Week 5 HW: Protein Design Part II
Important
HTGAA | Part A: SOD1 Binder Peptide Design (From Dr Pranam Chatterjee)
Retrieve the human SOD1 sequence (P00441) from UniProt, introduce the A4V mutation, use the PepMLM Colab to generate four 12–amino acid peptides conditioned on the mutant sequence, include the known binder FLYRWLPSRRGG for comparison, and record the resulting perplexity scores to assess model confidence.
Peptides generated using PepMLM (Target Sequence-Conditioned Generation of Peptide Binders via Masked Language Modeling)
| Binder | Pseudo Perplexity |
|---|---|
| HHSPAVAAEHGK (B1) | 11.3666 |
| WLYPATAAALKE (B2) | 9.7318 |
| WHYGPVGARHKX (B3) | 9.0100 |
| WHYGPVAVEWWE (B4) | 18.0932 |
| FLYRWLPSRRGG (Control) | For Comparison |
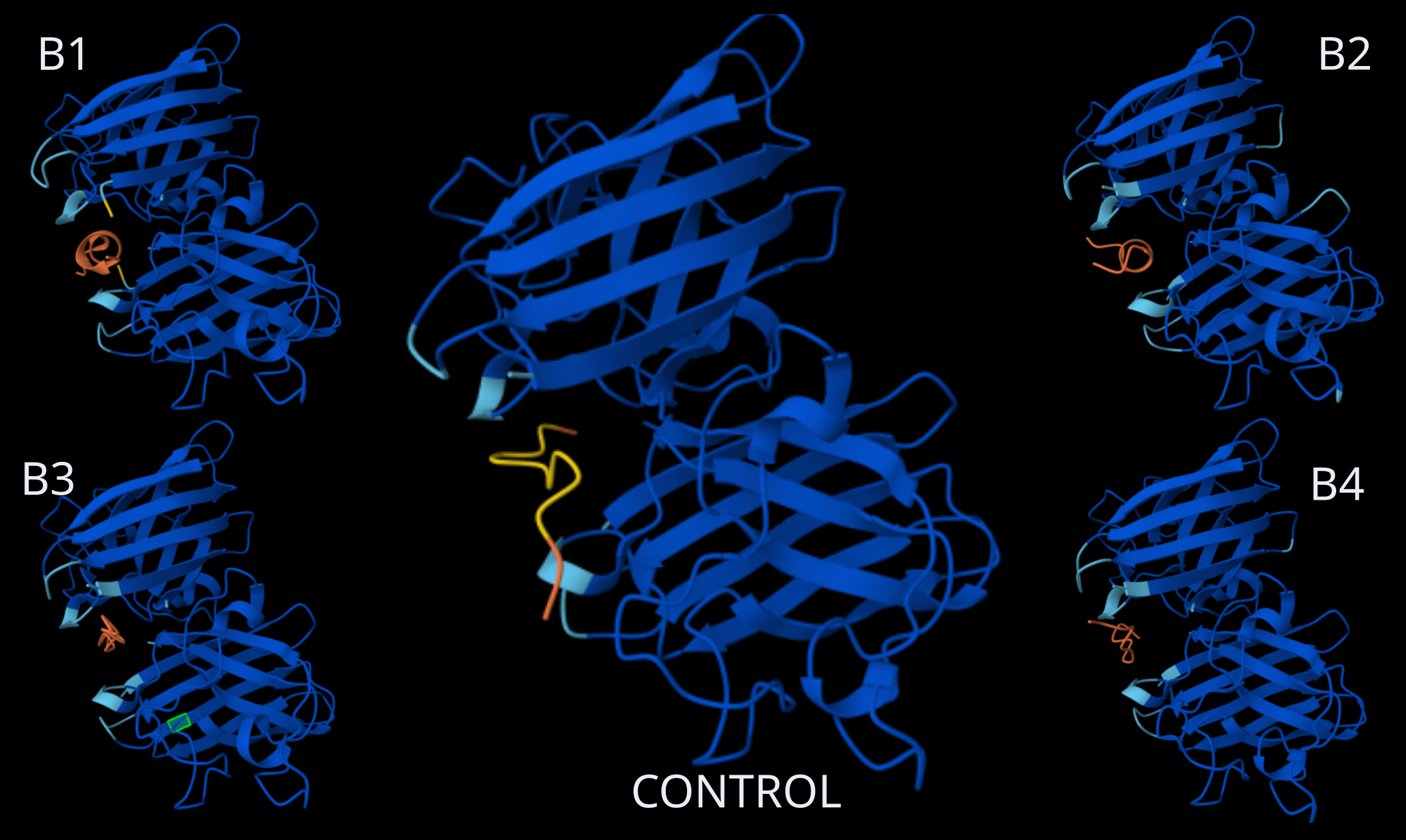
Homodimer Sod1 + Binder Peptides, including Control Peptide (Source: AlphaFold)
Binding Peptides ipTM scores
| Binder | ipTM score |
|---|---|
| HHSPAVAAEHGK (B1) | 0.87 |
| WLYPATAAALKE (B2) | 0.77 |
| WHYGPVGARHKX (B3) | 0.86 |
| WHYGPVAVEWWE (B4) | 0.82 |
| FLYRWLPSRRGG (Control) | 0.89 |
| Interaction Analysis and Binding Sites in SOD1 |
|---|
Looking at the AlphaFold3 structural predictions (see image above) for the five binder peptides (B1-B4 and the Control) and analyzing their confidence metrics (ipTM), all of the 12-amino acid peptides show a strong tendency to converge on the exact same binding pocket.
Where does the peptide appear to bind? All the peptides (in orange, with the control in yellow) position themselves right in the central cleft or groove formed between the two SOD1 monomers (in blue). This binding site is highly conserved across all five predictions, suggesting a strong structural affinity for this specific pocket.
Does it localize near the N-terminus where A4V sits? Yes. In the SOD1 structure, the N-terminus (where the Alanine to Valine mutation occurs at position 4) is a critical component of the dimerization interface. Since the peptides anchor right at the epicenter of the junction between the two monomers, they are located in the immediate vicinity of both the N-terminus and the A4V mutation site.
Does it engage the β-barrel region or approach the dimer interface? Its main feature is that it directly targets the dimer interface. To do this, the peptide inevitably interacts with the outer strands of the β-barrels from both subunits flanking that interface. The peptide essentially acts like a wedge between the two β-structures.
Does it appear surface-bound or partially buried? It appears partially buried. The peptides aren’t just resting on a convex, fully solvent-exposed surface; rather, they are tucked firmly inside the concave cavity of the dimer interface.
Notes on ipTM Scores
The ipTM metrics strongly back up what we are seeing visually. According to AlphaFold3 standards, an ipTM > 0.8 points to a high-quality, confident prediction for the protein-protein interface:
The Control (0.89), B1 (0.87), and B3 (0.86) show highly reliable interfaces, suggesting they fit very stably into that dimer interface cavity.
B2 (0.77), however, falls into the ‘gray zone’ (0.6 - 0.8). While AlphaFold3 places it in the same spot, we have to be more cautious here: the prediction is in a range where it could be correct or incorrect. The interaction of the WLYPATAAALKE sequence seems suboptimal compared to the others.