β-barrel region or approach the dimer interface?
Its main feature is that it directly targets the dimer interface. To do this, the peptide inevitably interacts with the outer strands of the β-barrels from both subunits flanking that interface. The peptide essentially acts like a wedge between the two β-structures.
Does it appear surface-bound or partially buried?
It appears partially buried. The peptides aren’t just resting on a convex, fully solvent-exposed surface; rather, they are tucked firmly inside the concave cavity of the dimer interface.
Notes on ipTM Scores
The ipTM metrics strongly back up what we are seeing visually. According to AlphaFold3 standards, an ipTM > 0.8 points to a high-quality, confident prediction for the protein-protein interface:
The Control (0.89), B1 (0.87), and B3 (0.86) show highly reliable interfaces, suggesting they fit very stably into that dimer interface cavity.
B2 (0.77), however, falls into the ‘gray zone’ (0.6 - 0.8). While AlphaFold3 places it in the same spot, we have to be more cautious here: the prediction is in a range where it could be correct or incorrect. The interaction of the WLYPATAAALKE sequence seems suboptimal compared to the others.